
The new phthalide derivative 3-[2′(R)-hydroxybutyl]-7-hydroxyphthalide (2) and the five known compounds (−)-3-butyl-7-hydroxyphthalide (1), isopatulin (3), m-hydroxybenzyl alcohol (4), cyclopenin (5), and cyclopeptine (6) were isolated from the marine isolate of the fungus Penicillium claviforme associated with the seagrass Zostera marina. The structures of the compounds were established using NMR spectroscopy and mass spectrometry. The absolute configuration of the C-2′ asymmetric center was determined using a modified Mosher method.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Recent research indicates that fungal micromycetes isolated from marine sources are of interest as producers of biologically active compounds [1–3]. In continuation of the search for secondary metabolites in extracts of marine isolates of mycelial fungi, we investigated strain Penicillium claviforme that was isolated from the surface of the seagrass Zostera marina (Peter the Great Gulf, Sea of Japan). Herein we present data for the isolation and structure identification of the new phthalide derivative 3-[2′(R)-hydroxybutyl]-7-hydroxyphthalide (2) and the five known compounds (−)-3-butyl-7-hydroxyphthalide (1), isopatulin (3), m-hydroxybenzyl alcohol (4), cyclopenin (5), and cyclopeptine (6).
The fungus was cultivated for 21 d in rice medium at 22°C. The dry EtOAc extract of the culture was separated sequentially over a column of SiO2 and then by reversed-phase HPLC to afford pure 1–6.
A comparison of mass spectrometric and PMR spectroscopic data for 1 (see Experimental) with the literature allowed its structure to be identified possibly as 3-butyl-7-hydroxyphthalide, which was isolated earlier from the soil fungus Penicillium vulpinum [4]. The structure of 1 was confirmed by an x-ray structure analysis (XSA) of a single crystal grown in hexane (Fig. 1, Table 1). The absolute configuration of C-3 as S was established by comparing the specific rotation angle of 1 with that of (−)-3-butyl-7-hydroxyphthalide [5] and paecilocin A isolated from the marine fungus Paecilomyces variotii [6].

Crystallographic asymmetric unit of 1 with atomic numbering.
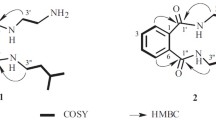
The ESI-MS of 2 contained a peak for the protonated molecule with m/z 223 [M + H]+. The molecular formula of 2 was defined as C12H14O4 based on ES-TOF high-resolution mass spectra and was confirmed by analyzing 13C NMR spectra. The PMR and 13C NMR spectra of 2 and DEPT and HSQC experiments showed that the molecule included one methyl (δH 0.88, δC 9.9), two methylenes [δH 1.91 (1H), 1.52 (1H), 1.41 (2H); δC 42.8, 30.6], and five methines, two of which were bonded to oxygen (δH 3.65, 5.57; δC 67.9, 77.1) and three of which were involved in a conjugated double-bond system (δH 6.87, 6.95, 7.51; δC 115.6, 112.2, 136.0). In addition, there were three quaternary C atoms on double bonds (δC 111.0, 153.1, 156.8) and one carbonyl C atom (δC 168.0) (Table 2).
HMBC correlations of H-3 (δ 5.57)/C-1 (δ 168.0), C-4a (153.1); H-4 (δ 6.95)/C-3 (δ 77.1), C-4a, C-6 (δ 115.6), C-7a (δ 111.0); H-5 (δ 7.51)/C-4a, C-7 (δ 156.8); and H-6 (δ 6.87)/C-1, C-4 (δ 112.2), C-7a and a comparison of the NMR spectra of 2 with those of 1 indicated that 2 contained a substituted 7-hydroxyphthalide moiety. The COSY and HSQC spectra were consistent with the proton sequence –CHO(3)–CH2(1′)–CHO(2′)–CH2(3′)–CH3(4′) in 2. These data and HMBC correlations of H-3 (δ 5.57)/C-1′ (δ 42.8), C-2′ (δ 67.9); H2-1′ (δ 1.92, 1.52)/C-3 (δ 77.1), C-4a (δ 153.1), C-3′ (δ 30.6); 2′-OH (δ 4.8)/C-1′, C-2′, C-3′; and H2-3′ (δ 1.41)/C-1′, C-2′, C-4′ (δ 9.9) were consistent with a 2′-hydroxybutyl group in the hydroxyphthalide 3-position. Thus, the full structure of 2 was elucidated. The compound was called 3-[2′-hydroxybutyl]-7-hydroxyphthalide. A modified Mosher method was used to determine the configuration of the C-2′ asymmetric center [7]. Esterification of 2 by (R)- and (S)-MTPA acid chlorides at the C-2′ hydroxyl formed the (S)- and (R)-MTPA esters 2a and 2b, respectively. The differences ∆δ S-R in chemical shifts in PMR spectra of the esters (Table 2, Fig. 2) were indicative of the 2′(R)-configuration.

Δδ S-R values for MTPA-esters of 2.
A comparison of the PMR and 13C NMR spectral data with the literature identified 3 and 4 as isopatulin and m-hydroxybenzyl alcohol; 5 and 6, as cyclopenin and cyclopeptine. Compounds 3 and 4 were isolated earlier from the terrestrial fungus P. vulpinum [8]; 5, from the terrestrial fungus Penicillium simplicissimum [9]; and 6, from the terrestrial fungus Penicillium cyclopium [10].
Thus, strain P. claviforme KMM 4665 represented a new source of the second metabolites with various structures. Compounds 1–6 were isolated for the first time from a fungal marine isolate.
EXPERIMENTAL
A low-temperature [200(2) K] XSA was performed on a Bruker Smart APEX2 diffractometer using the standard procedure (Mo Kα-radiation, graphite monochromator). Absorption of x-rays in the sample was calculated using equivalent reflections. Table 1 lists the crystallographic data and principal structure refinement parameters of 1.
The structure was determined by direct methods and refined by anisotropic least-squares method for non-hydrogen atoms. H atoms of the water molecule and carboxylic acid were located in a difference electron-density synthesis and refined with thermal parameters equal to 1.5 Ueq(O). Other H atoms were placed in geometrically calculated positions. Data collection and processing and refinement of unit-cell constants used the APEX2 programs [11]. All calculations for the structure determination and refinement were performed using the SHELXTL/PC programs [12].
A CIF file containing complete information about the structure of 1 was deposited in the CCDC under No. 1015369, from where it can be obtained for free by a request to the internet site www.ccdc.cam.ac.uk/data_request/cif.
Optical rotation was measured on a PerkinElmer 343 polarimeter (Germany). UV spectra were recorded on a Shimadzu UV-1601 PC spectrophotometer (Japan). Circular dichroism spectra were recorded on a Chirascan plus spectropolarimeter (Applied Photophysics, UK). PMR and 13C NMR spectra were measured vs. Me4Si internal standard on a Bruker DRX-700 spectrometer (700 and 175 MHz, respectively). Mass spectra were obtained on an Agilent 6510 Q-TOF LC mass spectrometer. Preparative HPLC was performed on a Beckman-Altex chromatograph (USA) over a Supelco Discovery C-18 column (250 × 4.6 mm, 5 μm). Column chromatography used silica gel L (40/100 μm, Chemapol, Czechoslovakia). TLC used plates with a fixed silica gel layer (5–17 μm, Sorbfil, Russia).
Cultivation of Fungus. The fungus P. claviforme was isolated from the surface of seagrass Z. marina and identified based on morphological signatures by Cand. Biol. Sci. N. N. Kirichuk, Pacific Institute of Bioorganic Chemistry (PIBOC). The strain was preserved in the Collection of Marine Microorganisms, PIBOC (Vladivostok, Russia) under the code KMM 4665. The fungus was cultivated on solid medium for 21 d at 22°C in 50 500-mL Ehrlenmeyer flasks, each of which contained medium consisting (g) of rice (15.0), KH2PO4 (0.001), MgSO4∙7H2O (0.001), sodium tartrate (0.001), FeSO4∙7H2O (0.0001), and sea water (20 mL).
Extraction and Isolation of Compounds. Fungus mycelium with medium was extracted with EtOAc (2 × 1.0 L). The extract was evaporated. The dry residue (5.5 g) was dissolved in EtOH–H2O (1:4). The resulting solution was extracted with hexane (3 × 100 mL) and EtOAc (3 ×100 mL). The EtOAc extract was evaporated to dryness. The resulting residue (4.0 g) was chromatographed over a column (5 × 12 cm) of silica gel using hexane–EtOAc (stepwise gradient, 1:0→2:1). Elution of the dry residue with hexane–EtOAc (95:5) produced fraction A (110 mg); by hexane–EtOAc (83:17), fraction B (265 mg); and by hexane–EtOAc (75:25), fraction C (130 mg). Crystallization of fraction A from hexane afforded 1 (100 mg). Fractions B and C were separated by reversed-phase HPLC over a Supelco Discovery C-18 column using MeOH–H2O (55:45). Fraction B yielded 2 (1.7 mg), 3 (2.7 mg), and 4 (3.6 mg); fraction C, 5 (1.7 mg) and 6 (1.7 mg).
3( S )-Butyl-7-hydroxyphthalide (1), white amorphous crystals, [α]D 20 –39.0° (c 0.10, MeOH). UV spectrum (0.01 mg/mL, CH3OH, λmax, nm) (log ε): 209 (3.36), 234 (2.75), 300 (2.51). CD spectrum (0.01 mg/mL, CH3OH, λmax) (Δε): 205 (−0.25), 215 (+0.82), 230 (−0.23), 238 (+0.88), 250 (−0.18), 299 (+0.53). 1H NMR spectrum (700 MHz, DMSO-d6, δ, ppm, J/Hz): 10.63 (1H, s, 7-OH), 7.51 (1H, t, J = 7.8, H-5), 6.96 (1H, d, J = 7.8, H-4), 6.87 (1H, d, J = 7.8, H-6), 5.44 (1H, dd, J = 4.0, 7.8, H-3), 1.98 (1H, m, H-1′a), 1.63 (1H, m, H-1′b), 1.34 (2H, m, H-2′), 1.31 (2H, m, H-3′), 0.85 (3H, t, J = 7.3, H-4′). 13C NMR spectrum (175 MHz, DMSO-d6, δ, ppm): 168.0 (C-1), 156.8 (C-7), 152.4 (C-4a), 136.8 (C-5), 115.6 (C-6), 112.3 (C-4), 111.3 (C-7a), 79.5 (C-3), 33.8 (C-1′), 26.5 (C-2′), 21.9 (C-3′), 13.8 (C-4′). ESI-MS, m/z 206 M+, C12H14O3.
3-[2′( R )-Hydroxybutyl]-7-hydroxyphthalide (2), white amorphous crystals, [α]D 20 +7.0° (c 0.10, MeOH). UV spectrum (0.01 mg/mL, CH3OH, λmax, nm) (log ε): 207 (3.40), 234 (2.74), 300 (2.53). CD spectrum (0.01 mg/mL, CH3OH, λmax) (Δε): 204 (−0.95), 214 (+0.64), 230 (+0.22), 238 (+0.55), 250 (−0.17), 298 (+0.22). ESI-HR-MS, m/z 223.0970 [M + H]+ (calcd for C12H15O4 223.0964). Table 2 lists the NMR spectra of 2.
Preparation of ( S )- and ( R )-MTPA Esters of 3-[2′-Hydroxybutyl]-7-hydroxyphthalide (2a and 2b). A solution of 2 (0.8 mg) in Py was treated with several crystals of 4-dimethylaminopyridine and (R)-MTPA-Cl (20 μL) and stirred for 1 h at room temperature. The course of the reaction was monitored by TLC. The solvent was evaporated. The residue was chromatographed over silica gel using hexane–EtOAc (20:1) to afford (S)-MTPA ester 2a (1.1 mg). (R)-MTPA ester 2b (1.2 mg) was synthesized by the same method using (S)-MTPA-Cl. Table 2 presents the PMR spectra of 2a and 2b.
References
J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro, and M. R. Prinsep, Nat. Prod. Rep., 30, 237 (2013).
O. I. Zhuravleva, M. P. Sobolevskaya, E. V. Leshchenko, N. N. Kirichuk, V. A. Denisenko, P. S. Dmitrenok, S. A. Dyshlovoy, A. M. Zakharenko, N. Y. Kim, and Sh. Sh. Afiyatullov, J. Nat. Prod., 77, 1390 (2014).
M. E. Rateb and R. Ebel, Nat. Prod. Rep., 28, 290 (2011).
M. Makino, T. Endoh, Y. Ogawa, K. Watanabe, and Y. Fujimoto, Heterocycles, 48 (9), 1931 (1998).
T. Ohzeki and K. Mori, Biosci. Biotechnol. Biochem., 67 (10), 2240 (2003).
J. Liu, F. Li, E. L. Kim, J. L. Li, J. Hong, K. S. Bae, H. Y. Chung, H. S. Kim, and J. H. Jung, J. Nat. Prod., 74, 1826 (2011).
T. Kuzumi, T. Ooi, Y. Ohkubo, and T. Yabuuchi, Bull. Chem. Soc. Jpn., 79, 965 (2006).
A. Mikami, T. Okazaki, N. Sakai, T. Ichihara, K. Hanada, and K. Mizoue, J. Antibiot., 49 (10), 985 (1996).
M. Kusano, H. Koshino, J. Uzawa, S. Fujioka, T. Kawano, and Y. Kimura, Biosci. Biotechnol. Biochem., 64 (12), 2559 (2000).
J. Framm, L. Nover, A. El Azzouny, H. Richter, K. Winter, S. Werner, and M. Luckner, Eur. J. Biochem., 37, 78 (1973).
Bruker, APEX2, Bruker AXS Inc., Madison, Wisconsin, USA, 2010.
G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 64, 112 (2008).
Acknowledgment
The work was performed under the auspices of Russian Science Foundation Project No. 14-14-00030.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 1, January–February, 2015, pp. 98–101.
Rights and permissions
About this article

Cite this article
Afiyatullov, S.S., Leshchenko, E.V., Sobolevskaya, M.P. et al. New 3-[2′(R)-Hydroxybutyl]-7-Hydroxyphthalide from Marine Isolate of the Fungus Penicillium claviforme . Chem Nat Compd 51, 111–115 (2015). https://doi.org/10.1007/s10600-015-1214-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-015-1214-y