

For the estimation of the biological affinity of nitrogen-containing π-conjugated heterocyclic systems toward amino acid residues in proteins, the fragment-to-fragment approach was proposed. Two mechanisms of complexation between the heterocycle molecule with different donor/acceptor properties and the amino acid residue in the active part of the protein biomolecule were considered. One of these mechanisms is the π-stacking interaction and the other is formation of hydrogen bonds with model amino acid residues. It was found that heterocycles with a π-conjugated electron-acceptor moiety form a more stable heterocycle–biomolecule complex with protein fragments. Nitrogen-containing conjugated heterocycles with several nitrogen atoms form poly-hydrogen-bonded complexes. The stabilization energy of complexes with two pyrimidine–biomolecule hydrogen bonds increases by 4–6 kcal/mol compared to similar complexes with one hydrogen bond. Hydrophobic interactions are much more sensitive to the donor-acceptor properties of heterocycles in the formation of hydrogen-bonded complexes than in the formation of π-stacked complexes. The hydrophobic effect in the fragment-to-fragment approach allows us to see the values of the stabilization energies of the heterocycle–biomolecule complexes as close as possible to the experimentally studied biological systems.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Conjugated nitrogen-containing heterocyclic systems form the basic core of some natural products such as amino acids and proteins, nucleic acids, vitamins, and some neurotransmitters. Therefore, these types of heterocyclic conjugated systems are favorite platforms for the discovery of new drugs and modeling their effectiveness. Among the different heterocyclic compounds, imidazole is better known due to its broad range of chemical and biological properties. Imidazole has become an important synthon in the development of new drugs.
Derivatives of 1,3-azoles show different biological activities such as antibacterial, antimycobacterial, antiinflammatory, antitumor, antidiabetic, anti-allergic, antipyretic, antiviral, antioxidant, anti-amoebic, anthelmintic, antifungal, and ulcerogenic.1,2 Pyrimidines and their derivatives are also considered important bases in medicinal chemistry. A large number of reported pyrimidine derivatives exhibit antimycobacterial, antitumor, antiviral, anticancer, and antimicrobial activities.3,4 Condensed pyrimidines continue to attract a lot of attention because of their great practical utility, primarily because of the very wide spectrum of biological activities. In recent years,5–7 pyridine- and pyrimidine-based anticancer drugs have been developed based on structural modification of these core structures. The in silico drug discovery pathway is especially important for identifying new active substances as it allows to use, at an early stage, fragment-based drug discovery strategy8,9 and fragment-to-fragment approach10–11 for the analysis of the interaction between a small heterocycle molecule,12 e.g., heterocycle (Het), and the amino acid residue in the active part of the protein biomolecule (BioM).13,14
The key condition for the biological action of any potential drug compound is the formation of a [Het–BioM] complex with the target protein molecule. The ability of heterocyclic systems to form such a [Het–BioM] complex is called affinity.15,16 In addition, hydrophobic interaction affects the energy of stabilization of such [Het–BioM] complex.
This paper presents the results of in silico studies of the stability of [Het–BioM] complexes of the simplest nitrogencontaining conjugated heterocyclic molecules that will be considered in detail, taking into account the dependence of the intermolecular interaction on the donor-acceptor properties of both the heterocycle and the biologically active molecule, using the fragment-to-fragment approach.
Here we will consider only the two most widely known heterocycles with two nitrogen atoms, which are used as a platform for many pharmacological drugs. A typical representative of donor heterocycles can be imidazole (1), and a typical heterocycle of acceptor nature is pyrimidine (2). In the 5-membered ring of compound 1, the first nitrogen atom (NH) with trtrtrπ2 configuration donates two electrons into the conjugated system, while the second nitrogen atom with tr2trtrπ configuration gives only one electron. The excess of π-electrons in comparison with the number of π-centers provides the donor property of molecule 1. In the 6-membered ring of pyrimidine (2), both nitrogen atoms have the tr2trtrπ configuration, and, therefore, each atom gives one electron. An electronically balanced π-system with two nitrogen atoms with higher electronegativity provides the acceptor properties of molecule 2. Both platforms are simple conjugated molecules and contain dicoordinated nitrogen atoms (one or two, respectively) with a lone electron pair (LEP), which can be used as an acceptor center for the formation of hydrogen bonds.
Of course, real drug molecules also contain various substituents in the heterocycle, but they are linked with the main platform by σ-bonds and, therefore, do not fundamentally affect the donor-acceptor properties of the substance and the possibility of hydrogen bonding at the LEP of dicoordinated nitrogen atoms.
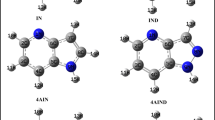
Only four amino acids contain conjugated residues: phenylalanine, tyrosine, tryptophan, histidine. They can form a [π,π] complex with “platform” 1 and 2; in addition, histidine can form a hydrogen bond with ligands at the LEP of the dicoordinated nitrogen atoms, and tyrosine can be a donor of the hydrogen atom by the OH group during the stabilization of the [Het–BioM] complex through the hydrogen bond mechanism. In this work, instead of real acids, their model molecules 3–6 will be used, the noncyclic fragment of the amino acid being replaced by methyl group, as shown in Figure 1. Furthermore, for comparison, the interaction with 2-methylbutane (7) as model leucine will be considered. In addition, peptide fragments are formed in the protein chain with their conjugated system, which includes electronic pairs of the amide C=O group, as well as LEP at the nitrogen atom; this system is modeled by molecule of acetamide (8) with the simplest conjugated fragment. The structures of these compounds are presented in Figure 1.

Chemical structures of drug-like heterocycles 1, 2, amino acid models 3–7, and the peptide bond model 8.
Some proteins contain functional groups OH, NH, SH which participate in the formation of hydrogen bonds as donor centers. The rest of the components were modeled by methyl groups, replacing the amino acid residue, optimistically assuming that the effect of the unconjugated portion of amino acids is insignificant. A biomolecule, or rather an amino acid in the [Het–BioM] complex, stabilized by a hydrogen bond, is modeled by quantum-chemical calculations on a simpler molecule: MeХ, where X = OH, NH, Н. It is unlikely that other amino acid fragments in protein molecules can form a stable complex with any π-conjugated systems. A [Het–BioM] complex between a nitrogen-containing conjugated heterocyclic system and a biomolecule is formed both due to the effective interaction of the π-system of both components (π,π interaction, π,π stacking, [π,π] complex) and due to hydrogen bonds between LEPs of dicoordinated nitrogen atoms of heterocycles and a proton of biomolecules ([HB] complex). It can be assumed that the boundary π-molecular orbitals (MO) and higher molecular orbitals (n-MO) play a decisive role in these interactions. Therefore, we will restrict ourselves to only these MOs, comparing the effect of the donor-acceptor property (or basicity) of both conjugated platforms 1 and 2.
According to the molecular docking results, compounds based on imidazole (1) or its hetero analogs and pyrimidine can form [Het–BioM] complexes with a peptide fragment: protein–ligand complex.5,14 This indicates a significant stacking interaction between the systems of both components of the complex. Naturally, the frontier molecular orbitals (the highest occupied MO (HOMO) and the lowest unoccupied MO (LUMO)) of both molecules contribute to the formation of the [π,π] complex. Calculations of the studied molecules 1 and 2 show that among π-MO are n-MO, similar in both nitrogen heterocycles 1 and 2. In the molecule of pyrimidine (2), there are two such n-MO one of which is a frontier orbital. Thus, the probability of complexation by the mechanism of hydrogen bonding with biomolecules for pyrimidine (2) increases significantly. There are also two centers of such complexation. The mutual arrangement and shape of the frontier MOs, as well as some of the nearest orbitals, are shown in Figure 2, while their energies are presented in Table 1.

Shape of the π-MO and n-MO in imidazole (1) and pyrimidine (2).
Figure 2 shows that the frontier molecular orbitals HOMO and LUMO of imidazole (1) are shifted upward compared to the corresponding molecular orbitals of pyrimidine (2). At the same time, the energy gap (the distance between the HOMO and LUMO) significantly decreases passing from molecule 1 to molecule 2. The n-MOs are located below the frontier orbital in the donor imidazole (1), while acceptor pyrimidine (2) contains two n-MO LEPs, they are cleaved and one of those is the HOMO.
It is to be noticed that the HOMO corresponds to the valence band top whereas the LUMO corresponds to the conductivity band bottom, as well as the distance between both bands is the energy gap; the relative position of the gap with respect to the Fermi level points directly to the donor-acceptor properties of the electronic system.
Table 1 shows that since n-MOs of compounds 1 and 2 (HOMO) are located at a level close to nonbonding, they can effectively form hydrogen bonds with the protons of functional groups of amino acid residues in proteins, such as OH, NH2, or SH. Moreover, this type of bond formation will be more typical for pyrimidine derivatives, since its n-MO occupies the HOMO, from which the first electronic transition occurs corresponding to the bond formation.
Previously,17 we proposed to estimate quantitatively the donor-acceptor properties of conjugated systems by parameter φ0 calculated by the following equation (1):
where Δ = εLUMO – εHOMO, εLUMO is the energy of the LUMO; εHOMO is the energy of the HOMO; α = –3.56 еV is the energy of Fermi level.18
For neutral conjugated molecules (for example, long unsubstituted polyenes or acenes), α corresponds to such disposition of the frontier MOs when the donor and acceptor properties are mutually balanced and φ0 = 0.5, i.e. the energy gap is situated symmetrically with respect to the imaginary level α. The shift of the energy gap (and hence increase of the parameter φ0 > 0.5) indicates mainly the donor nature of the conjugated molecule. On the contrary, if φ0 < 0.5, then the frontier MOs are shifted down, evidencing a predominately acceptor nature of the molecule.19 The calculated MO energies and parameter φ0 of the molecules studied 1–8 are collected in Table 2.
Firstly, the values obtained in Table 2 for the parameter φ0 indicate that imidazole (1) is a conjugated donor system, while pyrimidine (2) is a conjugated acceptor molecule. Moreover, in pyrimidine (2) HOMO is n-orbital, HOMO-1 is π-orbital. Thus, it is logical that a particular parameter φ0 can also be used for relative positions of n-MOs. As can be seen from Table 2, φ0 for n-MO (φ0n) is slightly smaller than φ0 for π-MO (φ0π), except for pyrimidine (2), which exhibits acceptor properties.
As for the conjugated fragments of model amino acids 3–6, the model phenylalanine 4 can be considered as a weak donor due to the inductive effect of the methyl group (φ0 = 0.53); the introduction of a hydroxyl group (model tyrosine 5) increases the donor properties (φ0 = 0.57); the basicity of model histidine 3 and model tryptophan 6 is higher and, therefore, the parameter is larger (φ0 = 0.64 and φ0 = 0.59, respectively); the model leucine 7 is the weakest donor φ0 = 0.54, although the nature of the MOs is different. The model peptide 8 molecule is also a weak donor, the parameter changes insignificantly on going from π-MO (φ0π = 0.56) to n-MO (φ0n = 0.54), φ0n is close to the value of 0.5.
Nitrogen-containing heterocyclic π-conjugated system [Het] and a biomolecule [BioM] form a [Het–BioM] complex (Scheme 1).

Scheme 1
The stability of the [Het–BioM] complex depends on the electronic environment of both components. We assumed that the complex and its components are neutral and hence no electron redistribution between the components upon complexation occurs.20 Stabilization energy of the complex (or binding energy Ebind) was estimated as the difference of the total energies of the complex components:
where EComplex is energy of [Het–BioM] complex after optimization of molecular geometry, while EComp(n) and EComp(n–1) are the energies of the individual components after optimization of molecular geometry.
Generally, according to the perturbation theory,21 the intermolecular interaction in the complex between two systems A and B which could represent the heterocycle and biomolecule, respectively, depends on the relative positions of the frontier MOs of both molecules, as well as on the overlap of their π- or n-MOs. It can be quantified using the following equation (3):
where εi and εj are energies of MO i and j; Ciμ and Cjν are MO coefficients at atoms μ and ν; indices i, μ pertain to system A, while indices j, ν pertain to system B; the first two sums run over all energy levels, whereas the second two sums run over all atoms.
In the ground state, all MOs in the valence band are occupied by electrons, whereas the vacant MOs in the conductivity band contain "holes". In the framework of molecular orbital theory,21 the occupied (or vacant) orbitals repel each other, while the vacant and occupied MOs attract each other pairwise. Therefore, it follows from equation (3) that the interaction between the components is inversely related to the distance between interacting levels and is proportional to the overlapping of both n- or π-systems.
As an illustration, the optimized [π,π] complex of pyrimidine (2) and toluene (4) as model phenylalanine are presented in Figure 3.

Optimized [π,π] complex between pyrimidine (2) and model phenylalanine 4 (visualized by the HyperChem package).
All pairwise interactions between conjugated components can be quantum-chemically calculated. According to equation (3), the stabilization energy of the complex should depend first of all on the distance between the frontier MO energy levels of the interacting components.
The calculated distances between the valence top of the first component (drug model heterocycle) and the conductivity bottom of the second component (amino acid model) for all possible [π,π] complexes are presented in Table 3.
The numerator in formula (3) points to the fact that interaction between two components of the [π,π] complex depends on the overlap of the MOs, whereas the denominator shows that the interaction is inversely on the values of the energies of molecular orbitals.
Calculations show that the interactions of the π-conjugated nitrogen-containing heterocycles 1 and 2 with π-systems of model fragments 3–6, and 8 are possible, thus such interactions should stabilize the [π,π] complex.
The calculated data in Table 3 indicate unambiguously that the distances between frontier levels in [π,π] complexes of heterocycles depend on the basicity of both conjugated components. Generally, the distances between the HOMO of heterocycle εHOMO(Het) and the LUMO of protein fragment εLUMO(Frag) are close for both molecules 1 and 2. In contrast, the corresponding distances between the HOMO of the protein fragment εHOMO(Frag) and the LUMO of the heterocycle εLUMO(Het) differ considerably for both platforms, i.e. they are essentially sensitive to the parameter φ0. Thus, the distances between the HOMO of the donor imidazole (1) and the LUMO of any conjugated model protein fragment are less than the corresponding values for the acceptor pyrimidine (2). At the same time, the distances between the HOMO of any protein fragment and the LUMO of the heterocycles are greater for the donor imidazole (1), compared with acceptor pyrimidine (2). The maximum values Δε1 and Δε2 are obtained for the model peptide 8 and model histidine 3 with their greatest energy gap. We could assume that decreasing the electron donor ability of the heterocycle should lead to the increase of the energy of stabilization of [π,π] complexes. Moreover, it is to be noted that the formation of the [Het–BioM] complexes is accompanied by MOs splitting of the components.
For the purpose of illustration, the shapes of the frontier and closest molecular orbitals of the complexes of both heterocycles 1 and 2 with the model phenylalanine 4 are pictured in Figure 4.

Shapes of the MOs in the [π,π] complexes [Het–BioM] between compounds a) 1, b) 2 and model phenylalanine 4.
One can see that the HOMO in the [Het–BioM] complex (Fig. 4a) is the LEP and hence it is mainly located at heterocycle 1, whereas the HOMO-1 is totally delocalized. The delocalized LUMO is concentrated at the model phenylalanine fragment 4. It has maximum overlap with HOMO-1, and, consequently, their interaction should be effective for the complex stabilization. In the [Het–BioM] complex (Fig. 4b), the frontier MOs are located at separated components. They should overlap effectively and hence stabilize the [π,π] complex. Analogically, the interaction of other orbitals could be also taken into consideration. Of course, the perpendicular overlap of the pz-orbitals of the different components in the [π,π] complex is lesser than the overlap in the planar conjugated system. Nevertheless, this interaction between the conjugated systems of both components provides the stabilization of the [π,π] complex. The calculated stabilization energies of the possible [π,π] complexes by formula (2) are presented in Table 4.
Table 4 shows that the calculated stabilization energies of all possible [π,π] complexes confirm the assumption that their stability depends on the donor-acceptor properties of the heterocycle molecule: all complexes with acceptor pyrimidine (2) are more stable than the corresponding complex with donor imidazole (1). We consider the formation of a [Het–BioM] complex according to the principle of n,π-stack interaction between the model leucine and heterocycles 1 and 2. As can be seen from Table 4, the [n,π] complex of leucine with acceptor pyrimidine (2) is more stable than with donor imidazole (1).
The difference in binding energy of [π,π] complexes also depends on the donor properties of the model fragments of proteins 3–8 and reaches a maximum for a pair of [1–3] and [2–3] complexes, while the calculated binding energies for the complex of both heterocycles 1 and 2 with model peptide 8 differ minimally.
Hydrophobic effects are considered very significant in biological processes for protein folding and ligand binding.22–24 They are associated with the interaction between water and dissolved compounds, which leads to the formation of a common surface of contact between the two components and lowers the energy barrier of the approach of molecules, thereby increasing the possibility of complexation. Figure 5 schematically shows the mechanism of the hydrophobic effect of water on the components of the [Het–BioM] complex.

Hydrophobic effect as the mechanism of complexation.
Here, we will treat the hydrophobic effect as an energetic advantage when two organic molecules generate the complex, and hence the water molecules between them are absent, in comparison with the separated components (as can be seen from Figure 5). Calculated hydrophobic effects for the investigated heterocycles with model fragments of protein molecules 3–8 are presented in Table 5. The calculated data indicate that the hydrophobic effects are strong enough and depend, first of all, on the area of overlap of both components in the [Het–BioM] complex. The dimensions of pyrimidine 2 are slightly larger than those of imidazole 1, and, therefore, the hydrophobic effects of its complex with biomolecules 3–8 are slightly larger. There is also a difference in the stabilization energies of the [π,π] complex. This is undoubtedly associated with the donor-acceptor properties of both heterocycles 1 or 2 and protein fragments 3–6.
Nevertheless, there is a clearly pronounced dependence of the stabilization energy of [Het–BioM] complexes under the influence of the hydrophobic effect on the basicity of heterocycles 1 and 2: increasing of the φ0 parameter is accompanied by an increasing of additional stabilization of the [π,π] complex due to the hydrophobic effect.
Another type of [Het–BioM] complexes is formed by the generation of hydrogen bonds between nitrogen atoms of heterocycles with LEP which are considered as electron acceptors, and amino acids in proteins containing functional groups such as NH2, OH, SH. In the case of model protein fragments 3–8, the non-conjugated fragment of model amino acids was modeled by methyl group. Therefore, the various conformations of the peptide chain are not taken into consideration, as we suppose optimistically that we could neglect them for our purpose. Besides, we will compare the stabilization energies of [HB] complex in the series of chemically similar compounds. In addition, a peptide fragment of a protein can give a proton to the NH group as a donor center, and give a pair of electrons to the oxygen atom of the C=O group as an acceptor center.
According to formula (3), the stability of possible [HB] complexes between molecules 1 or 2 and model proton donors depends on the distance between the n-level of nitrogen atoms of LEP in heterocycles 1–2 and on the LUMO of the model proton donor MeXH (were X = O, S, NH), which can be seen in Figure 6.

Shapes of the MOs in the [HB] complexes [Het–BioM] between compounds а) 1, b) 2 and model MeSH.
The possibility of stabilizing the complex between heterocycles 1 and 2 by the mechanism of hydrogen bonding also depends on their donor-acceptor properties. As can see from Figure 6a, imidazole is a better donor and its n-MO is on HOMO-1, then in the HOMO [HB] complex (which is responsible for the first electronic transition – the chemical bond formation energy) it belongs to the proton donor, the model SH group (MeSH molecule), and LUMO belongs to imidazole (1). Conversely, when a [Het–BioM] complex is formed by the hydrogen bond mechanism with a better acceptor pyrimidine (2) in the [HB] complex, HOMO belongs to pyrimidine (2), and LUMO belongs to the donor group, in this case SH (see Fig. 6b). It should also be noted that such properties of heterocycles 1 and 2 affect the stabilization energy values of their complexes, which are collected in Table 6.
First, the calculations give the binding energies of [HB] complexes, which are close to the stabilization energies of the [π,π] complexes. Of course, these energies are determined by the nature of the donor center: the highest energy is obtained for the [HB] complex with the OH group (model MeOH), especially in the [HB] complex with the acceptor pyrimidine platform 2.
In addition, a hydrogen bond can arise through the donor center of the three-coordinated nitrogen atom (proton of the NH group) of model tryptophan (6) and model histidine 3. The calculations shown in Table 6 show that the binding energies of the [HB] complexes with these protein fragments exceed the corresponding values for the [HB] complexes with the model MeXH (were X = O, S, NH). The stability of the [HB] complex of model tryptophan 6 and model acceptor of histidine 3 with pyrimidine (2) exceeds 2–5 kcal/mol.
It should be noted that [HB] complexes can theoretically be formed with the NH group in the model peptide 8. The calculated binding energies of such complexes are of the same order, and the [HB] complex with the acceptor is more stable than [π,π] complexes.
Finally, it must be considered that pyrimidine (2) allows two acceptor sites to form the [HB] complexes. Then even two hydrogen bonds can form simultaneously, forming a common [HB] complex. The energies of stabilization of such bis-complexes, as well as their comparison with the respective mono-complexes, are presented in Table 7.
As can be seen from Table 7, bis-[HB] complexes of pyrimidine (2) with model MeXH are more stable (by about 4–5 kcal/mol). The stabilization energy of such biscomplexes also depends on the nature of the X atom of the model proton donor and has the same tendency to change as mono-[HB] complexes. It should be noted that the expected increase of the binding energy of bis-complexes relative to the value of the stabilization energy of monocomplexes is not observed. However, it is noticeable that the stabilization of the bis-[HB] complex by a model MeSH molecule has the largest difference. Hydrophobic interactions induce some order in the surrounding water. Small hydrophobic units reduce the volume of configuration space available for hydrogen bonding. In the case of water-soluble compounds, the water molecules can adopt an orientation that allows the hydrogen bonding to orient the compounds relative to each other with minimal energy consumption (see Fig. 5). In this case, the probability of complexation increases significantly.
Let us consider the hydrophobic effect of water molecules on the stabilization energy of complexes formed between heterocycles 1 and 2 as electron pair donors and the model MeXH as proton donors, presented in Table 8.
Table 8 shows that hydrophobic interactions have a colossal effect on the energy of stabilization of complexes by the mechanism of hydrogen bond formation: they reduce the energy effect by several times compared to calculations in a vacuum, making the binding energy of [HB] complex formation as close as possible to biological systems.25,26 Interestingly, the hydrophobic effect on the stabilization energy of the bis-[HB] complexes of pyrimidine (2) is higher compared to mono-[HB] complexes, in contrast to the energy in vacuum. Moreover, hydrophobic effects are sensitive to the donor-acceptor properties of heterocycles 1 and 2: the stabilization energy of complexes with electron-donor imidazole (1) decreases about 2.5 times, and with electron-acceptor pyrimidine (2) – approximately 3.5 times relative to these values in vacuum.
Thus, a detailed in silico study using the fragment-tofragment approach shows that nitrogenous conjugated heterocyclic compounds can form a stable [Het–BioM] complex with the conjugated protein model fragments through a π-stack interaction, as well as due to the hydrogen bond between LEPs at the dicoordinated nitrogen atom as an acceptor center and the proton of functional groups NH, OH, and SH of amino acid residues. The relative position of the boundary MOs (φ0 parameter) is used to quantify the donor-acceptor properties of all components of the [Het–BioM] complex. It was found that the energy stabilization of the [Het–BioM] complex, formed both by the π-stack mechanism and by the mechanism of hydrogen bonding, is sensitive to the parameter φ0.
For nitrogen-containing conjugated heterocycles with several nitrogen atoms (several LEP), it is possible to form poly-[HB] complexes. In the example of pyrimidine, it was found that the stabilization energy of bis-[HB] complexes increases by 4–6 kcal/mol compared to similar mono-[HB] complexes, but not two times, as might be expected.
The influence of the hydrophobic effect on the stabilization energy of the [Het–BioM] complexes by quantumchemical calculations has been studied. Hydrophobic interactions are much more sensitive to the donor-acceptor properties of heterocycles in the formation of [HB] complexes than in the formation of [π,π] complexes. Thus, under the influence of the hydrophobic effect the stabilization energy of [π,π] complexes increases 3 times both in complexes with donor imidazole and in complexes with acceptor pyrimidine in comparison with similar calculations in a vacuum. Moreover, the stabilization energy of [HB] complexes with donor imidazole under the influence of the hydrophobic effect increases 2 times while in [HB] complexes with acceptor pyrimidine the stabilization energy of HB complexes increased 3 times in comparison with similar calculations in a vacuum. It should be noted that the hydrophobic effect has a much smaller effect on the stabilization energy of bis-[HB] complexes compared to mono-[HB] complexes.
The hydrophobic effect in the fragment-to-fragment approach allows us to see the values of the stabilization energies of the [Het–BioM] complexes as close as possible to the experimentally determined biological systems.
Computational details
The main characteristics of the electron structure (optimized molecular geometry, charge distribution, energies and shape of MOs) were calculated by DFT wB97XD/6-31G(d,p) method using the Gaussian 0927 software package. In the quantum-chemical modeling of the interactions between the model nitrogen-containing heterocycles and the model biomolecules for both types of complexation, the previous optimization of both components was performed. To simulate a [π,π] complex, the interacting molecules were located in parallel at a distance of 3.4 Å,28–30 the dipole moments of each component being oriented antiparallel to each other.31,32 To simulate [HB] complexes between nitrogen heterocycles and biomolecules by the mechanism of hydrogen bonding, we took into account the 2 Å distance25,26 between the LEP of the dicoordinated nitrogen atom of heterocycle and proton of the biomolecule. After this initial position, the geometry optimization procedure, π-stacking interaction between the heterocyclic system and the biomolecule, was studied according to the appropriate mechanisms. The hydrophobic effect was calculated using the polarized continuum model (PCM).
We would like to thank the Enamine Ltd. for the financial and technical support.
References
Siwach, A.; Verma, P. K. BMC Chem. 2021, 15, 12.
Waller, D. G.; Sampson, A. P. Medical Pharmacology & Therapeutics; Elsevier: Edinburgh, 2018, p. 581.
Mohana, K. N.; Prasanna K. B. N.; Mallesha, L. Drug Invent. Today 2013, 5, 216.
Prachayasittikul, S.; Pingaew, R.; Worachartcheewan, A.; Sinthupoom, N.; Prachayasittikul, V.; Ruchirawat, S.; Prachayasittikul, V. Mini-Rev. Med. Chem. 2017, 17, 869.
Velihina, Ye.; Scattolin, T.; Bondar, D.; Pil'o, S.; Obernikhina, N.; Kachkovskyi, O.; Semenyuta, I.; Caligiuri, I.; Rizzolio, F.; Brovarets, V.; Karpichev, Ye.; Nolan, St. P. Agents Helv. Chim. Acta 2020, 103, e2000169.
Velihina, Ye. S.; Obernikhina, N. V.; Pilyo, S. G.; Kachkovsky, O. D.; Brovarets, V. S. Curr. Org. Chem. 2021, 25, 1441.
Zhirnov, V. V.; Velihina, Ye. S.; Mitiukhin, O. P.; Brovarets, V. S. Chem. Biol. Drug Des. 2021, 98, 561.
Mortenson, P. N.; Erlanson, D. A.; de Esch, I. J. P.; Jahnke, W.; Johnson, C. N. J. Med. Chem. 2019, 62, 3857.
Ribeiro de Souza Neto, L.; Moreira-Filho, J. T.; Junior Neves, B.; Riveros Maidana, R. L. B.; Ramos Guimarães, A. C.; Furnham, N.; Horta Andrade, C.; Paes Silva, F. Front. Chem. 2020, 8, 93.
Obernikhina, N. V.; Kobzar, O. L.; Kachaeva, M. V.; Kachkovsky, O. D.; Brovarets, V. S. Curr. Comput.-Aided Drug Des. 2022, 18, 95.
Velihina, Y. S.; Obernikhina, N. V.; Pilyo, S. G.; Kachaeva, M. V.; Kachkovsky, O. D. Ukr. Bioorg. Acta 2021, 16(1), 34.
Obernikhina, N.; Zhuravlova, M.; Kachkovsky, O.; Kobzar, O.; Brovarets, V.; Рavlenko, O.; Kulish, M.; Dmytrenko, O. Appl. Nanosci. 2020, 10, 1345.
Marquez, B. L.; Watts, K. S.; Yokochi, A.; Roberts, M. A.; Verdier-Pinard, P.; Jimenez, J. I.; Hamel, E.; Scheuer, P. J.; Gerwick, W. H. J. Nat. Prod. 2002, 65, 866.
Kachaeva, M. V.; Hodyna, D. M.; Semenyuta, I. V.; Pilyo, S. G.; Prokopenko, V. M.; Kovalishyn, V. V.; Metelytsia, L. O.; Brovarets, V. S. Comput. Biol. Chem. 2018, 74, 294.
Jordan, M. Interactions 2010, 17(5), 6.
Obernikhina, N. V.; Nikolaev, R. O.; Kachkovsky, O. D.; Tkachuk, Z. Yu. Dopov. Nac. akad. nauk Ukr. 2019, (6), 75.
Obernikhina, N.; Kachaeva, M.; Shchodryi, V.; Prostota, Ya.; Kachkovsky, O.; Brovarets, V.; Tkachuk, Z. Polycyclic Aromat. Compd. 2020, 40, 1196.
Obernikhina, N.; Pavlenko, O.; Kachkovsky, A.; Brovarets, V. Polycyclic Aromat. Compd. 2021, 41, 2110.
Kachaeva, M. V.; Obernikhina, N. V.; Veligina, E. S.; Zhuravlova, M. Yu.; Prostota, Ya. O.; Kachkovsky, O. D.; Brovarets, V. S. Chem. Heterocycl. Compd. 2019, 55, 448.
Bissantz, C.; Kuhn, B.; Stahl, M. A J. Med. Chem. 2010, 53, 5061.
Rauk, A. Orbital Interaction Theory of Organic Chemistry; John Wiley & Sons: New York, 2001, 2nd ed., p. 34.
Cui, X.; Liu, J.; Xie, L.; Huang, J.; Liu, Q.; Israelachvili, J. N.; Zeng, H. Angew. Chem., Int. Ed. 2018, 57, 11903.
Davis, J. G.; Gierszal, K. P.; Wang, P.; Ben-Amotz, D. Nature 2012, 491, 582.
Berne, B. J.; Weeks, J. D.; Zhou, R. Annu. Rev. Phys. Chem. 2009, 60, 85.
Marx, D. Chem. Phys. Chem. 2006, 7, 1848.
Zheng, S.; Xu, S.; Wang, G.; Tang, Q.; Jiang, X.; Li, Z.; Xu Y.; Wang R.; Lin, F. J. Chem. Inf. Model. 2017, 57, 1535.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox D. J. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, 2016.
Kim, K. S.; Tarakeshwar, P.; Lee, J. Y. Chem. Rev. 2000, 100, 4145.
Churchill, C. D. M.; Rutledge, L. R.; Wetmore, S. D. Phys. Chem. Chem. Phys. 2010, 12, 14515.
Singh, S. K.; Das, A. Phys. Chem. Chem. Phys. 2015, 17, 9596.
Kouza, M.; Banerji, A.; Kolinski, A.; Buhimschi, I.; Kloczkowski, A. Molecules 2018, 23, 1995.
Gao, X.-C.; Hao, Q.; Wang, C.-S. J. Chem. Theory Comput. 2017, 13, 2730.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Geterotsiklicheskikh Soedinenii, 2022, 58(8/9), 412–420
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Obernikhina, N.V., Kachaeva, M.V., Kachkovsky, O.D. et al. In silico Study of Conjugated Nitrogen Heterocycles Affinity in their Biological Complexes. Chem Heterocycl Comp 58, 412–420 (2022). https://doi.org/10.1007/s10593-022-03107-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-022-03107-5