
The bacterium Paenibacillus sp. BG134 was capable of biotransforming the principal 20(S)-protopanaxadiol ginsenosides Rc, Rb2, Rd, and Rb1 into the corresponding minor glycosides C-Mc1, C-O, and F-2. The specificity of Paenibacillus sp. BG134 differed from that of several other microorganisms by cleaving only the terminal C-3 and C-20 β -D-glucose from their carbohydrate components.
Similar content being viewed by others
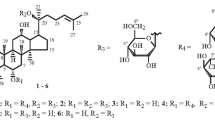
Avoid common mistakes on your manuscript.
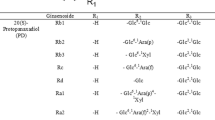
The therapeutic properties of Panax ginseng C. A. Meyer are due to a unique complex of biologically active compounds, the principal ones of which are dammarane-type triterpene glycosides or ginsenosides [1–3]. Ginsenosides exhibit immunostimulating, anti-inflammatory, anticancer, antitumor, antidiabetic, hepatoprotective, antistress, neuromodulating, and other properties [4,5]. The principal ginsenosides contained in the plant are Rc (1), Rb2 (2), Rd (3), and Rb1 (4), which have 20(S)-protopanaxadiol as the aglycon, and Re and Rg1, which are 20(S)-protopanaxatriol glycosides. These make up greater than 80% of the total ginsenosides [1,3]. The contents of P. ginseng minor glycosides such as F-2, C-O, or C-Mc1, which differ from the principal ginsenosides by fewer monosaccharides in the glycoside part, are less than tenths of a percent in most instances [2,3]. Furthermore, the minor glycosides in several instances have broader spectra of biological activity or higher activity than the principal ginsenosides. For example, ginsenosides F-2 and C-O inhibited the growth of HCT-116 and HT-29 human colorectal cancer cells [6] whereas F-2 induced apoptosis of breast cancer stem cells [7] and human stomach cancer cells [8]. Therefore, deglycosylation of the principal ginsenosides to produce related minor glycosides is considered a viable method for increasing the pharmacological activity of ginseng glycosides [9].
We reported earlier on the identification of new bacteria in soil taken from a ginseng field [10] and the use of several of them for biotransformation of the principal ginsenosides [12–14]. Strains belonging to the genus Paenibacillus were identified among the isolated bacteria [10,11]. Herein we present data on the biotransformation of the principal 20(S)-protopanaxadiol ginsenosides into minor glycosides through the action of Paenibacillus sp. BG134.
Paenibacillus sp. BG134 was isolated using the published method from a soil sample taken from a ginseng field [10]. The isolated strain BG134 was assigned to the genus Paenibacillus by comparing nucleotide sequences of the gene 16S pRNA in the GenBank2a by using published methods and programs [10,11]. The observation of β-glucosidase activity in the isolated bacterium and preliminary positive results on its ability to transform Rb1 prompted a detailed study of the action of Paenibacillus sp. BG134 on the principal ginseng glycosides. We used intact bacterial cells for the biotransformation and incubated them with the studied glycosides.
TLC and HPLC showed that compound 5 was formed by incubating the bacterium in medium with added Rc (1). The concentration of 5 increased in proportion to the incubation time whereas that of starting 1 decreased simultaneously. The Rf value on TLC (0.43) and the HPLC retention time (tR, 17.72 min) of 5 agreed with those of standard ginsenoside Mb [15], which is known more commonly by the name C-Mc1 [4]. This compound was isolated by preparative HPLC from the BuOH extract of cultivation medium after incubation for 72 h (67% yield) and was identified by spectral methods. Its mass, PMR, and 13C NMR spectra agreed fully with those of C-Mc1 that were published by us [14]. A comparison of these spectra for 1 and 5 during a study of the bacterium Sphingopyxis sp. BG97 showed that C-Mc1 was formed from Rc by cleavage of the β-D-glucose from the 20(S)-protopanaxadiol C-3 β-sophorose (Fig. 1) [14].

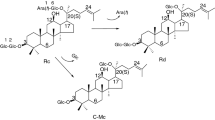
Transformation of ginsenosides Rc (1) and Rb2 (2) through the action of Paenibacillus sp. BG134.
The action of Paenibacillus sp. BG134 on Rc was similar to that described above for Sphingopyxis sp. BG97 [14] but differed from those of microorganisms such as Actinosynnema mirum [16] and Leuconostoc mesenteroides KFRI 690 [17], which transform Rc into C-Mc and C-K, respectively, through more complete deglycosylation.
Another principal glycoside, Rb2 (2), was affected analogously to the biotransformation of Rc by Paenibacillus sp. BG134. TLC and HPLC analysis showed that starting 2 (Rf 0.25, tR 14.84 min) and its transformation product 6 were present in samples taken from bacterial suspension incubated with added 2. The Rf on TLC (0.40) and HPLC retention time (tR 18.00 min) of 6 agreed with those of the standard ginsenoside known as C-O [16–19]. Like in the experiment with Rc, increasing the incubation time caused the concentration of starting 2 to decrease and the concentration of 6 to increase proportionally. The latter was isolated by preparative HPLC from the total compounds obtained by extraction of the culture medium after incubation for 72 h. It was identified by mass and NMR spectroscopy. Spectral and other characteristics of 6 agreed with those published for C-O [20]. The difference of the m/z values for the molecular ions [M + Na]+ of starting 2 (m/z 1101) and its transformation product 6 (m/z 940) corresponded to a β-D-glucose unit. This indicated glucose was cleaved from 2 during the biotransformation. The structure of 2 (Fig. 1) indicated that the cleavage should have occurred from the 20(S)-protopanaxadiol C-3 β-sophorose. In fact, the 13C NMR spectrum of 6 (Table 1) showed resonances for only three anomeric C atoms (98.1, 104.6, and 107.0 ppm). The C-3 carbohydrate C-22 resonance underwent a strong-field shift (75.9 ppm) with respect to that in the β-sophorose of 2 (83.3 ppm). This conclusion was also confirmed by the presence in PMR spectra of 6 and 2 of three resonances for anomeric protons with practically the same chemical shifts and the lack of a resonance for a fourth anomeric proton at 5.28 ppm (H-1″ in 2) in the spectrum of 6. Thus, the biotransformation product of 2 (Fig. 1) through the action of Paenibacillus sp. BG134 was C-O [20]. The deglycosylation of 2 was highly selective because no other ginsenosides were formed. Other microorganisms such as Terrabacter ginsenosidimutans [18] and A. mirum [16] are known to transform 2 into C-O. However, C-O was usually further deglycosylated to form C-Y or C-K, which hindered C-O production.
Ginsenoside Rd (3) was incubated with bacterial suspension. TLC and HPLC showed that samples taken from it contained starting 3 (Rf 0.38, tR 16.54 min) and its transformation product 7. The concentration of 3 decreased in proportion to the incubation time whereas the concentration of 7 increased proportionally. The Rf on TLC (0.64) and HPLC retention time (19.21 min) of 7 agreed with those of standard ginsenoside F-2. We confirmed this by isolating 7 using preparative HPLC of the BuOH extract of the incubation medium (74% yield) and identified it using mass and NMR spectroscopy. The spectral and other characteristics of 7 agreed fully with those of F-2 that were published by us [13]. A comparison of mass, PMR, and 13C NMR spectra of Rd and F-2, which were reported by us [13] during a study of the bacterium Sphingomonas sp. BG25, showed that the biotransformation of 3 through the action of Paenibacillus sp. BG134 involved cleavage of β-D-glucose from the C-3 β-sophorose of 3 and formation of minor glycoside F-2 (Fig. 2). Paenibacillus sp. BG134 affected 3 similarly to microorganisms such as Sphingopyxis alaskensis DSM 13593 [21] and Sphingomonas sp. 2F2 [22] but differed from aforementioned T. ginsenosidimutans [18] and A. mirum [16], which transformed 3 into C-K and ginsenoside 20(S)-Rh2, respectively.

Transformation of ginsenosides Rd (3) and Rb1 (4) through the action of Paenibacillus sp. BG134.
Thus, biotransformation of Rc, Rb2, and Rd through the action of Paenibacillus sp. BG134 involved in all instances cleavage of only β-D-glucose from the 20(S)-protopanaxadiol C-3 β-sophorose. This formed minor glycosides C-Mc1, C-O, and F-2, respectively, which did not under further deglycosylation.
Ginsenoside Rb1 (4) underwent a more complicated biotransformation pattern than the other three compounds. TLC and HPLC showed the presence in first (incubation for 36 h) and second (incubation for 72 h) samples of starting 4 (Rf 0.14, tR 13.31 min) and two transformation products 8 (Rf 0.34, tR 16.89 min) and 9 (Rf 0.64, tR 19.21 min). Furthermore, a sample taken after incubation for 6 h contained a compound that had Rf (0.38) and tR (16.54 min) values that corresponded to those of Rd. However, it was practically absent in other later samples. The accumulation dynamics of 4, 8, and 9 in the incubation medium showed that the concentration of 4 decreased in proportion to the incubation time and that of 8 increased, reaching a maximum after 30 h. Then, it gradually decreased and was accompanied by a proportional increase of the concentration of 9. This was clearly evident in samples taken from the second mixture after incubation for 30 h. The TLC Rf and HPLC tR values of 8 and 9 agreed with those of standard gypenoside XVII (Gyp XVII) and F-2. Both compounds were isolated from extracts by preparative HPLC and identified using mass and NMR spectroscopy in order to confirm this. The spectral and other characteristics of 8 agreed with those of Gyp XVII [23]; of 9, with those published by us for F-2 [23]. The yield of F-2 was 58%.
The accumulation dynamics of the ginsenosides indicated that 4 was biotransformed by Paenibacillus sp. BG134 first into 8, which was then transformed into 9. Thus, the transformation was Rb1 → Gyp XVII → F-2 (Fig. 2). This was confirmed by spectral data. The difference in the m/z values for the molecular ions [M + Na]+ of starting 4 (m/z 1131) [12] and its transformation product 8 (m/z 969) corresponded to a β-D-glucose unit. This indicated that it was cleaved from 4.
The PMR and 13C NMR spectra of 4 and 8 (Table 2) indicated that the cleavage occurred from the C-3 β-sophorose of 4. In fact, the 13C NMR spectrum of 8 contained resonances for only three anomeric C atoms (98.2, 105.4, and 107.0 ppm). The C-2′ resonance of the C-3 carbohydrate (75.8 ppm) underwent a strong-field shift with respect to that in the β-sophorose of 4 (83.5 ppm). Furthermore, the resonances of C-6‴, which formed the C-20 β-gentiobiose glycoside bond, differed in 4 and 8 by only 0.2 ppm. This indicated that the C-20 carbohydrate component was unchanged by the biotransformation. The PMR spectrum of 8 contained resonances for two anomeric protons of the C-20 disaccharide (H-1‴ at 5.09 ppm and H-1‴ at 5.04 ppm) and only one resonance for the anomeric proton of the C-3 carbohydrate (H-1′ at 4.90 ppm). This also confirmed that the biotransformation product was Gyp XVII with a β-D-glucose and β-gentiobiose on C-3 and C-20, respectively. These same spectra of Gyp XVII and 9 (Table 2) showed that the latter contained only one β-D-glucose in the C-20 position because C-6‴ underwent a strong-field shift (62.7 ppm) in 9 with respect to that in Gyp XVII (70.0 ppm). Therefore, 9 had one β-D-glucose each on C-3 and C-20 and was the aforementioned known F-2 [2,4]. The PMR and 13C NMR spectra of 9 were identical to those of F-2 that were previously published by us [13] and also confirmed this.
Thus, the biotransformation of 4 through the action of Paenibacillus sp. BG134 involved sequential cleavage of-D-glucose from the C-3 and C-20 carbohydrates of 4 (Fig. 2). Gyp XVII was formed faster from 4 than F-2. This led to initial accumulation of 8 in the medium and then a gradual decrease of its concentration as a result of the transformation into 9 with practically complete conversion of starting 4.
It was noted above that a compound with TLC and HPLC data identical to those of Rd (3) appeared in the incubation medium after 6 h. It was hypothesized that F-2 could be formed from 4 through the action of Paenibacillus sp. BG134 by formation of intermediate 3 as a result of initial cleavage of β-D-glucose from the C-20 β-gentiobiose of 4 (Fig. 2, dashed arrows). The faster transformation of 3 into F-2 than its formation from 4 probably hindered its observation during almost the whole incubation period. The contribution of this potential biotransformation pathway to the formation of the final product F-2 was insignificant because in general the increase of the concentration of 9 correlated with the corresponding decrease in the concentration of Gyp XVII in the incubation medium. Paenibacillus sp. BG134 affected 4 similarly to aforementioned Sphingomonas sp. 2F2 [22] but differed from Sphingopyxis alaskensis DSM 13593, the biotransformation by which stopped at Gyp XVII [21], and A. mirum, which deglycosylated completely 4 to form 20(S)-protopanaxadiol [16].
The results for the biotransformation of 1-4 indicated that Paenibacillus sp. BG134 cleaved selectively only the terminal β-D-glucose from the carbohydrate components of these glycosides and did not affect the α-L-arabinofuranose of 1 and the α-L-arabinopyranose of 2 or the glycoside bonds formed by β-D-glucose directly to C-3 and C-20 of 20(S)-protopanaxadiol, i.e., the inner β-D-glucoses. The terminal C-3 β-D-glucose was preferentially cleaved first in 4. This could be used to produce Gyp XVII from 4 by limiting the incubation time.
The results indicated that Paenibacillus sp. BG134 differed from other known microorganisms with respect to the specificity of action on the principal 20(S)-protopanaxadiol ginsenosides and could be used for their biotransformation in order to produce minor glycosides, in particular, F-2 and C-O, which have broader spectra of biological activity [6–8].
EXPERIMENTAL
PMR and 13C NMR spectra were recorded with TMS internal standard on a Bruker AM 500 spectrometer at operating frequencies 500 and 125 MHz, respectively. FAB mass spectra were obtained on a JMS-700 spectrometer (JEOL, Japan). Melting points were determined on a Fisher-Johns apparatus (Fisher Scientific Co.). Standard ginsenosides Rb1, Rb2, Rc, Rd, and F-2; compounds C-O and C-Mc1; and gypenoside Gyp XVII were purchased (Ambo Institute, Daejeon, South Korea).
Bacterium Paenibacillus sp. BG134 was isolated as before [10] from soil taken from a ginseng field in the vicinity of Pocheon (South Korea). The strain was identified by taxonomy as BG134 using phylogenetic analysis based on a comparison of nucleotide gene sequences coding 16S pRNA and the literature methods [10].
Screening for bacterial strains with β-glucosidase activity was performed on solid medium containing agar R2A (Difco, 15.2 g/L) with added esculin (1 g/L) and iron citrate (0.5 g/L). The appearance of a brownish-black color around the colonies indicated a positive result.
Isolation of Ginsenosides from P. ginseng Root. Glycosides 1-4 were isolated from six-year ginseng roots as described before [24] using in the final stage preparative HPLC over an Alltech Alltima C18 column (250 × 22 mm, 10 μm) and gradient elution by MeOH-H2O. The detector was set at 203 nm. The yields of Rb1, Rd, Rc, and Rb2 were 1.48, 0.17, 0.46, and 0.23%, respectively, calculated as the dry root weight. The spectral and other characteristics of Rb1, Rd, and Rc were reported earlier by us [12,25].
Biotransformation of Ginsenosides. Paenibacillus sp. BG134 was cultivated in Ehrlenmeyer flasks with R2A liquid medium (200 mL) for 48 h at 30°C with constant stirring. Then, bacterial suspension (100 mL) was mixed with an aqueous solution (100 mL, 1 mM) of one of the studied ginsenosides (Rb2, Rc, Rd, or Rb1). The mixture was incubated for 72 h at 30°C with constant stirring, collecting analytical samples every 6 h. Two mixtures were used in parallel for Rb1. The first was incubated for 36 h; the second, for 72 h. After the incubation, each mixture was extracted with water-saturated BuOH (200 mL). The resulting extract was evaporated in vacuo. The residue was used to isolate the biotransformation products.
TLC was performed on silica gel plates (TLC Silica gel plates 60 F254, Merck, Germany) using CHCl3-MeOH-H2O (65:35:10, lower phase). Detection used spraying with H2SO4 solution (10%) followed by heating at 110°C for 3-5 min until spots appeared.
Analytical HPLC was carried out on a YL9100 chromatograph (Young Lin Instrument Co. Ltd., South Korea) equipped with a vacuum de-gasser, four-channel gradient pump, column thermostat, and YL9120 UV/Vis spectrophotometric detector using a reversed-phase Hypersil ODS C18 column (250 × 4.6 mm, 5 μm). The mobile phase was a mixture of MeCN (A) and H2O (B) at flow rate 1 mL/min. Gradient elution used an A-B mixture (25:75, v/v, 10 min), 25-32% A (5 min), 32-55% A (5 min), 55-60% A (5 min), 60-100% A (2 min), 100% A (13 min), and 100-25% A (20 min). The detector was set at 203 nm. Ginsenosides were identified by retention times (tR) and comparison with those of standards. The concentrations of separate ginsenosides were determined using calibration curves constructed for each of the standards under the HPLC conditions given above.
Biotransformation products were isolated from the resulting extracts by preparative HPLC over a YMC-Pack Pro C18 column (250 × 20 mm, 10 μm, YMC, Japan) using gradient elution by an MeCN-H2O mixture at flow rate 10 mL/min. Absorption of the eluates was measured at 203 nm.
3-O-[β-D-Glucopyranosyl-(1→2)-β-D-glucopyranosyl]-20-O-[α-L-arabinopyranosyl-(1→6)-β-D-glucopyranosyl]-3β ,12β ,20β -trihydroxydammar-24-ene (2). C53H90O22. White powder, mp 200-202°C. 1Í NMR spectrum (500 MHz, Py-d5, δ, ppm, J/Hz): 0.80 (3H, s, CH3-19), 0.93 (3H, s, CH3-30), 0.94 (3H, s, CH3-18), 1.06 (3H, s, CH3-29), 1.25 (3H, s, CH3-28), 1.60 (3H, s, CH3-21), 1.61 (3H, s, CH3-27), 1.64 (3H, s, CH3-26), 4.85 (1H, d, J = 7.6, H-1 ), 4.93 (1H, d, J = 6.0, H-1‴), 5.07 (1H, d, J = 7.6, H-1‴), 5.28 (1H, d, J = 7.6, H-1″) (only characteristic proton resonances are given). Mass spectrum: FAB, m/z 1101 [M + Na]+.
3-O-(β-D-Glucopyranosyl)-20-O-[α-L-arabinopyranosyl-(1 → 6)-β-D-glucopyranosyl]-3β, 12β, 20β -trihydroxydammar-24-ene (6). Yield 69%. C47H80O17. White powder, mp 185-186°C. 1H NMR spectrum (500 MHz, Py-d5,δ, ppm, J/Hz): 0.86 (3H, s, CH3-19), 0.93 (3H, s, CH3-18), 0.98 (3H, s, CH3-30), 1.01 (3H, s, CH3-29), 1.22 (3H, s, CH3-28), 1.64 (3H, s, CH3-21), 1.66 (3H, s, CH3-27), 1.69 (3H, s, CH3-26), 4.85 (1H, d, J = 7.6, H-1′ ), 4.92 (1H, d, J = 6.0, H-1″″), 5.07 (1H, d, J = 7.6, H-1‴) (only characteristic proton resonances are given). Mass spectrum: FAB, m/z 939 [M + Na]+.
3-O-(β-D-Glucopyranosyl)-20-O-[β-D-glucopyranosyl-(1→6)-β-D-glucopyranosyl]-3β, 12β, 20β-trihydroxydammar-24-ene (8). C48H82O18. White powder, mp 179-181°C. 1H NMR spectrum (500 MHz, Py-d5,δ, ppm, J/Hz): 0.76 (3H, s, CH3-19), 0.92 (3H, s, CH3-18), 0.94 (3H, s, CH3-30), 0.95 (3H, s, CH3-29), 1.26 (3H, s, CH3-28), 1.57 (3H, s, CH3-26), 1.62 (6H, s, CH3-21, 27), 4.90 (1H, d, J = 7.6, H-1′), 5.04 (1H, d, J = 7.6, H-1″″), 5.09 (1H, d, J = 7.6, H-1‴) (only characteristic proton resonances are given). Mass spectrum: FAB, m/z 969 [M + Na]+.
References
K. T. Choi, Acta Pharmacol. Sin., 29, 1109 (2008).
L. P. Christensen, Adv. Food Nutr. Res., 55, 1 (2009).
H. S. Jee, K. H. Chang, S. H. Park, K. T. Kim, and H. D. Paik, Food. Rev. Int., 30, 91 (2014).
J. D. Park, D. K. Rhee, and Y. H. Lee, Phytochem. Rev., 4, 159 (2005).
K. W. Leung, Natural Products, K. G. Ramawat and J. M. Merillon (eds.), Springer, Berlin, 2013, p. 3498.
Y. Zheng, H. Nan, M. Hao, C. Song, Y. Zhou, and Y. Gao, Biomed. Rep., 1, 555 (2013).
T. T. Mai, J. Moon, Y. Song, P. Q. Viet, and P. V. Phuc, Cancer Lett., 321, 144 (2012).
Q. Mao, P. H. Zhang, Q. Wang, and S. L. Li, Phytomedicine, 21, 515 (2014).
F. Gao, J. M. Zhang, Z. G. Wang, W. Peng, H. L. Hu, and C. M. Fu, Asian Pac. J. Cancer Prev., 14, 5599 (2013).
L. N. Ten, S. H. Baek, W. T. Im, M. Lee, H. W. Oh, and S. T. Lee, Int. J. Syst. Evol. Microbiol., 56, 2677 (2006).
M. H. Yoon, L. N. Ten, and W. T. Im, Int. J. Syst. Evol. Microbiol., 57, 1810 (2007).
L. N. Ten, S. M. Chae, and S.-A. Yoo, Chem. Nat. Compd., 49, 773 (2013).
L. N. Ten, S. M. Chae, and S.-A. Yoo, Chem. Nat. Compd., 49, 1168 (2014).
L. N. Ten, S. M. Chae, and S.-A. Yoo, Chem. Nat. Compd., 50, 565 (2014).
X. Zhao, J. Gao, C. Song, Q. Fang, N. Wang, T. Zhao, D. Liu, and Y. Zhou, Phytochemistry, 78, 65 (2012).
C. H. Cui, S. C. Kim, and W. T. Im, Appl. Microbiol. Biotechnol., 97, 649 (2013).
S. J. Park, S. Y. Youn, G. E. Ji, and M. S. Park, Food Sci. Biotechnol., 21, 839 (2012).
X. F. Jin, H. S. Yu, D. M. Wang, T. Q. Liu, C. Y. Liu, D. S. An, W. T. Im, S. G. Kim, and F. X. Jin, J. Microbiol. Biotechnol., 22, 343 (2012).
C. S. Park, M. H. Yoo, K. H. Noh, and D. K. Oh, Appl. Microbiol. Biotechnol., 87, 9 (2010).
H. Koizumi, S. Sanada, Y. Ida, and J. Shoji, Chem. Pharm. Bull., 30, 2393 (1982).
K. C. Shin and D. K. Oh, J. Biotechnol., 172, 30 (2014).
L. Wang, Q. M. Liu, B. H. Sung, D. S. An, H. G. Lee, S. G. Kim, S. C. Kim, S. T. Lee, and W. T. Im, J. Biotechnol., 156, 125 (2011).
D. S. An, C. H. Cui, H. G. Lee, L. Wang, S. C. Kim, S. T. Lee, F. Jin, H. Yu. Y. W. Chin, H. K. Lee, W. T. Im, and S. G. Kim, Appl. Environ. Microbiol., 76, 5827 (2010).
M. C. Yang, D. S. Seo, J. Hong, S. H. Hong, Y. C. Kim, and K. R. Lee, Nat. Prod. Sci., 14, 171 (2008).
L. N. Ten, S. M. Chae, and S.-A. Yoo, Chem. Nat. Compd., 50, 562 (2014).
Acknowledgment
This work partially supported by the Brain Pool Program of 2014, Republic Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 4, July-August, 2014, pp. 598-603.
Rights and permissions
About this article

Cite this article
Ten, L.N., Chae, S.M. & Yoo, SA. Biotransformation of the Principal Ginsenosides of Panax ginseng Into Minor Glycosides Through the Action of Bacterium Paenibacillus sp. BG134. Chem Nat Compd 50, 691–696 (2014). https://doi.org/10.1007/s10600-014-1054-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-014-1054-1