
Abstract
There is an increasing awareness that the long-term viability of endemic island populations is negatively affected by genetic factors associated with population bottlenecks and/or persistence at small population size. Here we use contemporary samples and historic museum specimens (collected 1888–1938) to estimate the effective population size (N e) for the endangered yellow-eyed penguin (Megadyptes antipodes) in South Island, New Zealand, and evaluate the genetic concern for this iconic species. The South Island population of M. antipodes—constituting almost half of the species’ census size—is thought to be descended from a small number of founders that reached New Zealand just a few hundred years ago. Despite intensive conservation measures, this population has shown dramatic fluctuations in size over recent decades. We compare estimates of the harmonic mean N e for this population, obtained using one moment and three likelihood based-temporal methods, including one method that simultaneously estimates migration rate. Evaluation of the N e estimates reveals a harmonic mean N e in the low hundreds. Additionally, the inferred low immigration rates (m = 0.003) agree well with contemporary migration rate estimates between the South Island and subantarctic populations of M. antipodes. The low N e of South Island M. antipodes is likely affected by strong fluctuations in population size, and high variance in reproductive success. These results show that genetic concerns for this population are valid and that the long-term viability of this species may be compromised by reduced adaptive potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Untangling the relative roles of genetic and demographic factors that affect the persistence of endangered populations is a fundamental goal of conservation biologists and wildlife managers. In New Zealand, exotic mammalian predators have played a dramatic role in the decline and extinction of endemic fauna (Clout 2001; Duncan and Blackburn 2004), but intense conservation efforts have resulted in the eradication or control of these predators in localised mainland and offshore areas. Recent New Zealand conservation studies have also started to highlight the potential role of genetic factors in shaping the long-term viability of persisting endemic populations (Jamieson 2007; Jamieson et al. 2008). In particular, it is recognised that the loss of genetic diversity and increased levels of inbreeding—due to population bottlenecks and/or persistence at small population sizes—might have reduced mean population fitness and adaptive potential (Allendorf 1986; Lande and Shannon 1996; Frankham et al. 2002; Keller and Waller 2002).
Effective population size (N e), defined as the size of an ideal population experiencing the same rate of genetic drift as the actual population under consideration (Wright 1931; Frankham 1995; Willi et al. 2006; Palstra and Ruzzante 2008), is a key parameter in studies of genetic diversity. Historically, estimation of N e has been notoriously difficult, but this situation has been much improved by recent statistical developments facilitating the estimation of N e from temporal genetic samples (Nei and Tajima 1981; Pollak 1983; Waples 1989; Wang 2001; Berthier et al. 2002; Beaumont 2003; Wang and Whitlock 2003). These so-called temporal methods estimate the harmonic mean of a population’s variance effective size based on the change in allele frequencies over the time interval separating the temporally spaced samples. The use of museum specimens is particularly promising in the estimation of N e for species with long generation times (Wandeler et al. 2007). Here we use contemporary and historical samples to estimate N e for the endangered yellow-eyed penguin (Megadyptes antipodes) in South Island, New Zealand, and evaluate the genetic concern for this iconic species.
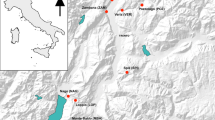
Megadyptes antipodes is thought to have expanded its range from the subantarctic islands to South Island, around 500 years ago, after the arrival of Polynesians but before settlement by Europeans and their commensals (Fig. 1; Boessenkool et al. 2009a). Based on low contemporary migration rates (<2%) between South Island (including surrounding islands such as Stewart Island) and subantarctic yellow-eyed penguins, and the relatively low levels of genetic variation of the current South Island population, it is thought that the South Island population descended from a small number of founders (Boessenkool et al. 2009b). Nevertheless, around 40% (~800 nests, ~2,200 individuals) of M. antipodes globally are now found on and around South Island (McKinlay 2001).

Map of the South and subantarctic islands of New Zealand. The dark grey line represents the current breeding range of Megadyptes antipodes. Arrows point to the geographical locations where samples were collected. Sample sizes for contemporary/historic samples are given in brackets. The dashed line symbolically represents the genetic split between South Island and subantarctic populations
By the 1980s, non-native predators—chiefly mustelids and cats, introduced by Europeans in the late 19th century—had caused major egg and chick predation (Darby and Seddon 1990) and prompted the implementation of intensive predator trapping around M. antipodes breeding areas. Despite these recent conservation measures, however, M. antipodes population sizes have remained highly unstable (McKinlay 2001; Moore 2001). This demographic instability has been attributed to changes in food supply (van Heezik and Davis 1990), climatic variations (Peacock et al. 2000) and disease epidemics (e.g. Gill and Darby 1993; Department of Conservation unpublished data). Regardless of their underlying causes, such fluctuations in population size are a primary factor leading to substantial reductions in N e (Frankham 1995).
Based on the suggested recent founding of South Island M. antipodes, with subsequent fluctuations in population size, conservation biologists hold genuine concerns for this population. In particular, the ongoing emergence of novel diseases (for example, a diphtheria-like disease linked to infection by a strain of Corynebacterium; Department of Conservation, unpublished data) suggests that the adaptive potential of this population may be limited, a concern that may become increasingly important with predicted climate change. In this study we use microsatellite analyses of contemporary and historic South Island samples to test for temporal changes in genetic diversity over the last century, and to provide genetic estimates of N e.
Materials and methods
Study area and sampling
Yellow-eyed penguin blood samples (N = 249) were collected between 2005 and 2008 at five breeding areas on and around South Island, including Stewart and Codfish Islands (Fig. 1). Together, these areas form the South Island yellow-eyed penguin population (Boessenkool et al. 2009b). A total of 101 additional samples were collected from the subantarctic Auckland and Campbell Islands (genotypes of these samples are used for N e estimates that allow for migration, see below). Details of blood sampling methods are described in Boessenkool et al. (2009b).
To facilitate sampling of historic yellow-eyed penguin specimens we contacted a total of 128 museums around the world. Toe pad samples were obtained from 35 specimens collected between 1888 and 1938 at several locations on the South Island and on Stewart Island (Fig. 1, for sample details see electronic supplementary material). These 35 samples included almost all yellow-eyed penguins specimens from the South Island with an explicit collection date (<1950) that are currently held in museum collections.
DNA extraction and genotyping
DNA from contemporary samples was extracted and purified using 40 μg proteinase K in 5% Chelex (Biorad: Walsh et al. 1991). All samples were genotyped at 12 microsatellite loci previously developed for yellow-eyed penguins (Man03, Man08, Man13, Man21, Man22, Man27, Man39, Man47, Man50, Man51, Man54, Man55; Boessenkool et al. 2008). Microsatellite primer sequences and polymerase chain reaction (PCR) conditions for contemporary samples are described in Boessenkool et al. (2008).
For DNA extraction of historic toe pad samples a ~1 × 2 mm piece was rehydrated by a 24 h wash in 1 ml 10 mM Tris–HCL (pH 8.0). Following rehydration, toepad samples were finely cut with a sterile scalpel blade and DNA was extracted using the Chargeswitch Forensic DNA Purification Kit (Invitrogen) or the DNeasy Tissue Kit (Qiagen) following manufacturers’ instructions. No differences were observed in extraction or amplification success between these two kits. Historic samples were amplified at the same 12 loci described above, with the exception of Man22 and Man27 which did not amplify consistently for the historic samples. These two loci were therefore omitted from all further analyses. PCR reactions (10 μl) contained 2 μl DNA, 0.5 μM of each primer, 0.5 U Taq DNA polymerase (Mango Taq, Bioline), 1× Taq buffer, 0.8 μM dNTP and 1.5 μM MgCl2, with the addition of betaine and DMSO (1.1 M and 2%, respectively) if necessary (see Boessenkool et al. 2008). The amplification profile was 2 min at 94°C, 35–50 cycles of 15 s at 96°C, 15 s at 45–50°C and 30 s at 72°C, followed by a 4 min final extension at 72°C.
To prevent contamination of historic DNA with exogenous DNA or PCR products, all DNA extractions and PCR set-up of historic samples were performed inside a UV hood in a laboratory where no contemporary yellow-eyed penguin DNA or vertebrate PCR products have ever been present. Standard precautions for the analysis of historic DNA were closely adhered to, including the use of filter tips, UV radiation and cleaning of materials with bleach and/or 70% ethanol before and after each laboratory session, and maintenance of a one-way flow from the historic DNA laboratory to the modern/post-PCR laboratory. Historic samples were extracted in small batches of nine samples and potential contamination was monitored by negative extraction and PCR controls. To minimise the risk of erroneous genotypes due to allelic dropout and the amplification of false alleles (Taberlet et al. 1996; Sefc et al. 2003), 2–7 successful amplifications were obtained for each historic sample before a genotype was scored, and genotypes were only scored when every allele was observed at least twice.
Disequilibrium and genetic diversity
Deviations from Hardy–Weinberg proportions and linkage equilibrium were tested using GENEPOP 4.0 (Rousset 2008) for contemporary South Island, contemporary subantarctic, and historic South Island samples separately. Markov chain parameters employed 10,000 dememorizations, 1,000 batches and 10,000 iterations. Significance levels were adjusted for multiple comparisons using Bonferroni corrections (Rice 1989). Genetic diversity was quantified for the ten loci that amplified consistently in contemporary and historic samples, using the total number of alleles and expected and observed heterozygosity calculated in GENETIX 4.05.2 (Belkhir et al. 1996–2004). Calculations of allelic richness were performed using FSTAT 2.9.3 (Goudet 2002) to adjust for sample size differences. Statistical significance of differences in genetic diversity between historic and contemporary South Island samples was tested with a Wilcoxon signed rank test in SPSS (α = 0.05). The difference in genetic diversity between subantarctic and contemporary South Island is discussed extensively in Boessenkool et al. (2009b), and subantarctic diversity is included here for comparative purposes only.
Effective population size
The quantification of N e using temporal methods requires an estimation of the number of generations (T) separating the temporally spaced sampling points. We calculated average generation time using the formula Σ(x l x b x )/Σ(l x b x ), where x is age, l x is the proportion of individuals surviving to age x and b x is the reproductive output at age x (Begon et al. 2006; see electronic supplementary material). Yearly adult survival of M. antipodes was set to 0.856 (Richdale 1957) and reproductive output set to 1.16 fledglings per pair (Darby and Seddon 1990). Maximum age was set to 20 years (Richdale 1975; Department of Conservation unpublished data) and variation in age at first breeding as estimated by Richdale (1957) was incorporated in the analysis. Using these estimates, average generation time of M. antipodes was calculated at 7.7 years (see electronic supplementary material). The time span between the collection year of contemporary samples (2006) and the weighted average collection year for historic samples (1901) was 105 years, resulting in T = 14. To account for uncertainty in T we also present estimates of N e using T = 12 and T = 16.
We used one moment-based and three likelihood-based approaches of the temporal method to obtain estimates of N e. These methods typically assume discrete generations, no selection, no mutation, and a closed panmictic population. Although our dataset violates the first of these assumptions, any bias due to overlapping generations can be minimised if samples are taken more than ten generations apart (Waples and Yokota 2007), which is the case in our study. The effects of migration are more complex (Wang and Whitlock 2003; Fraser et al. 2007a; Palstra and Ruzzante 2008), and we therefore included an estimator of N e that relaxes the assumption of a closed population (this estimator is referred to as N eOPEN, in contrast to the other estimators, which are referred to as N eCLOSED). Finally, previous research has shown that significant, albeit relatively low, F ST values within the South Island may indicate sub-structuring among several of the sampled breeding areas (Boessenkool et al. 2009b), violating the assumption of panmixia. This population genetic structure is very weak, however, and it has formerly been concluded that the South Island can be broadly regarded as a single population (Boessenkool et al. 2009b). This conclusion is further supported by the lack of any significant departure from Hardy–Weinberg proportions in the pooled South Island samples (see Results). We nevertheless evaluate the possible effect of substructuring on our estimates of N e (see Discussion).
First, we calculated the moment-based estimator from Waples (1989; extended from Nei and Tajima 1981 and Pollak 1983) using the program NeEstimator (Peel et al. 2004). Second, we applied the coalescent-based likelihood method from Beaumont (2003) as implemented in the program TMVP (which is based on the program TM3 from Berthier et al. 2002). We assumed no change in N e during the sampling interval and calculated N e as the mode of the posterior distribution. The MCMC simulation was performed with 50,000 updates of which ten percent were discarded as burnin. The size of importance sampling was 100, the thinning interval was 10 and the size of the proposal distribution of parameter updates was 0.5. Third, we estimated N e with the pseudo-likelihood based approach from Wang (2001) using the program MLNE. Finally, we applied the pseudo-likelihood method from Wang and Whitlock (2003; also implemented in MLNE) that relaxes the assumption of no migration by jointly estimating N e (N eOPEN) and the migration rate m. This method requires allelic data from the source population (the subantarctic population) and at least two samples from the focal population (the contemporary and historic South Island population). The method assumes migration is constant, that all sources are sampled and that the source population is sufficiently large that allele frequencies are temporally stable, although the method is relatively robust to violations of the latter assumption (Wang and Whitlock 2003). For all likelihood-based methods, maximum N e (N eMAX) was set to 1,000. Higher values of N eMAX affected only the upper bound of the 95% confidence interval (CI) when this fell above 1,000 in MLNE, but never influenced point estimates or the lower bound of the CI. For TMVP analyses, increasing N eMAX only lead to marginal increases of the upper bound of the CI (data not shown).
Results
All 350 contemporary samples amplified at all 12 microsatellite loci with the exception of six samples from South Island, which have missing genotypes for one (three samples), three (two samples) or five loci (one sample), respectively. Of the 35 historic samples, DNA was successfully extracted from 27 samples and a total of 249 genotypes were scored at ten loci (historic samples did not amplify at loci Man22 and Man27). Eight historic samples had missing genotypes at one (four samples), three (one sample), four (two samples) and six loci (one sample), respectively. Allelic dropout was encountered in 16 out of 224 PCR amplifications of confirmed heterozygous historic samples. These 16 cases of allelic dropout were restricted to four of the 27 historic samples, with most instances occurring multiple times in replicate amplifications of the same locus (e.g. for one sample, dropout was observed in five out of seven replicate amplifications of locus Man47). The amplification of a false allele was detected in just one out of a total of 634 successful PCRs.
Disequilibrium and genetic diversity
There was no evidence for linkage disequilibrium between any pairs of loci, and no loci showed significant departure from Hardy–Weinberg proportions. Eight out of ten loci were polymorphic in the contemporary South Island M. antipodes samples, and these same eight loci showed variation in the historic samples. In contrast, all ten loci were polymorphic in the subantarctic population. Genetic diversity estimators were slightly lower historically compared to estimates from contemporary samples of the South Island population, but these differences were not significant (all P values > 0.05, Table 1).
Effective population size
Point estimates of the harmonic mean of N e for the South Island population of M. antipodes varied between 128 and 656 (T = 14) for the different methods applied (Table 2). Wang’s pseudo-likelihood method gave the highest estimate with a large CI of which the upper bound was limited by our setting of N eMAX = 1,000. The moment-based estimator (Waples 1989) gave a slightly lower point estimate and, similar to Wang’s estimator, the CI were large (note that an upper bound cannot be set for the moment-based estimator). N e estimates from Beaumont’s (2003) likelihood-based method and the joint estimator of N eOPEN and m from Wang and Whitlock (2003) were similar with highly congruent CIs. The N e estimates are relatively robust to the number of generations (T) between sampling periods, showing only slight increases in N e with increasing T (Table 2). Estimates of m were low (m = 0.003, CI 0.002–0.007) and consistent for different values of T (Table 2).
Discussion
Effective population size estimates of South Island yellow-eyed penguins
Using microsatellite DNA analyses of historic (1888–1938) and contemporary samples we estimate the harmonic mean N e of South Island M. antipodes between 124 and 656 with lower bounds of the CI varying between 67 and 228 and upper bounds between 431 and >1,000. The evaluation of CIs in addition to point estimates of N e is essential, because CIs generated by different analytical methods are often more consistent than point estimates (Fraser et al. 2007a). Additionally, the lower bound of the CI gives important insight into the status of a population with respect to critical conservation thresholds (Hansen et al. 2002). In the current study, the four methods applied to estimate N e varied in their point estimates and their CIs (Table 2), a finding which raises questions about the relative accuracy of the different techniques.
Moment-based estimators such as the estimator from Waples (1989) are known to overestimate N e and have low precision (resulting in large CIs), particularly when populations experience rapid genetic drift and allele frequencies are skewed (Wang 2001; Berthier et al. 2002; Jorde and Ryman 2007; Palstra and Ruzzante 2008). Furthermore, the bias of this estimator seems to increase with increasing generations between samples (Tallmon et al. 2004). In contrast, simulations have shown that both Beaumont’s (Beaumont 2003) and Wang’s estimators (Wang 2001) show reduced bias when ten generations have passed between samples, with the former becoming very accurate and precise (Tallmon et al. 2004). Interestingly, our estimate of N e from Beaumont’s method, and in particular the associated CI (N e = 124, CI 67–504), was very similar to the joint estimator (N eOPEN = 196, CI 92–431) of Wang and Whitlock (2003), while Wang’s N e estimate (Wang 2001) was three times larger with an upper bound of the CI above 1,000. The N eOPEN estimator from Wang and Whitlock (2003) is considered to be superior to the closed population estimators (N eCLOSED) and expected to give more realistic values of N e, because it relieves the assumption of no migration.
Nevertheless, the effect of migration on N e is complex and should be addressed cautiously (Wang and Whitlock 2003; Fraser et al. 2007a; Palstra and Ruzzante 2008). Ignoring immigration can lead to either upward or downward biases of N e depending on 1) the extent of gene flow, 2) the sampling interval and 3) the genetic differentiation between focal versus source population(s) (Wang and Whitlock 2003; Fraser et al. 2007a). Many studies have found N eOPEN < N eCLOSED, particularly in cases where spatial genetic structuring is weak or moderate, and associated migration rates (sometimes unrealistically) high (Fraser et al. 2007a). In these scenarios it is thought that N eCLOSED estimates the N e of the entire metapopulation, rather than the N e of the population of interest (Wang and Whitlock 2003). With the exception of Beaumont’s estimator, we also find N eOPEN < N eCLOSED in the present study. This result may seem surprising given that M. antipodes has low migration rates (CI 0.002–0.007; see also Boessenkool et al. 2009b). Genetic differentiation between our two populations is strong, however, and our sampling interval was relatively long (14 generations). Consequently, migrants may have significantly altered genetic diversity on the South Island during the sampling interval, leading to an upward bias of estimated N eCLOSED. The above reasoning, however, does not explain the strong overlap between our estimates of N eOPEN and Beaumont’s N eCLOSED. Beaumont’s estimator is the only estimate that is based upon coalescent theory, and perhaps this estimator is affected differently by such migration patterns. The above issues further emphasize the complex interaction between N e and m, and reiterate that our understanding of the influence of m on N e and their estimators is currently incomplete (Fraser et al. 2007b).
Similar to the complex relationship between N e and m, the effect of population substructuring can bias estimates of N e either upwards or downwards. Specifically, whereas genetic differentiation among subpopulations will lead to an increase in the population-wide N e , estimate, variance in reproductive success or productivity among subpopulations will, conversely, reduce the estimate of N e (Whitlock and Barton 1997; Nunney 1999). Whether either of these effects has biased the findings of the present study is difficult to establish, but we believe that any bias due to substructuring will be weak because F ST values among South Island M. antipodes samples are low and only rarely significant (Boessenkool et al. 2009b). Furthermore, no departures from Hardy–Weinberg proportions were observed in either the modern or the historical samples. Although exploring the possible effects of substructuring would undoubtedly be interesting, there are too few historic samples to estimate N e for separate breeding areas within the South Island.
Although historic samples provide a valuable means to estimate N e using temporal genetic analyses, the inherent scarcity of such samples means that low sample sizes are unavoidable. Our sample size of 27 for the historic South Island population is substantially lower than the sample size of fifty that is typically recommended (Palstra and Ruzzante 2008), which may have compromised the precision of our estimates of N e . Furthermore, the historic samples were collected over a 50-year period. The analytical consequences of pooling samples across multiple generations have not, to our knowledge, been evaluated, but it is conceivable that such pooling could distort allele frequency estimation and thereby bias estimates of N e .
It is difficult to calculate the N e/N c (effective population size/census population size) ratio in M. antipodes because, in fluctuating populations, the harmonic mean N e is weighted towards the smallest values of N e during the sampling interval (Leberg 2005), and we cannot calculate the harmonic mean N c over the time interval used to calculate the harmonic mean N e. Dividing our point estimates of N e (124, 196, 277, 656) by the current census size (2,200 for the total South Island population, including surrounding islands) gives ratios of 0.06, 0.09, 0.13 and 0.30, respectively, but this may be a slight underestimate of the actual ratio as the harmonic mean N c over the time interval is likely to be less than the current census size. With the exception of 0.30, these estimates appear close to the average N e/N c ratios found in natural populations of vertebrate taxa (N e/N c = 0.10–0.11; Frankham 1995). Fluctuating population size is arguably the most important factor reducing this ratio (Frankham 1995). Indeed, close monitoring of yellow-eyed penguins on the South Island has revealed strong fluctuations in the total number of breeders during the last two decades, with the lowest population estimate recorded in the 1990/1991 season when as few as 140 pairs bred on the South Island (Gill and Darby 1993), versus approximately 500 breeding pairs on the South Island in more recent years (Department of Conservation, unpublished data). The second most important factor leading to low N e/N c ratios is variance in reproductive success (Frankham 1995). Such variance has been shown to exist in yellow-eyed penguins, and parental ‘quality’ is likely an important component determining this variation (Efford and Edge 1998; Bull 2005). Unfortunately, no comparable estimates of N e/N c exist for any other penguin species (Frankham 1995; Palstra and Ruzzante 2008). Long-term estimates of N e for Galápagos and Magellanic penguins were calculated by Akst et al. (2002) but these analyses are not directly comparable to our estimates for M. antipodes because they involve much longer time-scales. Indeed, application of the method applied by Akst et al. (2002) to South Island M. antipodes would yield non-credible N e estimates (data not shown)—values as high as the current census size.
Conservation implications
The minimum N e required to retain sufficient evolutionary potential is thought to approximate 500, although thresholds as high as 5,000 have been proposed (Franklin 1980; Franklin and Frankham 1998; Lynch and Lande 1998). Coping with certain environmental challenges, such as the introduction of disease and toxins, may require only an adaptive response at a few specific loci, and the population size needed to maintain sufficient genetic variation at such loci is more likely to lie in the thousands than in the hundreds (Willi et al. 2006). Not withstanding the limitations of our sampling, most of our N e estimates (and especially the lower bounds of the CI) for South Island M. antipodes are well below such critical thresholds required to maintain adaptive potential. This finding is particularly notable in the context of the regular disease epidemics experienced by this population. Furthermore, South Island M. antipodes already have low genetic diversity compared to the subantarctic population at neutral loci and immigration rates are sufficiently low for the population to be considered demographically isolated (Boessenkool et al. 2009b). The low effective population size estimates presented in the current study imply that the South Island population will likely experience the loss off genetic variation due to random drift, potentially eroding adaptive potential. Given predicted increases in rates of environmental variations due to climate change (NIWA 2008), the maintenance of adaptive genetic diversity in M. antipodes may become increasingly important. These results suggest that the South Island population of yellow-eyed penguins will remain vulnerable and unstable in the near future, and ongoing monitoring of the population, in addition to continued predator trapping, is therefore essential.
References
Akst EP, Boersma PD, Fleischer RC (2002) A comparison of genetic diversity between the Galapagos Penguin and the Magellanic Penguin. Conserv Genet 3:375–383
Allendorf FW (1986) Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol 5:181–190
Beaumont MA (2003) Estimation of population growth or decline in genetically monitored populations. Genetics 164:1139–1160
Begon M, Townsend CR, Harper LL (2006) Ecology: from individuals to ecosystems. Blackwell Publishing, Malden
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (1996–2004) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, Université de Montpellier II, Montpellier
Berthier P, Beaumont MA, Cornuet JM, Luikart G (2002) Likelihood-based estimation of the effective population size using temporal changes in allele frequencies: a genealogical approach. Genetics 160:741–751
Boessenkool S, King TM, Seddon PJ, Waters JM (2008) Isolation and characterization of microsatellite loci from the yellow-eyed penguin (Megadyptes antipodes). Mol Ecol Resour 8:1043–1045
Boessenkool S, Austin JJ, Worthy TH, Scofield P, Cooper A, Seddon PJ, Waters JM (2009a) Relict or colonizer? Extinction and range expansion of penguins in southern New Zealand. Proc R Soc Lond B Biol Sci 276:815–821
Boessenkool S, Star B, Waters JM, Seddon PJ (2009b) Multilocus assignment analyses reveal multiple units and rare migration events in the recently expanded yellow-eyed penguin (Megadyptes antipodes). Mol Ecol 18:2390–2400
Bull E (2005) Components influencing lifetime reproductive success in yellow-eyed penguins. Bachelor of Science (Honours) thesis. Department of Zoology, University of Otago, Dunedin
Clout M (2001) Where protection is not enough: active conservation in New Zealand. Trends Ecol Evol 16:415–416
Darby JT, Seddon PJ (1990) Breeding biology of the yellow-eyed penguin (Megadyptes antipodes). In: Davis LS, Darby JT (eds) Penguin biology. Academic Press Inc, San Diego, pp 45–62
Duncan RP, Blackburn TM (2004) Extinction and endemism in the New Zealand avifauna. Global Ecol Biogeogr 13:509–517
Efford MG, Edge KA (1998) Can artificial brood reduction assist the conservation of yellow-eyed penguins (Megadyptes antipodes)? Anim Conserv 1:263–271
Frankham R (1995) Effective population size/adult population size ratios in wildlife—a review. Genet Res 66:95–107
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Franklin IR (1980) Evolutionary change in small populations. In: Soule ME, Wilcox BA (eds) Conservation biology: an evolutionary—ecological perspective. Sinauer Associates, Sunderland
Franklin IR, Frankham R (1998) How large must populations be to retain evolutionary potential? Anim Conserv 1:69–70
Fraser DJ, Hansen MM, Ostergaard S, Tessier N, Legault M, Bernatchez L (2007a) Comparative estimation of effective population sizes and temporal gene flow in two contrasting population systems. Mol Ecol 16:3866–3889
Fraser DJ, Jones MW, McParland TL, Hutchings JA (2007b) Loss of historical immigration and the unsuccessful rehabilitation of extirpated salmon populations. Conserv Genet 8:527–546
Gill JM, Darby JT (1993) Deaths in yellow-eyed penguins (Megadyptes antipodes) on the Otago Peninsula during the summer of 1990. New Zeal Vet J 41:39–42
Goudet J (2002) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Insitute of Ecology, Lausanne
Hansen MM, Ruzzante DE, Nielsen EE, Bekkevold D, Mensberg KLD (2002) Long-term effective population sizes, temporal stability of genetic composition and potential for local adaptation in anadromous brown trout (Salmo trutta) populations. Mol Ecol 11:2523–2535
Jamieson IG (2007) Has the debate over genetics and extinction of island endemics truly been resolved? Anim Conserv 10:139–144
Jamieson IG, Grueber CE, Waters JM, Gleeson DM (2008) Managing genetic diversity in threatened populations: a New Zealand perspective. New Zeal J Ecol 32:130–137
Jorde PE, Ryman N (2007) Unbiased estimator for genetic drift and effective population size. Genetics 177:927–935
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17:230–241
Lande R, Shannon S (1996) The role of genetic variation in adaptation and population persistence in a changing environment. Evolution 50:434–437
Leberg P (2005) Genetic approaches for estimating the effective size of populations. J Wildl Manage 69:1385–1399
Lynch M, Lande R (1998) The critical effective size for a genetically secure population. Anim Conserv 1:70–72
McKinlay B (2001) Hoiho (Megadyptes antipodes) recovery plan 2000–2025. Department of Conservation, Wellington
Moore PJ (2001) Historical records of yellow-eyed penguin (Megadyptes antipodes) in southern New Zealand. Notornis 48:145–156
Nei M, Tajima F (1981) Genetic drift and estimation of effective population size. Genetics 98:625–640
NIWA (2008) Climate change: projections for New Zealand. National institute of water & atmospheric research, Wellington, New Zealand. Available from www.niwascience.co.nz/ncc/clivar/scenarios. Downloaded on: 15 March 2009
Nunney L (1999) The effective size of a hierarchically structured population. Evolution 53:1–10
Palstra FP, Ruzzante DE (2008) Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol 17:3428–3447
Peacock L, Paulin M, Darby JT (2000) Investigations into climate change influence on population dynamics of yellow-eyed penguins Megadyptes antipodes. New Zeal J Zool 27:317–325
Peel D, Ovenden JR, Peel SL (2004) NeEstimator: software for estimating effective population size, version 1.3. Queensland Government, Department of Primary Industries and Fisheries, Australia
Pollak E (1983) A new method for estimating the effective population size from allele frequency changes. Genetics 104:531–548
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Richdale LE (1957) A population study of penguins. Clarendon Press, Oxford
Rousset F (2008) GENEPOP ‘007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Sefc KM, Payne RB, Sorenson MD (2003) Microsatellite amplification from museum feather samples: effects of fragment size and template concentration on genotyping errors. Auk 120:982–989
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24:3189–3194
Tallmon DA, Luikart G, Beaumont MA (2004) Comparative evaluation of a new effective population size estimator based on approximate Bayesian computation. Genetics 167:977–988
van Heezik Y, Davis L (1990) Effects of food variability on growth rates, fledging sizes and reproductive success in the yellow-eyed penguin Megadyptes antipodes. Ibis 132:354–365
Walsh PS, Metzger DA, Higuchi R (1991) Chelex-100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Wandeler P, Hoeck PE, Keller LF (2007) Back to the future: museum specimens in population genetics. Trends Ecol Evol 22:634–642
Wang JL (2001) A pseudo-likelihood method for estimating effective population size from temporally spaced samples. Genet Res 78:243–257
Wang JL, Whitlock MC (2003) Estimating effective population size and migration rates from genetic samples over space and time. Genetics 163:429–446
Waples RS (1989) A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics 121:379–391
Waples RS, Yokota M (2007) Temporal estimates of effective population size in species with overlapping generations. Genetics 175:219–233
Whitlock MC, Barton NH (1997) The effective size of a subdivided population. Genetics 146:427–441
Willi Y, Van Buskirk J, Hoffmann AA (2006) Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst 37:433–458
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Acknowledgments
We are very grateful to the Auckland Museum, American Museum of Natural History, Australian Museum, Canterbury Museum, Museum of Comparative Zoology, Natural History Museum Geneva, Natural History Museum Tring, Natural History Museum Paris, Museum of New Zealand Te Papa Tongarewa, Natural History Museum Vienna, Swedish Museum of Natural History, Otago Museum, South Australian Museum, Smithsonian Institution, Museum für Naturkunde Berlin and Craig Millar for supplying tissue samples of historic specimens. We thank the New Zealand Department of Conservation for help with collecting contemporary samples. We are indebted to Tania King for guidance and advice in the laboratory and we thank Stein Are Sæther for helpful discussions about analyses. We would further like to thank the ESF Science Networking Programme ConGen for organising the Conservation Genetics conference in Trondheim in May 2009 and for this special issue of Conservation Genetics. This research was supported by the Department of Zoology, University of Otago, including PBRF Research Enhancement Grants to PJS and JMW. Samples were collected under Department of Conservation permits SO-17933-FAU and OT-19097-RES and University of Otago Animal Ethics Approval 69/06.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Boessenkool, S., Star, B., Seddon, P.J. et al. Temporal genetic samples indicate small effective population size of the endangered yellow-eyed penguin. Conserv Genet 11, 539–546 (2010). https://doi.org/10.1007/s10592-009-9988-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-009-9988-8