
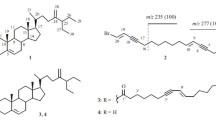
Chemical investigation of an Okinawan marine sponge resulted in the isolation of two new spongian-class diterpenes 1 and 2 together with two known compounds, chromodorolide B (3) and chromodorolide C (4). Compound 1, named chromodorolide D, is an example of a diterpenoid with a highly rearranged chromodorane carbon skeleton, while compound 2 retains the open side chains. The structures of the new compounds were elucidated on the basis of extensive spectroscopic analysis and chemical conversion. Compounds 1–4 exhibited significant cytotoxicity against NBT-T2 rat bladder epithelial cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A growing number of diterpenes of the spongian type have been reported from various marine sponge species. In addition, isolation of several rearranged spongian metabolites from spongivorous nudibranchs of the genus Chromodoris implies undiscovered sponge sources of these diterpenes. These diterpenes can be related biosynthetically to metabolites described previously. Some examples are chromodorolides, dendrillolides, shahamines, norrisolide, macfarlandins, polyrhaphins, chelonaplysins, and aplyviolenes, which have been isolated form both sponges and nudibranchs [1–12]. Aplyviolenes [10–12], polyrhaphin C [10], chelonaplysin B and chelonaplysin C [11], and macfarlandin D and macfarlandin E [9] have been reported to show antimicrobial activities while chromodorolide A displayed both cytotoxic and antimicrobial activities [1, 2]. Chromodorolide C was also reported to exhibit significant cytotoxicity [3]. In the course of our studies on bioactive metabolites of marine invertebrates, we isolated two new spongian-class diterpenes 1 and 2 together with the known chromodorolides B (3) and C (4) from a sponge collected along the coast of Okinawa. In this paper we describe the isolation, structure elucidation, and bioactivities of these compounds.

As part of our ongoing investigations of biologically active metabolites of marine invertebrates, a cytotoxic screening assay on 24 crude extracts (both EtOAc and methanolic extracts) of Okinawan marine specimens was performed. The crude EtOAc extract of the sponge displayed significant cytotoxic activity (1 μg/mL). On the basis of the biological activity, subsequent chemical investigations were carried out on this extract. Repetitive normal phase HPLC separation of the active fractions afforded two new bioactive compounds 1, 2 together with two bioactive known members 3, 4. The structures of the known compounds chromodorolide B (3) and chromodorolide C (4) were assigned on the basis of their spectroscopic data and comparison with those of the reported compounds [2, 3].
The molecular formula of chromodorolide D (1), C22H32O7, was determined by analysis of its mass spectral and NMR data. The seven degrees of unsaturation in its molecular formula can be accounted for by an acetate carbonyl (δ 169.4) and a lactone carbonyl (δ 176.8), and to satisfy the remaining degrees of unsaturation chromodorolide D (1) had to be pentacyclic like chromodorolide B (3) and C (4) [2, 3]. The similarity of the 1H and 13C NMR data of chromodorolide D (1) to those of chromodorolide C (4) showed that the two molecules were closely related, and 1 differed from 4 only in its acetylation pattern. The C12H21 hydrocarbon fragment in 4 could also be identified in 1 by a detailed analysis of the COSY, HMQC, and HMBC data. The remaining fragment of C10H11O7 had to be tricyclic and it had to contain the acetate and lactone functionalities. The resonances at δ 6.46 br.s, δ 98.2 and 6.00 d, δ 103.6 revealed that there are two acetal functionalities present in this fragment. HMBC correlations connected the acetal methine proton at δ 6.46 br.s (H-15) to C-14 at 45.9, and H-14 at 2.92 t to C-15 at 98.2. COSY correlations then linked H-14 to H-13 (δ 3.54 dd), which was further coupled to the second acetal methine resonance at 6.00 d (H-16). The methine resonance at 2.92 t (H-14) showed an additional COSY correlation to a methine resonance at 2.31 dt (H-8), which was in turn correlated to a deshielded methine resonance at 3.93 dd (H-17) and to a resonance at 1.72 m which had already been assigned to H-9 of the hydrocarbon fragment (Table 1). The placement of the acetate functionality at C-15 was confirmed from the HMBC correlations from H-15 (δ 6.46 br.s) to the acetate carbonyl at 169.4, and from the methyl signal at 2.06 s to the acetate carbonyl at 169.4 and C-15 (δ 98.2). Difference NOE correlations (H-14/H-13, 8; H-16/H-13; H-20/H-17, H-15/H-17) were in complete agreement with the proposed structure of 1 for chromodorolide D and the relative stereochemistry of 1 (Fig. 1). Therefore, the chromodorolide D (1) also had the chromodorane skeleton with a bisacetal-oxalone ring fused 3, 4 to a cyclopentane ring (C-8 and C-12 to C-17). The acetylation of 1 with acetic anhydride in pyridine provided an acetate product whose 1H NMR spectrum was identical to that of natural chromodorolide B (3). This transformation further confirmed the structure and stereochemistry of chromodorolide D (1).

NOE correlations observed for 1.
Compound 2 gave the molecular formula C25H40O7 from 13C NMR and mass measurements. The 13C NMR resonances at δ 170.8 s, 170.9 s, 173.1 s, 51.7 q, 21.0 q (2C) along with IR bands at 1741 and 1227 cm−1 indicated the presence of two acetates and one methyl ester. Comparison of 13C NMR and 1H NMR data to known compounds [7, 10] suggested the same perhydroazulene subunit for the hydrocarbon portion of the molecule. In addition, 1H NMR signals at δ 4.82 br.s, 4.91 (d, J = 1.9 Hz), and 2.83 (d, J = 8.8 Hz) were characteristic of the exocyclic methylene protons and the H-9 ring junction proton in the perhydroazulene ring system, respectively (Table 1). The C-11/C-16 subunit was revealed by interpretation of COSY and HMBC data. COSY connectivities linked H-16 (δ 4.02 dd, 4.07 dd) to H-13 (2.94 br.q), H-13 to H-12 (2.21 dd and 2.52 dd), and H-14 (δ 1.84 br.d) to H-15 (3.98 dd, 4.36 dd). HMBC correlations from H-13 to C-14, 8, 15, H-15 to C-8, 14, and H-17 (δ 0.85 s) to C-8, 14 revealed that the C-11/C-16 subunit must be joined at C-14 to C-8 of the perhydroazulene subunit. The connection of an acetate functionality at C-15 was confirmed from the HMBC correlations from H-15 (δ 3.98 dd, 4.36 dd) to the acetate carbonyl at 170.8 and from methyl signal at 2.05 s to the acetate carbonyl at 170.8 and C-15 (63.6). Another acetate functionality could be connected at C-16 from the HMBC correlations from H-16 (4.02 dd, 4.07 dd) to the acetate carbonyl at 170.9, and from the methyl signal at 2.07 s to the acetate carbonyl at 170.9 and C-16 (δ 68.4). The methyl ester was linked to H-12 by the HMBC correlations from H-12 and C-11 of the ester carbonyl at 173.1, and from the methyl signal at 3.68 s to the ester carbonyl at 173.1. The clear NOE correlation observed between H-17 and H-13 but the absence of NOE correlation between H-17 and H-14 show that both H-13 and H-14 are in trans-position to each other, which confirms the relative stereochemistry at C-13 and C-14. Due to their common biosynthetic ancestry, it seems reasonable to assign the same relative stereochemistry to 2 as that found in the reported compounds [7].
Compounds 1–4 were evaluated with respect to cytotoxicity using NBT-T2 rat bladder epithelial cells. IC50 values for 1–4 were 5.6, 12, 3.4, and 3.8 μg/mL, respectively.
Experimental
Optical rotations were measured on a Jasco P-1010 polarimeter. The 1H, 13C, difference NOE, and all 2D NMR were recorded on a Jeol A500 NMR spectrometer using TMS as internal standard. Carbon multiplicities were determined using DEPT-135 and DEPT-90 sequences. Atom connectivities were determined using HMQC, HMBC, and COSY data. The chemical shifts are given in δ (ppm) and coupling constants in Hz. MS and IR spectra were obtained on a LTQ Orbitrap mass spectrometer and on a Varian FTIR FTS3000 spectrometer. HPLC was performed on a Hitachi L-6000 pump equipped with a Shodex RI-101 monitor and a Hitachi L-4000 UV detector using a Mightysil Si 60 column and a Develosil Si 60-3 column. Merck silica gel was used for vacuum flash chromatography. All solvents used were reagent grade. A Tecan sunrise microplate reader was used for cytotoxicity testing.
Animal Material. A sponge specimen was collected at a depth of 40 m at cape Manza, Okinawa Island by hand using Scuba and was kept frozen until extraction.
Extraction and Isolation. The sponge (7 g, wet) was cut into pieces and extracted by steeping in acetone (300 mL) for one day, and the extraction process was repeated three times. The combined extracts were concentrated under reduced pressure, and the residue was partitioned between EtOAc and water. The organic extract (133 mg) was separated by vacuum flash chromatography on silica gel to give four fractions. The second fraction (99 mg) was further separated on HPLC (silica, n-hexane–EtOAc, 2:3) to give 13 subfractions. The eighth subfraction (17.8 mg) was subjected to repetitive separation on HPLC (silica, n-hexane–EtOAc, 7:2) to give new compound 1 (5.3 mg). Similarly, the seventh subfraction (8.1 mg) yielded the new compound 2 (2.5 mg), and the ninth subfraction (15.9 mg) gave chromodorolide B (3, 3.3 mg) and chromodorolide C (4, 2.7 mg), both of which showed identical NMR data as those reported [2, 3].
Chromodorolide D (1). Colorless needles, \( \left[ \upalpha \right]_{\text{D}}^{{26}} \) –11.2° (c 0.28, CH2Cl2). IR (neat, νmax, cm−1): 3466, 2946, 1731, 1225. HR-MS m/z 409.2222 [M + H]+ (calcd for C22H33O7, 409.2219). 1H and 13C NMR data, see Table 1.
Compound (2). Colorless oil, \( \left[ \upalpha \right]_D^{{26}} \) +10.6° (c 0.20, CH2Cl2). IR (neat, νmax, cm−1): 2947, 1741, 1685, 1227. HR-MS m/z 453.2841 [M + H]+ (calcd for C25H41O7, 453.2846).1H and 13C NMR data, see Table 1.
Acetylation of 1. Acetic anhydride (0.2 mL) was added to a solution of compound 1 (1.3 mg, 3.2 × 10−3 mmol) in dry pyridine (1.3 mL). The mixture was stirred at room temperature overnight. The reaction mixture was partitioned between EtOAc and water. The organic layer was washed with brine and dried over Na2SO4. After concentration, the crude product (1.8 mg) was separated by HPLC (silica, n-hexane–EtOAc, 7:2) to give a product (0.8 mg, 62%) that was identical with natural chromodorolide B (3, TLC and 1H NMR) [2].
Cytotoxicity Assay. NBT-T2 cells (BRC-1370) were purchased from Riken Bioresource Center and cultured under a standard protocol using Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum and antimicrobials. Cultured cells were inoculated into each well (96-well plate) with 200 μL of the medium. After preincubation (24 h, 37°C, 5% CO2), aliquots of test compounds in MeOH were added to culture wells in duplicate. After incubating the sample wells for 2 days (24 h, 37°C, 5% CO2), the toxic effect of the compounds was observed under a microscope. The IC50 values were measured by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) colorimetric method. MTT solution (15 μL, 5 mg/mL in PBS) was added to each well after removal of the medium by aspiration and the culture was incubated for 3 h. The residual formazan was dissolved in 100 μL of dimethyl sulfoxide (DMSO). The absorbance was measured at 560 nm with a Tecan sunrise microplate reader. The IC50 values were estimated by plotting the absorbance values against concentrations [13].
References
E. J. Dumdei, E. D. de Silva, R. J. Anderson, M. I. Choudhary, and J. Clardy, J. Am. Chem. Soc., 111, 2712 (1989).
S. A. Morris, E. D. de Silva, and R. J. Anderson, Can. J. Chem., 69, 768 (1991).
W. Rungprom, W. Chavasiri, U. Kokpol, A. Kotze, and M. J. Garson, Mar. Drugs, 2, 1011 (2004).
B. Sullivan and J. Faulkner, J. Org. Chem., 49, 3204 (1984).
S. C. Bobzin and J. Faulkner, J. Org. Chem., 54, 5727 (1989).
E. D. de Silva, S. A. Morris, S. Miao, E. J. Dumdei, and R. J. Anderson, J. Nat. Prod., 54, 993 (1991).
S. Carmely, M. Cojocaru, Y. Loya, and Y. Kashman, J. Org. Chem., 53, 4801 (1988).
J. E. Hochlowski, J. Faulkner, G. K. Matsumoto, and J. Clardy, J. Org. Chem., 48, 1141 (1983).
T. F. Molinski, J. Faulkner, H. Cun-heng, G. D. V. Duyne, and J. Clardy, J. Org. Chem., 51, 4564 (1986).
S. C. Bobzin and J. Faulkner, J. Org. Chem., 54, 3902 (1989).
S. C. Bobzin and J. Faulkner, J. Nat. Prod., 54, 225 (1991).
T. W. Hambley, A. Poiner, and W. C. Taylor, Tetrahedron Lett., 27, 3281 (1986).
M. H. Uddin, N. Hanif, A. Trianto, Y. Agarie, T. Higa, and J. Tanaka, Nat. Prod. Res., 25, 585 (2011).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2012, pp. 373–375.
Rights and permissions
About this article
Cite this article
Uddin, M.H., Hossain, M.K., Nigar, M. et al. New cytotoxic spongian-class rearranged diterpenes from a marine sponge. Chem Nat Compd 48, 412–415 (2012). https://doi.org/10.1007/s10600-012-0262-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-012-0262-9