
Four known (1, 2, 4, and 5) and two new (3 and 6) steroidal glycosides were isolated from Polygonatum glaberrimum K. Koch (Convallariaceae) rhizomes by fractionation of the total extracted compounds. Their structures were determined as (25R)-spirost-5-en-3β,17α-diol 3-O-[α-L-rhamnopyranosyl-(1→2)]-β-Dglucopyranosyl-(1→4)- β-D-xylopyranoside (3) and 26-O-β-D-glucopyranosyl-(25R)-furost-5-en-3β,17α,22α,26-tetraol-3-O-[2″-OAc-α-L-rhamnopyranosyl-(1→2)]-β-D-xylopyranosyl-(1→3)-β-Dglucopyranoside (6).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The present work focused on steroids from rhizomes and basal stems of Polygonatum glaberrimum K. Koch (Convallariaceae) collected in May–June in the vicinity of Kojori and Saguramo (Georgia).
We reported previously [1] the isolation of four sapogenins from rhizomes of P. glaberrimum. Herein we present results for the structures of six steroidal glycosides from this plant.
Total saponins obtained by exhaustive extraction of air-dried raw material (400 g) by hot MeOH were evaporated to a resinous residue and then suspended in H2O by vigorous stirring. The insoluble precipitate was separated from the supernatant liquid and chromatographed repeatedly over columns of silica gel to afford five pure glycosides that were called 1–5 in increasing order of polarity. They belonged to the spirostanol class according to color reactions and IR spectroscopy [2, 3].

The supernatant liquid was extracted with n-BuOH. The solvent was evaporated to dryness. The residue was dissolved in EtOH. The glycosides were precipitated by Me2CO to isolate another glycoside 6 that was a furostanol.
The structures of the glycosides were studied using chemical transformations and GC, IR, mass, and NMR methods. These established the structures of two new (3 and 6) and four known glycosides (1, 2, 4, and 5).
Glycoside 1 contained diosgenin and one β-D-glucopyranose; C33H52O8; FAB-MS m/z 599 [M + Na]+, 437 [M + Na – hexose]+; J{13C–1H} (Hz) 160.0 (Glc-β); HMBC: H-1 Glc–C-3 of the genin; i.e., it was (25R)-spirost-5-en-3β-ol 3-O-β-Dglucopyranoside or trillin [4].
Glycoside 2 consisted of yamogenin, β-D-glucopyranose (terminal), and α-D-xylopyranose; C38H60O12; FAB-MS m/z 731 [M + Na]+, 569 [M + Na – hexose]+, 437 [M + Na – biose]+; J {13C–1H} (Hz) 159.4 (Glc-β), 170.0 (Xyl-α); HMBC: H-1 Xyl–C-3 of the genin, H-1 Glc–C-4 of Xyl. It was identified as (25S)-spirost-5-en-3β-ol 3-O-β-D-glucopyranosyl-(1→4)-α-D-xylopyranoside [5].
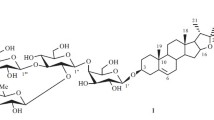
Glycoside 3 underwent acid hydrolysis to the aglycon pennogenin and the carbohydrate part that contained D-glucose, L-rhamnose, and D-xylose in a 1:1:1 ratio according to TLC and GC. Its IR spectrum showed absorption bands characteristic of a spiroketal group. Sannie reagent turned it yellow [3]. GC analysis agreed well with fragmentation of the carbohydrate chain in its FAB-MS (m/z 893 [M + Na]+, 747 [M + Na – deoxyhexose]+, 731 [M + Na – hexose]+, 453 [M + Na – triose]+). The nature of the carbohydrate chain fragmentation showed that it contained two terminal sugars, i.e., a deoxyhexose (rhamnose) and a hexose (glucose).
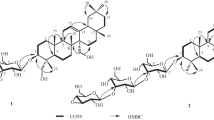
Next, the structure of 3 was studied using PMR and 13C NMR spectra. Resonances were assigned by interpreting COSY, HOHAHA, HMQC [6], and HMBC [7] spectra (Tables 1 and 2). Thus, interpretation of HOHAHA and HMQC spectra identified clearly proton resonances of the three sugars (glucose, xylose, and rhamnose). The SSCC of the glucose and xylose anomeric protons fell in the range 7.6–7.8 Hz and indicated that these sugars had β-glycoside bonds. The rhamnose anomeric proton appeared as a broad singlet. Its C-5 atom resonated at δ 69.5. This indicated unambiguously that its glycoside bond had the α-configuration. All sugars were found in the pyranose form [8] according to correlations between H-1 and H-5 of each of them in the NOESY spectrum [9] and the lack of resonances in the range δ 82–89.
The HMBC spectrum showed cross-peaks for through-space coupling of H-1 (δ 4.80) of Xyl and C-3 (δ 78.1) of the aglycon; H-1 (δ 6.25) of Rha and C-2 (δ 77.3) of Xyl; and H-1 (δ 4.98) of Glc and C-4 (δ 79.3) of Xyl. These data led to the conclusion that the carbohydrate part of 3 was bonded to C-3 of the aglycon and was branched. The branching center was xylose, which was substituted on C-2 and C-4 by rhamnose and glucose, respectively.
Thus, 3 was (25R)-spirost-5-en-3β,17α-diol 3-O-[α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranosyl-(1→4)-β-D-xylopyranoside.
Glycoside 4 had pennogenin as the genin and contained two terminal 2″-OAc-α-L-rhamnopyranose and β-D-xylopyranose. The branching center was β-D-glucopyranose. C46H72O18; FAB-MS (m/z 935 [M + Na]+, 893 [M + Na – Ac]+, 803 [M + Na – pentose]+, 747 [M + Na – Ac – deoxyhexose]+, 453 [M + Na – triose]+; J {13C–1H} (Hz) 159.0 (Glc-β), 158.1 (Xyl-β), 172.2 (Rha-α); HMBC: H-1 Glc–C-3 of the genin, H-1 Xyl–C-3 Glc, H-1 Ac-Rha–C-2 Glc, H-2″ Ac-Rha–C = O acetyl. The structure was determined as (25R)-spirost-5-en-3β,17α-diol 3-O-[2″-OAc-α-L-rhamnopyranosyl-(1→2)]-β-D-xylopyranosyl-(1→3)-β-D-glucopyranoside [10].
Glycoside 5 contained diosgenin, three β-D-glucopyranoses (one terminal), and β-D-galactopyranose; C51H82O23; FAB-MS (m/z 1085 [M + Na]+, 923 [M + Na – hexose]+, 761 [M + Na – biose]+, 599 [M + Na – triose]+, 437 [M + Na – tetraose]+; J {13C–1H} (Hz) 160.0 (Glc-β), 159.2 (Gal-β), 159.6 (Glc-β), 161.0 (Glc-β); HMBC: H-1 Glc–C-3 of the genin, H-1 Gal–C-3 Glc, H-1 Glc–C-4 Gal, H-1 Glc–C-3 Glc. The structure corresponded to (25R)-spirost-5-en-3β-ol 3-O-β-D-glucopyranosyl-(1→3)-β-D-glucopyranosyl-(1→4)-β-D-galactopyranosyl-(1→3)-β-D-glucopyranoside [11].
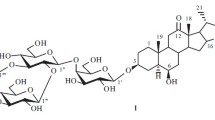
Glycoside 6 was an amorphous powder and appeared as two TLC spots with a slight difference in mobility ΔR f ~ 0.1. Its IR spectrum lacked absorption bands for a spiroketal. One broad band was observed at 890 cm–1. Ehrlich reagent colored it pinkish-red. All these characteristics identified 6 as a furostane glycoside. In fact, enzymatic hydrolysis by β-glucosidase gave a prosapogenin that was identical to that of 4, i.e., 6 was its furostanol analog. The hydrolysate from acid hydrolysis of 6 contained the aglycon pennogenin. TLC and GC detected in the carbohydrate part D-glucose, L-rhamnose, and D-xylose in a 2:1:1 ratio. This was also confirmed by a peak for the molecular ion and fragmentation of the carbohydrate part in the FAB-MS (m/z 1115 [M + Na]+, 1073 [M + Na –Ac]+, 983 [M + Na – pentose]+, 935 [M + Na – hexose – H2O]+, 927 [M + Na – Ac – deoxyhexose]+, 453 [M + Na – triose – hexose – H2O]+.
Next, the structure of 6 was studied using PMR and 13C NMR methods analogous to those used for 3. Interpretation of HOHAHA and HMQC spectra identified proton resonances of the four sugars as two glucoses, xylose, and 2″-OAc-rhamnose. The SSCC of the anomeric protons of the two glucoses and xylose fell in the range 7.4–7.8 Hz. Therefore, these sugars had the β-configuration for the glycoside bonds. The rhamnose anomeric proton resonated as a broad singlet; its C-5, at δ 69.5. This indicated unambiguously that it had an α-glycoside bond. All sugars were found in the pyranose form [8] according to correlations between H-1 and H-5 of each sugar in the NOESY spectrum [9] and the lack of resonances in the range δ 82–89.
The HMBC spectrum exhibited cross-peaks for through-space correlations that indicated unambiguously the attachment sites of the sugars to each other and to the aglycon. These were H-1 (δ 4.94) Glc–C-3 (δ 78.3) of the aglycon; H-1 (δ 6.18) AcO-Rha–C-2 (δ 77.3) 3-O-Glc; H-1 (δ 5.06) Xyl–C-3 (δ 81.4) 3-O-Glc; H-1 (δ 4.81) 26-O-Glc–C-26 (δ 75.1) of the aglycon; and H-2″ (δ 6.0) Rha–C=O (δ 177.8) of the acetyl.
Thus, all spectral data indicated that 6 had the structure 26-O-β-D-glucopyranosyl-(25R)-furost-5-en-3β,17α,22α,26-tetraol-3-O-[2″-OAc-α-L-rhamnopyranosyl-(1→2)]-β-D-xylopyranosyl-(1→3)-β-D-glucopyranoside.
Experimental
General Comments. TLC used Kieselgel 60 F254 (Merck) and Silufol UV 254 plates. Column chromatography used KSK silica gel (<63 and 63–100 μm) and solvent systems CHCl3–MeOH–H2O (65:15:2, a; 65:22:4, b; 65:35:8, c) (1) and CHCl3–MeOH (10:1, a; 50:1, b) (2). GC was performed on a Chrom-5 instrument. Monosaccharides were chromatographed as trimethylsilyl ethers of methylglycosides over a column (3 m × 4 mm) with Chromaton N-AW containing silicone SE-30 (5%) thermostatted at 190°C with He carrier gas at flow rate 45 mL/min.
Methylglycosides of methylated sugars were produced by refluxing (4 h) methyl esters in anhydrous MeOH with HCl (5%). The resulting products were chromatographed over a column (1.2 m × 3 mm) with cellite containing 1,4-polybutanediol succinate (20%) thermostatted at 160°C with He carrier gas at flow rate 50 mL/min.
Mass spectra were taken from a glycerin matrix on a Kratos MS 50 RF instrument. IR spectra were recorded from KBr pellets on a UR-20 instrument. PMR and 13C NMR spectra were obtained in Py-d5 with HMDS internal standard on an AMX-500 spectrometer (Bruker) at operating frequency 500.11 MHz for 1H and 125.76 MHz for 13C.
Isolation of Glycosides. Ground air-dried rhizomes (0.5 kg) were macerated in Et2O for 1.5 h in order to remove lipophilic substances. The resulting material was extracted with MeOH (70°) in a Soxhlet apparatus for 2 h. The extract was filtered, evaporated to a resinous residue, and suspended in H2O with vigorous stirring. The insoluble precipitate was separated by decanting the supernatant liquid, which then was extracted with n-BuOH.
The extract was condensed. The precipitate was dissolved in EtOH. Glycosides were precipitated by Me2CO and dried. The resulting total glycosides were chromatographed over a column of silica gel using systems 1b and 1c to afford a mixture of 6 and its 22-O-methyl ether. Refluxing the mixture in H2O (2 h) produced chromatographically homogeneous 6 (0.55 g, 0.11 mass% of air-dried raw material).
The insoluble precipitate was chromatographed over a column of silica gel (systems 1a and 1b). Fractions with chromatographically homogeneous glycosides were collected. Fractions containing more than one compound were combined and rechromatographed using the same solvent systems. This afforded 3 (0.67 g, 0.13 mass% of air-dried raw material).
(25 R )-Spirost-5-en-3 β ,17 α -diol 3- O -[ α -L-rhamnopyranosyl-(1→2)]- β -D-glucopyranosyl-(1→4)- β -Dxylopyranoside. Crystalline white powder, C44H70O17, mp 251–253°C, [α] 20D –62° (c 0.42, Py). FAB-MS spectra were presented above. IR spectrum (KBr, νmax, cm–1): 3400–3600 (OH), 984, 925, 896, 860 (intensity 896 > 925 – 25-R-spiroketal). 1H NMR spectrum (Py-d5, δ, ppm, J/Hz): 5.32 (1H, br.d, H-6), 4.46 (1H, dd, J = 7.2, 6.1, H-16), 3.86 (1H, m, H-3), 3.52 (1H, dd, J = 10.5, 2.9, H-26a), 3.50 (1H, dd, J = 10.5, 10.5, H-26b), 1.22 (3H, d, J = 7.0, Me-21), 1.09 (3H, s, Me-19), 0.96 (3H, s, Me-18), 0.68 (3H, d, J = 5.4, Me-27).
26- O - β -D-Glucopyranosyl-(25 R )-furost-5-en-3 α ,17 α ,22 α ,26-tetraol-3- O -[2′- O Ac- α -L-rhamnopyranosyl-(1→2)]- β -D-xylopyranosyl-(1→3)- β -D-glucopyranoside. Amorphous white powder, C52H84O24, mp 193–196°C, [α] 20D –52.4° (c 0.36, Py). FAB-MS spectra were presented above. IR spectrum (KBr, νmax, cm–1): 3450–3650 (OH), 890. 1H NMR spectrum (Py-d5, δ, ppm, J/Hz): 5.26 (1H, br.d, J = 3.8, H-6), 4.75 (1H, br.d, J = 4.6, H-16), 3.94 (1H, dd, J = 9.3, 7.2, H-26a), 3.76 (1H, m, H-3), 3.61 (1H, dd, J = 9.3, 5.8, H-26b), 1.36 (3H, d, J = 6.9, Me-21), 1.06 (3H, s, Me-19), 1.01 (3H, d, J = 6.8, Me-27), 0.99 (3H, s, Me-18).
Acid Hydrolysis. Compounds 3 and 6 (100 mg each) were dissolved in aqueous MeOH (15 mL, 50%) containing conc. H2SO4 (0.6 mL), refluxed for 8 h, cooled, and diluted with H2O. The resulting precipitates were filtered off and purified by recrystallization from MeOH to afford colorless crystals (48.7 and 34.8 mg, respectively) of the aglycon, C27H42O4, mp 230–232°C, [α] 20D –105° (c 1.0, CHCl3). EI-MS m/z 430 [M]+. IR spectrum (KBr, νmax, cm–1): 3550 (OH), 960, 925, 900, 870 (intensity 900 > 925, 25-R-spiroketal). Table 2 presents the 13C spectra. The aglycon was identified as pennogenin by comparison with the literature [12].
Enzymatic Hydrolysis. Glycoside 6 (70 mg) was dissolved in H2O (30 mL), treated with β-glucosidase (10 mg), left at room temperature for 10 h, and extracted (3 ×) with n-BuOH. The organic layer was evaporated. The enzymolysis products were chromatographed over a column of silica gel using system 1c to afford a spirostane glycoside (35.2 mg) that was identified as 4 according to physicochemical constants, 13C NMR spectra, and chromatographic mobility together with an authentic sample.
References
L. N. Gvazava and V. S. Kikoladze, Chem. Nat. Compd., 48, 917 (2012).
E. Stahl (ed.), Thin-Layer Chromatography: A Laboratory Handbook (translated from the German), Academic, New York, 1965.
C. Sannie, S. Heitz, and H. Lapin, Compt. rend., 233, 1670 (1951).
O. Espejo, J. C. Llavot, H. Jung, and F. Gilal, Phytochemistry, 21, 413 (1982).
V. K. Saxena and A. Shalem, J. Chem. Sci., 116 (2), 79 (2004).
M. F. Summers, L. G. Marzilli, and A. Bax, J. Am. Chem. Soc., 108, 4285 (1986).
A. Bax, A. Azolos, Z. Dinya, and K. Sudo, J. Am. Chem. Soc., 108, 8056 (1986).
A. S. Shashkov and O. S. Chizhov, Bioorg. Khim., 437 (1976).
J. D. Stevens and H. G. Fletcher, J. Org. Chem., 33, 1799 (1986).
H. F. Dall, J. Zhou, Z. T. Ding, J. Xiong, and N. H. Tan, Chin. Chem. Lett., 10, 901 (2000).
P. K. Kintya, A. I. Stamova, L. B. Bakinovskii, and V. V. Krokhmalyuk, Chem. Nat. Compd., 14, 290 (1978).
T. Nohara, K. Miyahara, and T. Kawasaki, Chem. Pharm. Bull., 22, 1772 (1974).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2014, pp. 423–426.
Rights and permissions
About this article

Cite this article
Gvazava, L.N., Kikoladze, V.S. Steroidal Saponins from Polygonatum glaberrimum Rhizomes. Chem Nat Compd 50, 489–493 (2014). https://doi.org/10.1007/s10600-014-0994-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-014-0994-9