
The CH2Cl2-MeOH extract of the Red Sea sponge Xestospongia sp. afforded two new compounds (1 and 2) and two previously identified C-30 sterols (3 and 4). The chemical structures of the isolated compounds were deduced by examining their NMR and MS spectral data. The antimicrobial activity evaluation of the isolated compounds showed that all the compounds have moderate to good antibacterial effects against the tested bacteria, while compound 1 displayed reasonable activity towards two pathogenic fungi, Aspergillus niger and Candida albicans. Moreover, metabolite 1 has excellent antitumor activities against lymphoma and Ehrlich cell lines with IC50 values of 0.05 and 0.08 mM, respectively. No toxicity was recorded for 1 and 2 (LC50 = 30 mM) against Artemia salina as test organism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Natural acetylenic molecules are widespread among terrestrial plants of the families Compositae/Asteraceae and Umbellifera/Apiaceae, marine organisms (algae, sponges, and tunicates), and several other sources including fungi, insects, frogs, moss, lichens, and humans [1,2,3]. These chemical skeletons are generally created by extra desaturation of carbon-carbon double bonds of lipid-derived structures, increase in chain length, conjugation, instability, and bromination, thus prompting researchers to study their biosynthesis and biochemical and ecological importance [1,2,3,4,5,6]. Barrel sponges or species of the genus Xestospongia (Desmospongia, Haplosclerida, Petrosiidae) are known for production of steryl esters and brominated polyunsaturated lipids that have great importance to chemists and biochemists due to their unique chemical structures and biological applications [7,8,9,10,11]. In the current work, a specimen of the Red Sea marine sponge Xestospongia sp. has been collected, extracted, and sequentially processed by applying several chromatographic techniques, including normal column chromatography and preparative thin-layer chromatography. Four compounds (1–4) were isolated and identified. The antimicrobial and antitumor effects of the isolated metabolites were estimated.
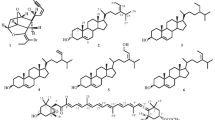
Fractionation of the sponge extract employing a Si gel column followed by preparative TLC gave four pure compounds (1–4) as shown in Fig. 1.

Structures of compounds 1–4 and mass fragmentations of compound 2.
Compound 1 has a molecular formula of C32H52O2, as deduced from HR-ESI-MS, which showed a molecular ion peak at m/z 468.3959 (equivalent to seven unsaturation sites). The 13C NMR spectrum displayed five signals assigned to two C=C and a C=O ester, which indicated the presence of a tetracyclic ring structure. The infrared data indicated absorbance bands at 3075 cm–1 assigned to =CH, at 1639 cm–1 assigned to a carbon-carbon double bond, and a band at 1732 cm–1, which was assigned to an ester group. The DEPT, HSQC, and 13C NMR showed five quaternary carbons (δ 37.0, 42.6, 139.9, 152.9, and 173.2 ppm), eight methine carbons (δ 28.2, 35.8, 50.2, 50.4, 56.2, 56.9, 73.7, and 122.8), 13 methylene carbons (δ 21.4, 24.9, 26.5, 26.7, 26.7, 27.8, 28.2, 29.2, 34.3, 34.9, 37.2, 40.0, and 108.7), and six methyl carbons (δ 12.2, 12.1, 12.3, 19.0, 19.5, and 22.9) (Table 1). The methylation pattern, especially of those of the two tertiary methyls (Me-18 and 19), and the chemical shifts (δH/δC) of C-6, C-10, C-13, and C-21 determined the presence of a tetracyclic structure similar to that of cholestane. Investigation of the HMBC spectrum confirmed the previous assumption through the correlations between H-19 and C-5 and C-9, H-6 (δ 5.38) and C-5 and C-7, and H-18 and C-12, C-14, and C-17. The tetracyclic skeleton consists of 19 carbon atoms and two carbons due to the acetyl moiety (CH3CO), leaving 11 carbon atoms for the side chain. The presence of two primary methyls (CH3-29 and CH3-30) and an olefinic methylene carbon (resonating at δH/δC 4.78, and 4.70/108.7) confirms the presence of a xestosterol-like structure, which was co-isolated with compound 1 [12]. However, the difference between xestosterol and compound 1 is the absence of the signal at δH 3.53 (H-3, R-CHOH) and the presence of the signal at δH 4.63 (H-3, R-CHOCOR), as shown in the1H NMR spectral data. The presence of an ester moiety instead of hydroxyl moiety was supported by the IR absorption band at 1732 cm–1. The signal at δC 173.2 ppm and the tertiary methyl resonating at δH/δC 2.19/22.9 compared to the typical value of acid at δC 179 ppm indicates that the carbonyl is an ester group. The stereochemistry of compound 1 was determined from the NOESY spectrum and analysis of the coupling constants. The large coupling constant of H-3 (dddd, J = 11.4, 11.4, 5.4, and 5.4 Hz) is attributed to the β-configuration of the acetate function; the α-configuration of CH3-21 was deduced based on the absence of correlations with CH3-18 or CH3-19 in the NOESY spectrum (Me-18 and Me-19 are biogenetically β-oriented). Based on the previous discussion, the new compound 1 was named 26,27-dimethylergosta-5,24(28)-dien-3-yl acetate.
Compound 2 was isolated as a gummy material. The HR-ESI-MS displayed two pseudo-molecular ion peaks at 377.1347 and 379.1327 with relative intensity (1:1), suggesting the presence of a Br atom. Moreover, the odd mass number indicates the presence of a nitrogen atom. Compound 2 displayed 20 signals in the 13C NMR which, along with obtained molecular mass, suggested the molecular formula C20H28BrNO. The structure and key MS fragmentation pattern are shown in Fig. 1. The IR data showed absorption bands characteristic of the =CH stretch (3018 cm–1), triple bond absorbance (2212 cm–1), and a carbonyl group (1707 cm–1). The 13C NMR, DEPT, and HSQC spectra showed resonances due to four acetylenic carbon signals [δC 77.4 (s), 79.3 (s), 88.5 (s), and 92.7 (s)], four olefinic carbons (δ 110.2, 117.1, 117.9, and 142.6), two methylene carbons α to triple bonds (δ 19.1 and 19.2), one allylic methylene (δ 32.3), two methylene carbons α and β to the carbonyl group at δ 33.8 and δC 24.2, a carbonyl signal due to the amide resonating at δ 176.0, and six methylene carbons represented by signals resonating at δ 27.7, 27.9, 28.2 × 2, and 28.4 × 2) (Table 2). The 13C NMR, DEPT, and HSQC data confirmed the presence of the three substructures given in Fig. 1.
The 1H NMR showed signals resonating at δ 6.60 (d, J = 14.4 Hz, H-20) and δ 6.19 (d, J = 14.4 Hz, H-19) were attributed to a trans-disubstituted double bond neighboring an acetylenic function and a Br atom. The downfield δH of this double bond H-19 and H-20 signals strongly supports the position of the Br atom at the terminal carbon.
The remaining three degrees of unsaturation together with the signals at δH 6.04 (dt, J = 15.0, 7.2 Hz, H-10) and δH 5.47 (d, J = 15.0 Hz, H-9) suggested a trans-disubstituted double bond with a methylene carbon in one side and an acetylene in the other side. The 13C NMR values of these triple bond signals at δ 88.5 and 79.3 support the presence of the ene-yne substructure.
The 1H–1H COSY data showed three correlation systems (H-20, H-19), (2H-11, H-10, and H-9), and (2H-3, 2H-2), which supports the presence of the substructure shown in Fig. 1.
These data were confirmed by comparison of the spectral data of compound 2 with the substructures published in [8]. The new compound named 20-bromo-(9E,19E)-icosa-9,19-diene-7,17-diyne (2).
Compound 3 (xestosterol ester of 18-bromooctadeca-(9E,17E)-diene-7,15-diynoic acid); its physical and spectral data are identical to those published in [11].
Compound 4 (xestosterol); its physical and spectral data are identical to those published in [12].
The antimicrobial activities of the isolated compounds from Xestospongia sp. were studied (Table 3). a) Compounds 1 and 4 showed good antibacterial activity against Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus, Staphylococcus epidermidis, and Streptococcus pneumonia with diameters of inhibition zone ranging from 12 to18 mm, and moderate activity against Acinetobacter sp. with diameter of inhibition zone of 11 mm; b) compounds 2 and 3 showed good antimicrobial activities against E. coli, P. aeruginosa, S. aureus, S. epidermidis, and S. pneumoniae with diameter of inhibition zone ranging from 12 to 16 mm, and moderate activity against Acinetobacter sp. and K. pneumoniae with diameter of inhibition zone ranging from 10 to 11 mm; c) the minimal inhibitory concentrations of the four compounds were recorded for P. aeruginosa and S. aureus. Only compound 1 showed significant antimicrobial activity against P. aeruginosa and S. aureus with IC50 value of 20 mg/L. On the other hand, compounds 2–4 exhibited IC50 of 30–50 mg/L against P. aeruginosa and S. aureus (data not shown).
Compound 1 showed reasonable antifungal activity against Aspergillus niger and Candida albicans with diameter of inhibition zone 19.7 and 11.9 mm, respectively (Table 4). Compounds 2–4 showed negligible antifungal activities (data not shown). Moreover, compounds 1 and 2 showed no toxicity against Artemia salina (at 30 mM, Table 4). The antitumor activity of the purified products was determined against two cell lines, and the IC50 was calculated. Compound 1 showed significant activity with IC50 of 0.05 and 0.08 mM against lymphoma and Ehrlich cell lines, respectively (Table 4).
Experimental
General. Silica gel GF 254 (Merck, Darmstadt, Germany) was used for analytical thin-layer chromatography (TLC). Preparative thin-layer chromatography (PTLC) was performed on aluminum oxide plates (20 × 20 cm) of 250 μm thickness. Electron impact mass spectra were determined at 70 eV on a Kratos (Manchester, UK) MS-25 instrument. 1D and 2D NMR spectra were recorded using Bruker (Karlsruhe, Germany) Avance III WM 600 MHz spectrometers, and 13C NMR was recorded at 150 MHz. Plates were sprayed with 50% H2SO4 in MeOH and heated at 100°C for 1–2 min.
Animal Material. Kingdom: animals; porifera: sponges; class: Demospongiae; order: Haplosclerida; family: Petrosiidae; genus: Xestospongia. The animal material was collected using scuba diving on November 2016 from Sharm Obhur 21°42′34′′N 39°05′45′′E and A voucher specimen (XeC-2016-11) was deposited in the Faculty of Marine Sciences of KAU.
Isolation of the Major Compounds. The soft coral material, Xestospongia sp., was dried by squeezing and then soaked in a mixture of CH2Cl2–MeOH (1:1, two times) at room temperature. The extract was concentrated under reduced pressure to obtain 24.9 g of viscose oil. This material was chromatographed on a column of silica gel. A silica gel column (500 g, 75 × 2 cm) was used for separation. The residue (15.0 g) was homogenized with silica gel and poured onto the top of the Si-gel column. Elution started with petroleum ether and then the polarity was increased gradually with diethyl ether (DEE) and ethyl acetate; 50-mL fractions were collected. The course of fractionation was followed by TLC using the solvent system DEE–n-hexane, DEE–EtOAc, and MeOH–CHCl3. Methanol/sulfuric acid was employed as spraying reagent. Preparative TLC was applied for further purification.
26,27-Dimethylergosta-5,24(28)-dien-3-yl Acetate (1). Fraction A (108.0 mg), eluted with DEE–n-hexane (3:7), was purified by PTLC using the solvent system n-hexane–DEE (7:3). The band with Rf 0.23 (brown color with sulfuric acid/ methanol reagent) was taken to give 2.4 mg of gummy material. IR (λmax, cm–1): 3075, 2936, 2865, 1732, 1639. HR-ESI-MS m/z 468.3959 [M]+ (calcd for C32H52O2, 468.3967); EI-MS (relative intensity): 468 (5) [M]+, 431 (10), 281 (20), 203 (30), 119 (70), and 68 (100). 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3), see Table 1.
20-Bromo(9 E ,19 E )-icosa-9,19-diene-7,17-diyne Amide (2). Fraction D (40.0 mg) eluted with EtOAc–n-hexane (3:7) was purified by preparative TLC using the solvent system EtOAc–n-hexane (3:7). The band with Rf 0.67 (brown color with sulfuric acid/methanol reagent) was taken to give 3.0 mg of gummy yellow material. IR (λmax, cm–1): 3547, 3018, 2932, 2860, 2212, 1707. HR-ESI-MS m/z 377.1347 and 379.1327 [M]+ (calcd for C20H28BrNO, 377.1354 and 379.1334). EI-MS (relative intensity): 377, 379 (1:1) [M, C20H28BrNO]+, 333/335 (5:5) [M – C1H2NO]+, 277/279 (1:1) [M – C5H10NO]+, 290 (17), 262 (35), 235 (100) [M – C5H4Br]+, 205 (30), 189 (23), 134 (24), 91 (25). 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3), see Table 1.
Xestosterol Ester of 18-Bromooctadeca-(9 E ,17 E )-diene-7,15-diynoic Acid (3). Fraction B (50.0 mg) eluted with DEE–n-hexane (4:6) was purified by PTLC using the solvent system DEE–n-hexane (3:7). The band with Rf 0.57 was taken to give 2.0 mg of compound 3.
26,27-Dimethylergosta-5,24(28)-dien-3-ol (xestosterol, 4). Fraction C (51.0 mg) eluted with EtOAc–n-hexane (3:7) was purified by PTLC using the solvent system EtOAc–n-hexane (3:7). The band with Rf 0.35 was collected to give 3.7 mg of compound 4.
Antimicrobial Activities. The antimicrobial activities of the isolated compounds 1–4 from Xestospongia sp. were detected against some Gram positive and negative bacteria, obtained from King Fahd General Hospital, Jeddah, Saudi Arabia using the agar well diffusion method of Holder and Boyce [13].
Lethality Assay. The brine shrimp lethality test was used to determine the toxicity of varying concentrations of the tested compounds (10–30 mM) in DMSO using brine shrimp larvae in seawater as test organism [14]. DMSO was used as negative control, and after 24 h of incubation at room temperature the number of the survived larvae was determined and LC50 was calculated.
Anticancer Activity. The first inoculum of Ehrlich carcinoma and lymphoma cell lines were purchased from Tanta Cancer Institute (Tanta, Egypt). The activity of compounds 1–4 as antitumor agents against the above two cell lines was determined and compared to bleomycin (antitumor control agent). The tumor cells were grown in RPMI 1640 medium (Sigma, USA) with 10% fetal calf serum (Gibco, USA). Cells were treated with different concentrations of compounds 1–4 (0.001–3.0 mM), incubated at 37°C in 95% air and 5% CO2 for 48 h, centrifuged for 2 min at 1500g, and counted using a hemacytometer and trypan blue; the IC50 was then calculated [11].
References
L. P. Christensen, Molecules, 25, 2568 (2020).
V. M. Dembitsky and D. O. Levitsky, Nat. Prod. Commun., 1, 405 (2006).
R. E. Minto and B. J. Blacklock, Prog. Lipid Res., 47, 233 (2008).
M. Taniguchi, Y. Uchio, K. Yasumoto, T. Kusumi, and T. Ooi, Chem. Pharm. Bull., 56, 378 (2008).
D. V. Kuklev, A. J. Domb, and V. M. Dembitsky, Phytomedicine, 20, 1145 (2013).
V. M. Dembitsky, Lipids, 41, 883 (2006).
X. Zhou, J. Sun, W. Ma, W. Fang, Z. Chen, B. Yang, and Y. Liu, Pharm. Boil., 52, 187 (2013).
S. Hirsh, S. Carmely, and Y. Kashman, Tetrahedron, 43, 3257 (1987).
L. F. Ling, T. Wang, Y. S. Cai, W. F. He, P. Sun, Y. F. Li, and Q. Huang, Eur. J. Med. Chem., 79, 290 (2014).
A. D. Patil, W. C. Kokke, S. Cochran, T. A. Francis, T. Tomszek, and J. W. Westley, J. Nat. Prod., 55, 1170 (1992).
N. B. Pham, M. S. Butler, J. N. A. Hooper, R. W. Moni, and R. J. Quinn, J. Nat. Prod., 62, 1439 (1999).
W. Kokke, C. Tarchini, D. B. Stierle, and C. Djerassi, J. Org. Chem., 44, 3385 (1979).
I. A. Holder and S. T. Boyce, Burns, 20, 426 (1994).
B. Meyer, N. Ferrigni, J. Putnam, L. Jacobsen, D. Nichols, and J. McLaughlin, Planta Med., 45, 31 (1982).
Acknowledgment
The authors wish to thank Mr. Kamal Al-dahodi (Faculty of Maritime Studies, King Abdulaziz University) for sample collection and identification.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 2, March–April, 2022, pp. 261–264.
Rights and permissions
About this article

Cite this article
Angawi, R.F., Alamri, M.M., Ayyad, SE.N. et al. Two New Bioactive Metabolites from Xestospongia sp. with Antimicrobial and Anticancer Activities. Chem Nat Compd 58, 301–306 (2022). https://doi.org/10.1007/s10600-022-03664-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-022-03664-3