
Abstract
Wild soybean individuals were sampled from ten habitat sites in Beijing region, China and were assessed using 36 SSR markers for the genetic variation among the habitat subpopulations. AMOVA analysis showed 57.46 % inter-population and 42.54 % intrapopulation genetic variation. The genetic variation had geographical regionality. The drought-stressed and founder subpopulations intensively reduced genetic diversity, and along-river system habitats appeared to have closer genetic similarity. The bottleneck impact of drought stress appeared to be inferior to the founding effect on subpopulation genetic diversity but superior on genetically geographical grouping. Here, all the subpopulations were found to contain unique alleles. The phenotypic and genetic diversities had similarly fluctuated patterns across the subpopulations. These results here suggest that a conservation strategy should be taken: theoretically as many as possible populations are sampled to maximize the genetic diversity in ex situ conservation of wild soybean within an area in China. Spatial distance should be considered for isolating wild soybean populations when genetically modified soybeans are cultivated in China.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The progenitor of cultivated soybeans [Glycine max (L.) Merrill] (Fukuda 1933; Hymowitz 1970), wild soybean (Glycine soja Sieb. & Zucc.) as an genetic resource for soybean breeding, is widely distributed in eastern Asia. This species is an herbaceous, annual and self-pollenating plant with a twining or procumbent stems, and commonly grows in fields such as watersides, roadsides, sparse woodlands, brushes, tussocks, abandoned farmlands, ditches, sunny hillsides, and around villages. Over the past 30 years a great number of wild soybean populations are unfortunately extinct or have been significantly reduced in China. Now this species has been placed in the protected plant catalogue because of its great significance to breeding and its serious loss of distribution habitats in China.
Many studies of genetic diversity related to the conservation of wild soybean have been conducted. Choi et al. (1999) reported a geographic pattern of SSR variation of wild soybean along five main Korean rivers. Tozuka et al. (1998) demonstrated the clinal distribution of mitochondrial DNA-RFLP in Japanese wild soybean. Wang and Takahata (2007) compared the geographic distribution of SSR variation between China and Japan. Chinese wild soybean has been studied for geographic diversity in morphology (Dong et al. 2001) and for geographical variation in chloroplast and mitochondrial DNA (Shimamoto et al. 1998; Xu et al. 2002). Isozyme analysis was carried out for a few natural populations in Korean, Japan (Kiang et al. 1992; Fujita et al. 1997) and China (Li et al. 1995).
The conservation of genetic diversity in wild soybean is actualized usually through both in situ and ex situ approaches. Effective conservation of the genetic diversity of natural populations is essential, which is involved in the cost, worthiness and validity of the conservation. Jin et al. (2003a) has suggested a sampling strategy for a single natural population of wild soybean through an ISSR analysis in a natural population case. However, another important sampling strategy is worthy of consideration, that is that how all the subpopulations in a region should be treated in in situ or ex situ conservation. It is important to understand the degree of genetic diversity, and its distribution patterns, and subpopulation genetic structures under various habitats or ecogeographical environments. In China, facing the fact that natural populations of wild soybean have been reducing in number and area, how to effectively protect wild soybean resource has become pressing. Our objective of this work were to assess the subpopulation genetic diversity in different habitats all over a region and understand how the subpopulations in a geographical or ecological region should be dealt with when conserving high genetic diversity with respect to sampling strategy for situ or ex situ conservation within a region.
Wild soybean is sensitive to environmental selection (Wang and Li 2012), and different ecogeographical locations and habitats would result in population genetic differentiation and emergence or elimination of specific alleles among populations in vast areas. To avoid overlarge ecogeographical effect because of vast regional areas, we selected an area-suitable ecotope, Beijing region as the studying simulation region.
Materials and methods
Sampling
We collected geographical site subpopulations of wild soybean in Beijing region (Table 1; Fig. 1). The sampling sites were selected considering representative geographic limits, most common habitats and practical distribution status. These sites were about 13–100 km apart (beeline distance) and separated in four areas: north mountain area (sites 1, 2 and 3 were distributed along the same river valley), east hill area (sites 4 and 5), central plain area (sites 6, 7, 8 and 9) and western mountain area (site 10, located in the eastern edge of this area, near the central plain area). Site 1 grew on a semiarid sandy soil close to a lake. Site 2 was a newly generated founder population of a small area with homogeneous morpha on a waterside. Sites 3, 5 and 8 had fertile habitat conditions. Site 4 was situated on an arid sandy soil, presenting dwarfish forms of plants with small leaves. Sites 6 and 7 were along an arroyo of a river. Site 9 grew in a semi-moist riverbank. Site 10 lay to roadsides on the foot of a mountain. Seeds of total 294 individual plants were gathered from 28 to 30 individuals at least 5–15 m apart from each other in each site based on Sjögren and Wyöni (1994)’s suggestion on plant sampling and Jin et al. (2003a)’s sampling strategy within a wild soybean natural population for conservation of genetic diversity. Twenty-eight Beijing cultivated soybeans were used for identification of the origin of the big seed individuals that appeared in a subpopulation.
Survey of subpopulation phenotypic characters
To identify morphological characters of the sampled plants, the seed samples from 294 individual plants were grown as 294 individual families by ten seeds each family in a condition-uniform experimental field and measured for main morphological traits. Each family line was harvested (6–9 plants survived in families) and measured for the mean values of quantitative traits (plant height, aboveground dry matter weight, yield per plant and 100-seed weight), and the population mean values were calculated by the arithmetic average of all family lines in subpopulations (Table 3).
SSR marker analysis
Total DNA was extracted from 20-d old leaves of the 294 family seedlings. Thirty-six public SSRs over 20 genetic linkage groups (Cregan et al. 1999) were applied to Polymerase chain reaction (PCR) amplification for these samples (Table 2). These markers were selected for their stability, as described in two reports on SSR assessment of genetic diversity in Chinese soybeans (Xie et al. 2003; Wang et al. 2003). PCR amplification was carried out in a volume of 20 μl reaction mixture containing 100 ng of template DNA, 10 pmol of each primer, 100 μM of each of the four dNTPs, 1 U Taq polymerase, and 2 μL of 10 × PCR buffer. Cycling consisted of 30 s denaturation at 94 °C, 30 s annealing at 47 °C, 30 s extension at 72 °C for 30 cycles. The amplified products were run on a 6 % gel (SDS-PAGE) and the bands were developed with silver staining.
Data analysis
Each pair of primers as a locus and each variant (bands) as an allele were recorded. POPGENE1.32 (Yeh et al. 2000) was used to estimate the observed total number of alleles (Na), Shannon–Weaver index (I, Shannon and Weaver 1949), and Nei’s gene diversity statistics (H S : intra-subpopulation genetic diversity; D ST : inter-subpopulation genetic diversity; G ST : amount of inter-subpopulation gene differentiation, Nei 1973) (Table 2). A clustering analysis for site subpopulations was performed in software PowerMarker version 3.25 (Liu and Muse 2005) using UPGMA with the software NEIGHBOR of PHYLIP version 3.5c (Felsenstein 1993) based on Nei’s genetic distance (Nei and Takezaki 1983). An AMOVA analysis was used to assess within-population genetic variation and population genetic parameter estimates (Table 3) using PowerMarker 3.25 (Liu and Muse 2005). Arlequin 3.1 (Excoffier et al. 2005) was used to calculate overall variation of subpopulations. A principal component analysis (PCA) based SSR variation was used to identify genetic differentiation among the subpopulations using NTSYScp version 2.1 (Rohlf 2000). A significance test was made for the population mean values of four quantitative traits (100-seed weight, aboveground dry matter per plant, yield per plant, and plant height) to determine the subpopulation morphological differentiation. Shannon–Weaver index was also used to estimate the population phenotypic diversity. For this estimate, the morphological quantitative traits (mean measured values of field-cultivated plants for each single-sampled individual) were each divided into several graded-samples and their frequencies were calculated. Plant height (m): seven grades of every 0.50 m interval from ≤1.00 to >3.50 m; aboveground dry matter weight (g): ten grades of every 10.00 g interval from ≤10.00 to >90.00 g; yield per plant: ten grades of every 5.00 g interval from ≤5.00 to >45.00 g and 100-seed weight: six grades of every 0.50 g interval from ≤1.00 to >3.00 g. A PCA of morphology was used to identify the natural big-seeded plants (G. gracilis) and the typical wild plants in a mixed subpopulation using the software NTSYScp version 2.1. Six morphological characters [100-seed weight (g), plant dry weight (g), plant height (cm), single-plant yield (g), leaf length (mm), and leaf width (mm)] were used for the PCA.
Results
SSR-polymorphic variation
In total, 454 alleles were detected at the 36 loci, ranging from 2 (satt197 and satt345) to 19 alleles (satt530) in the samples, with an average of 12.61 alleles for loci, 45.40 for sites and 1.26 for each locus per site, respectively (Table 2). The numbers of alleles at loci among site subpopulations varied from 1 to 8 bands (Table 2). Some loci appeared polymorphic at some sites but monomorphic at other sites. Locus satt197 was monomorphic at nine sites. At sampling sites, G ST values indicated that the degrees of gene differentiation occurred at different extents between loci with a mean value of 0.559 (range from 0.247 at locus Satt197 to 0.870 at locus Satt334) for loci in sites (Table 2).
Genetic diversity in relation to subpopulation establishment history
The genetic parameters in the sites were estimated (Table 3). The values indicated that all parameters drastically varied between the subpopulations. Subpopulation polymorphic loci (P) averaged a 72 % value, ranging from 13.89 to 97.22 %. The mean number of alleles per locus was 2.85, ranging from 1.14 to 4.39 in sites. The relationship between the genetic diversity and the history of the subpopulation establishment was revealed in the case of site 2. This site grew alone on a small area of no more than about 400 m2 at a crook of a montane rivulet without serious environmental stresses and bore similar morpha among the individuals and had the lowest genetic diversity (Na = 41, P = 13.89 %, A p = 1.139, h = 0.019, and I = 0.034) (Tables 2, 3). The AMOVA analysis showed the lowest intrapopulation variation in the site 2 subpopulation (Table 3). This site subpopulation was inferred to be a newly formed founder population generated by migration of the upriver lesser seeds during a short period so as to contain such low level of genetic diversity.
Genetic diversity in relation to ecological habitats
The AMOVA analysis indicated that higher genetic variation existed among subpopulations (=57.46 %, d.f. = 9; P = 0.000001) and within subpopulation (=42.54 %, d.f. = 576; P = 0.000001). The levels of subpopulation genetic diversity were basically related to the environmental conditions of habitats, as shown in Fig. 2, the founder and drought subpopulations at sites 2 and 4 with low levels of genetic diversity were congregated in narrow spaces, respectively; while subpopulations at sites 3, 5, and 8 with high levels of genetic diversity were scattered in broad spaces. Obviously, good habitat conditions would generate high levels of genetic diversity in wild soybean populations. For example, site 4 subpopulation at a drought habitat had the second lowest total number of alleles (Na = 44) (Table 2), percentage of polymorphic loci (P = 25), number of alleles per locus (A p = 1.306), gene diversity (h = 0.043) and Shannon Index (I = 0.076) (Table 3), while other subpopulations, particularly those at sites 3, 5 and 8 located in moist and fertile soil habitats, had higher values (Na = 140–158, P = 91.67–97.22, A p = 3.861–4.389, h = 0.554–0.608, and I = 1.006–1.145) (Tables 2, 3) and three river system subpopulations had medium–high genetic diversity (Na = 69–97, P = 52.78–86.11, A p = 1.917–2.722, h = 0.207–0.384, and I = 0.342–0.640) (Tables 2, 3). The site 4 subpopulation survived a drought selection, making the genetic background simplified.
Geographic regionality of subpopulation clustering
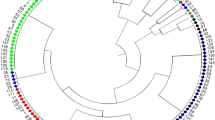
Genetically geographical structures of the site subpopulations were found to be related to habitats and formative history. The dendrogram obtained from the 36 SSR markers for the ten sites (Fig. 3) showed a geographic and ecological or environmental stress-depending clustering of site subpopulations and contained four divergent groups, i.e. respectively belonging to north-east mountain-hill areas (sites 1, 2, 3 and 5), central plain (sites 7, 8 and 9), west mountain area (site 10), and a drought habitat (site 4). Sites 1, 2 and 3 clustered into one wide group (about 63 km between sites 1 and 2 and about 64 km between sites 2 and 3 along the river). Although sites 6 and 7 were separated by 17.5 km along an arroyo, unexpectedly only site 6 deviated from its geographical area of the central plain and fell into a distant geographic area of clustering (Fig. 3).
Unique alleles in different habitat subpopulations
All subpopulations were found to contain alleles unique to the entire regional population (Table 4). However, the most distinct characterization for the subpopulations was that there were fewer unique alleles and fewer loci containing unique alleles in the founder population (site 2), the drought habitat (site 4) and the river system subpopulations (sites 6, 7 and 9) than in other subpopulations (Table 4). The founder population had smaller number of alleles than the drought-stress subpopulation.
Phenotypic diversity in subpopulations
Significance test of subpopulation phenotypic characters showed that morphologically, nine subpopulations one another had significant differences in one to three quantitative traits (Table 5), and only the site 4 drought subpopulation had significant differences with all other subpopulations in the four quantitative traits measured, suggesting that the ecological environments of the habitats could impact their respective subpopulation morpha (Table 5). Shannon–Weaver indexes (I) in the ten subpopulations also showed that the drought subpopulation at site 4 had the lowest phenotypic diversity (Table 3) and the smallest phenotypic values in seed size (1.20 g), aboveground dry matter (17.82 g), yield per plant (6.03 g), and plant height (106 cm). Most water system subpopulations (sites 1, 3, 6, and 7) excepting the semiarid site 9 had greater seeds (1.93–2.09 g) and high Shannon indexes (I) in morphology (I = 1.270–1.553). Site 9 subpopulation had relatively lower phenotypic diversity (I = 0.913), this might be the cause of its growing in a semiarid sandy soil so as to bear adaptive convergence of morphology although it was located on a riverbank. The subpopulations (sites 3, 5 and 8) with good habitat conditions had high phenotypic indexes (I = 1.328–1.553), particularly site 3 of the best habitat conditions gave the highest I value. While the drought subpopulation (site 4) and the founder population (site 2) showed two lowest I values, 0.795 and 1.093, respectively (Table 3). The phenotypic diversity and genetic diversity had similarly fluctuated patterns in Shannon index (I) (Fig. 4), with a significant value of r = 0.719.
Occurrence of big-seeded plants in site 1 subpopulation
Twenty-three or 63.89 % loci and 9 subpopulations were observed to have natural crossing (Tables 2, 3). The heterozygosity (H o) was detected to be mean 0.005 and range 0.000–0.017 in the sites (Table 3). A big-seeded type of over 3.0 g 100-seed weight is called semi-wild soybean (G. gracilis by Skvortzow 1927). Plants with 3.38–4.47 g in 100-seed weight were found in the site 1, they grew closely together within about 6 m2. These big-seeded plants were 2.08–3.25 fold seed mass of the typical G. soja plants and also very different from the typical G. soja type in other morphological traits at significance levels of differences (P < 0.01) (Table 6), were absolutely different from the typical wild soybean plants in morphology (Fig. 5). The genetic differentiation between the big-seeded and typical G. soja plants were characterized by the phylogenetic tree performed with the SSR loci (Fig. 6).
The SSR data showed that the big-seeded plants had so many rarely unique alleles or special genotypes at 10 loci (i.e. allele M at satt300, C at satt197, B and C at satt556, G at satt386, J at satt185, J at satt268, A at satt334, K at satt352, I at satt239, B and C at satt530), which did not appear in all the typical G. soja plants (Table 7). These unique alleles were inferred to be introgressive alleles from cultivated soybeans around the site because usually, only crossing between wild and cultivated soybeans can cause big-seeded offspring as a result of the gene effect of seed size that is involved in many genes with minor effect in the subgenus Soja (Liu et al. 2007).
We also observed that heterozygosity at satt346 (heterozygote EA) in the big-seeded type, which was caused by the segregation of the offspring between an AA (or EE) wild soybean plant and an EE (or AA) cultivated soybean. Loci satt556 and satt530 had two special genotypes BB and CC at their respective locus in the big-seed plants. There were two possible explanations for each locus case: one, the cultivated soybean donor plant originally was a BC heterozygote and it crossed with a typical G. soja plant to segregate BB and CC big-seeded plants; second, a cultivated donor with BB or CC genotype hybridized a typical G. soja plant with a genotype (CC or BB) but the CC and BB genotypes of typical G. soja were not collected in the samples from this site 1 in this study. The gene flow took place from cultivated into wild soybean, thereby generating the hybrid plants between wild and cultivated soybeans. So many unique alleles in these big-seeded plants could not come from mutations synchronously occurring in these big-seeded plants. In other words, the typical G. soja plants within this site were not the donors of these unique alleles in the big-seeded plants, which must be transferred from nearby soybean(s) around the site.
Discussion
Ecogeographic regionality of wild soybean subpopulations
Wild soybean had a variable level of genetic diversity in geographic sites (Table 3). In this study, a mean value (G st = 0.559) of gene differentiation. Isozyme research has indicated that riverbank populations of wild soybean would have very low genetic differentiation in Japan (Kiang et al. 1992; Fujita et al. 1997), which appears to be due to the “water system effect” in the along-river system populations. The clustering of site subpopulations studied here suggested that there was a genetic similarity for geographical ecotopes in wild soybean (Fig. 3); the subpopulations were distinctly clustered as corresponding geographical groups (Fig. 1), the north-east mountain-hill area, the central plain area, and the west mountain area, with the exception of drought site 4 and riverside site 6. Sometimes genetic differentiation between subpopulations may be rapid, just as shown in Fig. 3, where sites 6 and 7 were isolated in a distance of 17.5 km apart along a wadi but showed larger genetic differences between them.

Sampling sites of wild soybean subpopulations studied in Beijing region. Sites 1, 2 and 3 were distributed along a river valley in the northern mountain area. Sites 4 and 5 were located in the eastern foothill. Sites 6, 7, 8 and 9 were on the central plain; but sites 6 and 7 were distributed along a wadi. Site 10 lay on the eastern edge of the western mountainous, near the central plain

Principal component analysis for ten subpopulations of wild soybean in Beijing region based SSR markers. Site 2 founder and site 4 drought subpopulations with low levels of genetic diversity were distinctly congregated in narrow spaces, respectively. Eight big-seeded plants in site 1 were distinguished from other plants. Subpopulations at sites 3, 5, and 8 with high levels of genetic diversity were scattered in broad spaces

UPGMA dendrograms based Nei’s on (1983) genetic distance estimated by using 36 SSR markers indicating relationship between ten site subpopulations. The numbers on clades are genetic distance
Correlation between ecological environments or habitats and genetic diversity
Autogamous plants commonly have lower within-population genetic variation and higher among-population genetic variation (Loveless and Hamrick 1984). In the Beijing regional natural populations, the within-population genetic diversity (H S ) was 0.35 and the among-population diversity (D ST ) was 0.45. Nevertheless, large geographical-divided regions have very high intra-region genetic diversity and very low inter-region genetic variation (Wang and Takahata 2007; We et al. 2009) owing to the adjoining effect between regions and the large genetic heterogeneity within vast regional regions.
Our results indicated that the genetic diversity was related to ecological environments, habitats. A significant accordance between genetic and morphological variations in the subpopulations was observed, as demonstrated by the Fig 4, where similarly-fluctuated patterns of the I values between the phenotypic and genetic diversities were illustrated across the subpopulations, with a significant value of r = 0.719. Fertile habitats showed higher genetic diversity; such as site 3 as an extreme case of best environmental conditions correspondingly had the highest genetic and the phenotypic diversities. Commonly along-river system habitat subpopulations would have relatively lower genetic differences (Tables 3, 4).

Distribution patterns of genetic and phenotypic Shannon–Weaver indexes across ten subpopulations of wild soybean sampled in Beijing region. The phenotypic and genetic diversities had similarly fluctuated patterns

Principal component analysis for the site 1 subpopulation of wild soybean. Eight big-seeded natural hybrid plants were identified from the other 22 typical wild soybean of this site in six morphological traits (Table 6). Two first CP explain over 85 % of variability. Black circle typical wild soybean plants (nos. 1–22); Black Triangle big-seeded hybrid plants (nos. 23–30)

An UPGMA tree obtained by software NTSYSpc version 2.1 using 36 SSR markers. Eight big seed lines nos. 23–30 were clustered into a group distinctly differentiated from other wild soybean lines within site 1 subpopulation
Impacts of drought and founder bottlenecks on genetic structure variation
Evolutionary approaches such as the population founder effect and natural selection can affect the level of genetic variation within soybean species (Hyten et al. 2006). In wild barley, selection has played a role in molding the geographic structure of genetic diversity at some loci and loci with very little diversity and homogenous geographic distributions may have experienced a selective sweeping (Morrell et al. 2003). The impact of the two genetic bottlenecks on genetic structure and diversity was observed in the wild soybean subpopulations studied here. The two subpopulations, founder population at site 2 and arid sandy soil population at site 4, showed low genetic variation (Table 3). Our genetic diversity analysis and geographical UPGMA clustering suggested that the bottleneck impact of drought stress appeared be inferior to the founding effect on subpopulation genetic diversity but superior on genetically geographic grouping (Table 3; Fig. 3). Although site 2 founder population also had far fewer polymorphic loci and low genetic diversity, it still showed regionality within the north-east mountain-foothill ecotope (Fig. 3).
Geographical and genetic distances
Genetic distance reflects the identity of genetic variation or the interpopulation correlations within a region or ecotope. Kuroda et al. (2006) found clinal spatial structure of genetic variation by SSRs, with a positive correlation within a range of about 200 km in Japan. Fujita et al. (1997) analysed enzymatic genetic structures of four populations of wild soybean along a river reach of 50 km and found a significant correlation between genetic and geographical distances. Our results indicated that geographical beeline distance and genetic distance had a positive but nonsignificant correlation (r = 0.307, P > 0.05) within a limit of about 100 km. Seeds can spread a long distance in water or by animals, human or other natural factors (Cain et al. 2000). Seed dispersal distances of 0.2–12.4 km were observed in Japanese wild soybean (Kuroda et al. 2006). However, no long-distance seed dispersal was detected in this study. As a self-pollinated wild soybean, near subpopulations not always imply having close genetic distance and similar genetic variation, for example sites 6 and 7 along a wadi in the center plain area, although they were more similar to each other than to site 5 of the east hill area in numbers of alleles and unique alleles, genetic parameters and phenotypic diversity (Tables 3, 4), site 6 had genetic distance closer to site 5 (Fig. 3). This could be attributed to the similar genetic structure between sites 5 and 6, involved in the more similar categories and frequencies of alleles at the loci, showing genetic drift of natural populations in genetic structure.
The origin of G. gracilis type wild soybean
Big-seeded wild soybean was thought as a species G. gracilis (Skvortzow 1927). Hermann (1962) and Hymowitz (1970) suggested that G. gracilis should be a variant of G. max and originated from introgressive hybridization between wild and soybean (Hymowitz 1970). There have been some inferential studies based on morphology (Broich and Palmer 1980) and DNA variations (Sisson et al. 1978; Shoemaker et al. 1986; Close et al. 1989; Abe et al. 1999). In this study, allochthonous genes from cultivated soybeans in these big-seeded type plants, showing them to be hybrids between wild and cultivated soybeans. In this way, this study provided indirect population evidence for the hybridization origin of semi-wild soybean G. gracilis (Table 6).
Regional ex situ conservation strategy
Wild soybean has been decreasing in total distribution area, density, and number of populations and how to conserve this genetic resource is a pressing issue in China. Several studies using one single populations for sampling strategy of wild soybean in populations proposed that sampling should be conducted at intervals of at least 10 m (Jin et al. 2003b) and contain 30–45 individual plants (Jin et al. 2003a; Zhao et al. 2006) across the entire population to maximize the amount of genetic diversity.
However, the strategy of selecting subpopulations across a geographical or ecological or special area to conduct collecting samples of seeds is important for maximizing level of the genetic diversity in ex-situ conservation. Hu and Wang (1985) propounded setting up a sampling site and sampling 100 individual plants at every >10 km or several 10 km in a region, while Li et al. (1995) suggested collecting as many individual plants as possible in an area, and Fu et al. (2002) deemed it appropriate to collect samples from as many subpopulations as possible within areas and an appropriate number of plants in each population. Our results showed higher genetic variation between subpopulation (57.46 %) and within subpopulation (42.54 %) based AMOVA analysis, and that all individual subpopulations contained unique alleles, even the founder and the drought-stressed subpopulations (Table 4). So, theoretically all subpopulations should be sampled and as many individual plants as possible should be collected in consideration of such relatively high within-subpopulation genetic variation if conserving as high genetic diversity as possible in ex situ conservation. However, we think that founder subpopulations and environmental stress habitat subpopulations could be moderately reduced in sampling size to save conserving cost because of their low genetic diversity. In addition, it could be considered that number of sampling populations could be reduced along a river system considering seed dispersal between the upper and lower reaches.
Considering biosafety of the wild soybean gene pool
Despite no evidence that transgenic soybeans escape their genes into natural wild soybean populations in the fields, currently several artificial designed field experiments confirmed that transgenes could escape from transgenic soybeans to wild soybean in field conditions (Nakayama and Yamaguchi 2002; Chen et al. 2004; Mizuguti et al. 2009, 2010; Liu et al. 2012). In addition to this study, natural hybrids between wild and cultivated soybeans have been also found in eastern Asia (Kuroda et al. 2010; Wang et al. 2010). Visible direct evidence has been provided in our recent investigations of pollen flow from cultivated soybeans into wild soybean species (Wang and Li 2011, 2013). These documents suggest that transgenes can introgress into wild soybean like traditional soybean genes, which might lead to “genetic pollution” to this species and thereby incurr unknown ecological environments. The release of genetically modified (GM) soybeans appears to be only a matter of time in China. Therefore, some measures of spatial distance isolation should be considered to isolate wild soybean populations from GM soybeans in China.
References
Abe J, Hasegawa A, Fukushi H, Mikami T, Ohara M, Shimamoto Y (1999) Introgression between wild and cultivated soybean of Japan revealed by RFLP analysis for chloroplast DNA. Econ Bot 53:285–291
Broich SL, Palmer RG (1980) A cluster analysis of wild and domesticated soybean phenotypes. Euphytica 29:23–32
Cain ML, Milligan BG, Sterand AE (2000) Long-distance seed dispersal in plant populations. Am J Bot 87:1117–1227
Chen X, Yan JY, Gao B (2004) Preliminary study on roundup ready soybean’s round-up ready gene move to wild soybean. Chinese J Oil Crop Sci 26(2):89–91
Choi IY, Kang JH, Song HS, Kim NS (1999) Genetic diversity measured by simple sequence repeat variations among the wild soybean, Glycine soja, collected along the riverside of five major rivers in Korea. Genes Genet Syst 74:169–177
Close PS, Shoemaker RC, Keim P (1989) Distribution of restriction site polymorphism within the chloroplast genome of the genus Glycine, subgenus Soja. Theor Appl Genet 77:768–776
Cregan PB, Jarvik T, Bush AL, Shoemaker RC, Lark KG, Kahler AL, Kaya N, Vantoai TT, Lohnes DG, Chung J, Specht JE (1999) An integrated genetic linkage map of the soybean genome. Crop Sci 39:1464–1490
Dong YS, Zhuang BC, Zhao LM, Sun H, He MY (2001) The genetic diversity of annual wild soybean grown in China. Theor Appl Genet 103:98–103
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Felsenstein J (1993) PHYLIP (Phylogeny inference package) version 3.5c. University of Washington Press, Seattle
Fu YL, Qian J, Ma YH, Li J, Zheng SZ (2002) Genetic differentiation research on populations of wild soybeans in different scales. (China). Acta Ecol Sin 22:176–184
Fujita R, Ohara M, Okazaki K, Shimamoto Y (1997) The extent of natural cross-pollination in wild soybean (Glycine soja). J Hered 88:124–128
Fukuda Y (1933) Cytogenetical studies on the wild and cultivated Manchurian soybeans (Glycine L.). Japan J Bot 6:489–506
Hermann FJ (1962) A revision of the genus Glycine and its immediate allies. USDA Tech Bull 1268:1–79
Hu ZA, Wang HX (1985) Genetic structure of natural population of wild soybean (Glycine soja) on the Beijing region. (China). Acta Bot Sin 27:599–604
Hymowitz Y (1970) On the domestication of the soybean. Econ Bot 24:408–421
Hyten DL, Song QJ, Zhu YL, Choi IY, Nelson RL, Costa JM, Specht JE, Shoemaker RC, Cregan PB (2006) Impacts of genetic bottlenecks on soybean genome diversity. Proc Natl Acad Sci USA 103:16666–16671
Jin Y, Zhang WJ, Fu DX, Lu BR (2003a) Sampling strategy within a wild soybean population based on its genetic variation detected by ISSR markers. (China). Acta Bot Sin 45:995–1002
Jin Y, He TH, Lu BR (2003b) Fine scale genetic structure in a wild soybean (Glycine soja) population and the implication for conservation. New Phytol 159:513–519
Kiang YT, Chiang YC, Kaizuma N (1992) Genetic diversity in natural populations of wild soybean in iwate prefecture, Japan. J Hered 83:325–329
Kuroda Y, Kaga A, Tomooka N, Vaughan DA (2006) Population genetic structure of Japanese wild soybean (Glycine soja) based on microsatellite variation. Mol Ecol 15:959–974
Kuroda Y, Kaga A, Tomooka N, Vaughan D (2010) The origin and fate of morphological intermediates between wild and cultivated soybeans in their natural habitats in Japan. Mol Ecol 19:2346–2360
Li J, Tao Y, Zheng SZ, Zhou JL (1995) Isozymatic differentiation in local population of Glycine soja Sieb. & Zucc. (China). Acta Bot Sin 37:669–676
Liu K, Muse SV (2005) PowerMarker: Intergrated analysis environment for genetic marker data. Bioinformatics 21:2128–2129
Liu BH, Fujita T, Yan ZH, Sakamoto S, Xu DH, Abe J (2007) QTL mapping of domestication-related traits in soybean (Glycine max). Ann Bot 100:1027–1038
Liu J, Zhou B, Yang CY, Li YR, Jiang LX, Zhang MC, Tao B, Qiu LJ (2012) Gene flowing of genetically modified glyphosate-resistant soybean with EPSP. Soybean Sci 31:517–521
Loveless MD, Hamrick JK (1984) Ecological determinants of genetic structure in plant populations. Annu Rev Ecol Syst 15:65–95
Mizuguti A, Yoshimura Y, Matsuo K (2009) Flowering phenologies and natural hybridization of genetically modified and wild soybeans under field conditions. Weed Biol Manage 9:93–96
Mizuguti A, Ohigashi K, Yoshimura Y, Kaga A, Kuroda Y, Matsuo K (2010) Hybridization between GM soybean (Glycine max (L.) Merr.) and wild soybean (Glycine soja Sieb. et Zucc.) under field conditions in Japapn. Environ Biosafety Res 9:13–23
Morrell PL, Lundy KE, Clegg MT (2003) Distinct geographic patterns of genetic diversity are maintained in wild barley (Hoedeum vulgare ssp. spontaneum) despite emigration. Proc Natl Acad Sci USA 100:10812–10817
Nakayama Y, Yamaguchi H (2002) Natural hybridization in wild soybean (Glycine max ssp. soja) by pollen flow from cultivated soybean (Glycine max ssp. max) in a designed population. Weed Biol Manage 2:25–30
Nei M (1973) Analysis of gene diversity in subdivide populations. Proc Natl Acad Sci USA 70:3321–3323
Nei M, Takezaki N (1983) Estimation of genetic distances and phylogenetic trees from DNA anlysis. Proc. 5th World Cong. Genet Appl Livstock Prod 21:405–412
Rohlf FJ (2000) NTSYS 2.1: Numerical Taxonomic and Multivariate Analysis System. Exeter Software. New York
Shannon CE, Weaver W (1949) The mathematical theory of communication. University of Illinois Press, Ubana
Shimamoto Y, Fukushi H, Abe J, Kanazawa A, Gai JY, Gao Z, Xu DH (1998) Genet Resour Crop Evol 45:433–439
Shoemaker RC, Hatfield PM, Palmer RG, Atherly AG (1986) Chloroplast DNA variation in the genus Glycine subgenus Soja. J Hered 77:26–30
Sisson HD, Brim CA, Levings CS (1978) III. Characterization of cytoplasmic diversity in soybeans by restriction endonuclease analysis. Crop Sci 18:991–996
Sjögren P, Wyöni PI (1994) Conservation genetics and detection of rare alleles in finite populations. Conserv Biol 8:267–270
Skvortzow BW (1927) The soybean-wild and cultivated in Eastern Asia. Proc Manchurian Res Soc Publ Ser A 22:1–8
Tozuka T, Fukushi H, Hirata T, Ohara M, Kanazawa A, Mikami T, Abe J, Shimamoto Y (1998) Composite and clinal distribution of Glycine soja in Japan revealed by RFLP analysis of mitochondria DNA. Theor Appl Genet 96:170–176
Wang KJ, Li XH (2011) Interspecific gene flow and the origin of semi-wild soybean revealed by capturing the natural occurrence of introgression between wild and cultivated soybean populations. Plant Breed 130:117–127
Wang KJ, Li XH (2012) Genetic characterization and gene flow in different geographical-distance neighbouring natural populations of wild soybean (Glycine soja) Sieb. & Zucc.) and implications for protection from GM soybeans. Euphytica 186:817–830
Wang KJ, Li XH (2013) Pollen dispersal of cultivated soybean (Glycine max) into wild soybean (G. soja) under natural conditions. Crop Sci 53:2497–2505
Wang KJ, Takahata Y (2007) Apreliminary comparative evaluation of genetic diversity between Chinese and Japanese wild soybean (Glycine soja) germplasm pools using SSR markers. Genet Resour Crop Evol 54:157–165
Wang B, Chang RZ, Tao L, Guan RX, Yan L, Zhang MH, Feng ZF, Qiu LJ (2003) Identification of SSR primer numbers for analyzing genetic diversity of Chinaese cultivated soybean. (China). Mol Plant Breed 1:82–88
Wang KJ, Li XH, Zhang JJ, Chen H, Zhang ZL, Yu GD (2010) Natural introgression from cultivated soybean (Glycine max) into wild soybean (Glycine soja) with the implications for origin of populations of semi-wild type and for biosafety of wild species in China. Genet Resour Crop Evol 57:747–761
We ZX, Ding YL, Zhao TJ, Gai JY (2009) Genetic diversity and peculiarity of annual wild soybean (G. soja Sieb. et Zucc.) from various eco-regions in China. Theor Appl Genet 119:371–381
Xie H, Chang RZ, Cao YS, Zhang MH, Feng ZF, Qiu LJ (2003) Selection of core SSR loci by using Chinese autumn soybean. (China). Sci Agri Sin 36:360–366
Xu DH, Abe J, Gai JY, Shimamoto Y (2002) Diversity of chloroplast DNA SSRs in wild and cultivated soybeans: evidence for multiple origins of cultivated soybean. Theor Appl Genet 105:645–653
Yeh FC, Yang RC, Boyle T (2000) POPGENE software package version 1.32 for population genetic analysis, University of Alberta
Zhao RZ, Chen Z, Lu BR (2006) Estimate of genetic diversity and sampling strategy based on molecular markers in one single population of wild soybean. Chinese Sci Bull 51:1042–1048
Acknowledgments
This work was supported by projects “Crop Germplasm Resources Protection (Item no. 2130135-09)” and “Environmental and Biosafety Assessment of Transgenic Maize, Soybean and Wheat (Item no. 2014ZX08011-003)” of Agricultural Sciences from the Ministry of Agriculture of the People’s Repubic of China and “Sci & Teck Innovation Program of Chinese Academy of Agricultural Sciences”.
Author information
Authors and Affiliations
Corresponding author
Additional information
Xiang-Hu L is the joint first author.
Rights and permissions
About this article
Cite this article
Wang, KJ., Li, XH. & Yan, MF. Microsatellite markers reveal genetic diversity of wild soybean in different habitats and implications for conservation strategies (Glycine soja) in China. Conserv Genet 15, 605–618 (2014). https://doi.org/10.1007/s10592-014-0564-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-014-0564-5