
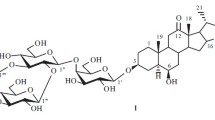
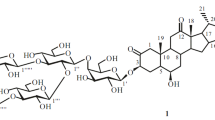
New steroidal glycoside 1 was isolated by fractionation of total extracted compounds from rhizomes of Polygonatum verticillatum (Convallariaceae). Chemical transformations, physical constants, and spectral data characterized its structure as (25S)-spirost-5-en-3β-ol 3-O-β-D-glucopyranosyl-(1→3)-[β-Dfucopyranosyl-(1→2)]-β-D-glucopyranosyl-(1→4)-β-D-galactopyranoside.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Biologically active compounds were isolated earlier by us from three Polygonatum species, i.e., P. glaberrimum, P. multiflorum, and P. polyanthemum [1–3]. Herein, the chemical structure of new steroidal glycoside 1 from P. verticillatum is elucidated.

Compound 1 was a 25S-spirostane spiroketal according to a positive Sannie–Lapin color test [4] and a characteristic absorption in the IR spectrum [5]. Acid hydrolysis of 1 produced an aglycon that was identified as yamogenin from its physicochemical constants and spectral data and those of an authentic sample [6].
Methanolysis of 1 followed by GC analysis of the sugars [7] found that its carbohydrate part consisted of galactose, glucose, and fucose in a 1:2:1 ratio.
The FAB mass spectrum of 1 showed key peaks with m/z values of 1069 [M + Na]+, 923 [M + Na – deoxyhexose]+, 907 [M + Na – hexose]+, 437 [M + Na – tetraose]+.
The 13C NMR spectra (Tables 1 and 2) was taken with full proton decoupling and contained 51 lines, of which 27
belonged to the aglycon and 24, to the sugars. Four lines were observed in the range of anomeric C resonances. The strongfield part showed resonances for five methyls. Obviously, the fifth methyl could belong only to one of the sugars. Thus, a comparison of the FAB mass spectrum of yamogenin (m/z 437 [M + Na]+) and the published 13C NMR spectrum [8] with those of 1 considering the GC data led to the conclusion that the carbohydrate part consisted of four sugars, one of which was a deoxyhexose.
Hakomori methylation [9] of 1 produced its permethylate derivative. The completeness of the methylation was monitored using the hydroxyl IR absorption band and the molecular peak in the FAB mass spectrum for an ion with m/z 1237 [M + Na]+.
The permethylate underwent methanolysis to afford yamogenin and total methylated sugars, which were identified by GC as 2,3,4,6-tetra-O-methyl-D-glucopyranose (terminal), 2,3,4-tri-O-methyl-D-fucopyranose (terminal), 4,6-di-O-methyl-D-glucopyranose (C-2 and C-3 disubstituted), and 2,3,6-tri-O-methyl-D-galactopyranose (C-4 substituted).
PMR and 13C NMR methods were used to study further the structure of 1. Lines were assigned by interpreting COSY, HOHAHA, HMQC [10], and HMBC [11] spectra (Tables 1 and 2).
Thus, proton resonances of four sugars were clearly identified by interpreting HOHAHA spectra in combination with HMQC spectra. These were two glucoses, fucose, and galactose. The SSCC of the anomeric protons of all sugars fell in the range 7.5–7.8 Hz. Therefore, all sugars had β-glycoside bonds and adopted the pyranose form [12]. This was consistent with correlations between H-1 and H-5 of each sugar in the NOESY spectrum [13] and the lack of resonances with δ = 82–89.
Cross-peaks for correlations in the HMBC spectrum represented through-space couplings and indicated unambiguously the bonding sites of the sugars to each other and to the aglycon. They were found between Gal H-1′ (δ 4.82) and aglycon C-3 (δ 78.2); Glc H-1′′ (δ 5.12) and Gal C-4′ (δ 79.9); Fuc H-1′′′ (δ 4.62) and Glc C-2′′ (δ 81.6); Glc H-1′′′′ (δ 5.36) (terminal) and Glc C-3′′ (δ 87.1).
Thus, 1 was (25S)-spirost-5-en-3β-ol 3-O-β-D-glucopyranosyl-(1→3)-[β-D-fucopyranosyl-(1→2)]-β-Dglucopyranosyl-(1→4)-β-D-galactopyranoside.
Experimental
General Comments. TLC used Kieselgel 60 F254 (Merck) and Silufol UV 254 plates. Column chromatography used KSK silica gel (<63 and 63–100 μm). Solvent systems were CHCl3–MeOH–H2O (1a, 65:15:2; 1b, 65:35:8) and CHCl3–MeOH (2a, 10:1; 2b, 50:1).
GC was performed on a Chrom-5 instrument. Monosaccharides were chromatographed as trimethylsilyl ethers of methylglycosides over a column (3 m × 4 mm) with Chromaton N-AW coated with SE-30 5% silicone at thermostat temperature 190°C and He carrier gas at flow rate 45 mL/min.
Methylglycosides of methylated sugars were prepared by refluxing (4 h) methyl ethers in anhydrous MeOH containing HCl (5%). The products were chromatographed over a column (1.2 m × 3 mm) of Celite containing 1,4-polybutanediol succinate (20%) at thermostat temperature 160°C and He carrier gas at flow rate 50 mL/min.
Mass spectra were recorded from a glycerin matrix on a Kratos MS 50 RF; IR spectra, in KBr on a UR-20 instrument; PMR and 13C NMR spectra, in Py-d5 with HMDS internal standard on an AMX-500 spectrometer (Bruker) at operating frequency 500.11 MHz for 1H and 125.76 MHz for 13C.
Isolation of Glycoside 1. Ground air-dried rhizomes of P. verticillatum were macerated for 1.5 h in Et2O in order to remove lipophilic substances. The resulting material was extracted with hot (70°C) MeOH (2 × 1.0 L). The extract was evaporated. The residue was extracted first with n-BuOH and then H2O. The BuOH extract was condensed and chromatographed over a column of silica gel with elution by 1a and 1b to give 12 fractions. Fraction 9 contained homogeneous glycoside 1 (108 mg). Fractions 8 and 10 showed two spots on TLC. They were combined and rechromatographed to isolate an additional 34 mg, which gave a total of 142 mg (0.035%) calculated for air-dried raw material.
(25 S )-Spirost-5-en-3 β -ol 3- O - β -D-Glucopyranosyl-(1→3)-[ β -D-fucopyranosyl-(1→2)]- β -D-glucopyranosyl-(1→4)- β -D-galactopyranoside. White crystals, C51H82O22, mp 246–249°C, [α] 20D –118° (c 0.75, CHC13 + MeOH 1:1). IR spectrum (KBr, νmax, cm–1): 3450–3650 OH), 2930 (CH3), 1620 (C=C), 1040 (C–O–C), 985, 915, 895, 850 (intensity 915 > 895, 25S-spiroketal). 1H NMR spectrum (Py-d5, δ, ppm, J/Hz): 5.36 (1H, m, Í-6), 4.62 (1H, ddd, J = 9.4, 6.9, 5.0, H-16), 3.93 (1H, m, H-3), 1.54 (3H, br.s, Me-6′′′), 1.13 (3H, d, J = 6.9, Me-21), 1.09 (3H, d, J = 7.1, Me-27), 0.93 (3H, s, H-19), 0.82 (3H, s, H-18).
Acid Hydrolysis. Compound 1 (50 mg) was dissolved in aqueous MeOH (50%, 15 mL) containing conc. H2SO4 (0.6 mL), refluxed for 8 h, cooled, and diluted with H2O. The resulting precipitate was filtered off and purified by recrystallization from MeOH to afford colorless crystals (23.8 mg) of the aglycon, C27H42O3, mp 180–182°C, [α] 20D –125° (c 0.75; CHC13). FAB-MS m/z 437 [M + Na]+. IR spectrum (KBr, νmax, cm–1): 3400 (OH), 2920 (CH3), 1640 (C=C), 1215 (C–O), 1030 (C–O–C), 980, 910, 890, 850 (intensity 910 > 890 – 25S-spiroketal). 13C NMR spectrum (Py-d5, δ, ppm): 37.8 (C-1), 32.5 (C-2), 71.2 (C-3), 43.4 (C-4), 142.0 (C-5), 121.0 (C-6), 32.3 (C-7), 31.8 (C-8), 50.5 (C-9), 37.0 (C-10), 21.2 (C-11), 40.0 (C-12), 40.5 (C-13), 56.7 (C-14), 32.2 (C-15), 81.2 (C-16), 62.7 (C-17), 16.4 (C-18), 19.7 (C-19), 42.5 (C-20), 14.9 (C-21), 109.8 (C-22), 27.5 (C-23), 26.2 (C-24), 26.4 (C-25), 65.1 (C-26), 15.9 (C-27). The aglycon was identified as yamogenin by comparing the results with the literature [6].
Methanolysis of 1. Glycoside 1 (10 mg) was dissolved in anhydrous MeOH (4 mL) containing HCl (5%), refluxed for 12 h, cooled, and treated with an equal volume of H2O. The resulting precipitate was filtered off and demonstrated to be identical to yamogenin (TLC, system 2a). The filtrate was neutralized with Ag2CO3, filtered, and evaporated to dryness. GC detected D-galactose, D-glucose, and D-fucose in a 1:2:1 ratio.
Methylation of 1. Glycoside 1 (50 mg) was dissolved in DMSO (10 mL), treated slowly with NaH (50 mg), stirred vigorously for 1 h, treated slowly with CH3I (1.5 mL), stirred for another 3 h, poured into H2O (0.1 L), and extracted with CHCl3 (5 × 10 mL). The combined CHCl3 extracts were worked up with Na2S2O3 and evaporated to dryness. The solid was chromatographed over a column of silica gel (system 2b). Recrystallization from MeOH afforded the permethylate (44 mg), which underwent acid hydrolysis as described above. The precipitated aglycon was separated and identified as yamogenin.
The aqueous solution of methylated sugars was refluxed for 6 h, neutralized with anion exchanger EDE-10P, and evaporated to dryness. The aforementioned methylglycosides were identified using the GC results.
References
L. N. Gvazava and V. S. Kikoladze, Chem. Nat. Compd., 48, 917 (2012).
L. N. Gvazava and V. S. Kikoladze, Chem. Nat. Compd., 49, 173 (2013).
L. N. Gvazava and V. S. Kikoladze, Chem. Nat. Compd., 50, 489 (2014).
Ch. Sannie and H. Lapin, Bull. Soc. Chim. Fr., 19 (11), 1080 (1952).
N. Jones, E. Katzenellenbogen, and K. E. Dobriner, J. Amer. Chem. Soc., 75, 158 (1958).
O. Espeji, K. Llavot, H. Jung, and F. Gital, Phytochemistry, 21, 413 (1982).
T. G. Gorovits, Khim. Prir. Soedin., 263 (1970).
P. K. Agrawal, D. C. Jain, R. K. Gupta, and R. S. Thakur, Phytochemistry, 11, 2479 (1985).
S. Hakomori, J. Biochem. (Tokyo), 55, 205 (1964).
M. F. Summers, L. G. Marzilli, and A. Bax, J. Amer. Chem. Soc., 108, 4285 (1986).
A. Bax, A. Azolos, Z. Dinya, and K. Sudo, J. Amer. Chem. Soc., 108, 8056 (1986).
A. S. Shashkov and O. S. Chizhov, Bioorg. Khim., 2, 437 (1976).
J. D. Stevens and H. G. Fletcher, J. Org. Chem., 33, 1799 (1986).
Author information
Authors and Affiliations
Corresponding author
Additional information
translated from Khimiya Prirodnykh Soedinenii, No. 6, November–December, 2016, pp. 905–907.
Rights and permissions
About this article

Cite this article
Gvazava, L.N., Skhirtladze, A.V. Steroidal Saponin from Polygonatum verticillatum . Chem Nat Compd 52, 1052–1055 (2016). https://doi.org/10.1007/s10600-016-1859-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-016-1859-1