
Abstract
At least four species of New Zealand snipes (Coenocorypha) became extinct following the introduction of predatory mammals, and another two species suffered massive range reductions. To investigate species limits and population differentiation in six of the seven remaining offshore populations, we assayed variation in nine microsatellite loci and 1,980 base pairs of four mitochondrial DNA (mtDNA) genes. Genetic diversity in all populations except the largest one on Adams Island in the Auckland Islands was very low in both genomes. Alleles were fixed at many microsatellite loci and for single mtDNA haplotypes, particularly in the populations in the Chathams, Snares, Antipodes and Campbell Islands. Strong population structure has developed, and Chathams and Snares Islands populations are effectively genetically isolated from one another and from the more southern island populations. Based on reciprocal monophyly of lineages and their morphological distinctiveness we recommend that three phylogenetic species should be recognized, C. pusilla in the Chatham Islands, C. huegeli in the Snares Islands and C. aucklandica in the southern islands. The populations of C. aucklandica in the Auckland Islands, Antipodes Island and Campbell Island may warrant recognition as subspecies, and all should be managed as separate conservation units.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Coenocorypha snipe once ranged across the New Zealand region and offshore islands, and as far north as Norfolk Island, Fiji, and possibly New Caledonia (Holdaway et al. 2001; Worthy 2003). Only a few of the New Zealand taxa have persisted to the present, and are of great conservation concern due to their small population sizes, restricted distributions, and extreme vulnerability to introduced mammalian predators. They are still found on some outlying islands in the Chatham Islands, and on the subantarctic Snares, Antipodes, Auckland and Campbell Islands. Snipe became extinct on the three main islands of New Zealand and from the main Chatham Island following introduction of Pacific rats (Rattus exulans) by Polynesians (Worthy and Holdaway 2002). More recent extinctions and range contractions of five taxa have followed introductions of cats (Felis catus), pigs (Sus scrofa), Norway rats (Rattus norvegicus), rabbits (Oryctolagus cuniculus), cattle (Bos taurus), sheep (Ovis aries), dogs (Canis familiaris), weka (Gallirallus australis), and ship rats (Rattus rattus) on various islands (Miskelly 1987, 1988, 2000; Aikman and Miskelly 2004; Miskelly et al. 2006). The only population that currently coexists with an introduced mammal (house mouse, Mus musculus) is on Antipodes Island.
Given the persistence of New Zealand snipe on small isolated islands, their presumably small effective population sizes and vulnerability to predation, an important component of population recovery planning is to estimate species limits and evolutionary significant units (ESUs) or conservation management units (CMUs). There is agreement that the extant population of the Chatham Islands is a valid species (C. pusilla), but the taxonomic status of the remaining populations is less certain. Populations from the Auckland Islands, Snares Islands, Little Barrier Island, Stewart Island and the Antipodes Islands were listed in the 1990 Checklist of New Zealand birds (Turbott 1990) as separate subspecies of the species originally described from the Auckland Islands (C. aucklandica). However, Worthy et al. (2002) suggested that separate species status was warranted for each of the populations on the Auckland Islands, Snares Islands, Little Barrier Island and islands off Stewart Island, based on morphological, plumage and behavioural differences between them.
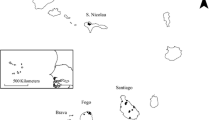
Two of the five snipe taxa that we sampled in this study still occupy their entire historic ranges: the Snares Island snipe on the tiny 333 ha Snares Islands, and the Antipodes Island snipe on the larger (2,111 ha) but more remote Antipodes Islands (Fig. 1; Miskelly et al. 2001, 2006). The three remaining taxa have suffered human-induced population size reductions of varying, and in some cases alarming extremes. Following the introduction of cats and pigs to 50,990 ha Auckland Island, and a variety of predatory and browsing mammals to Enderby Island (710 ha) and Rose Island (75 ha), Auckland Island snipe became confined to Adams Island (10,119 ha) in the south, Disappointment Island (566 ha) in the west, and Ewing Island (57 ha) in the Port Ross sample in the north (Fig 1, Miskelly et al. 2006). Chatham Island snipe became extinct on Chatham Island (78,438 ha) and Pitt Island (6,190 ha) between 1550 and 1870 (Millener 1999), and from 113 ha Mangere Island about 1900 following introduction of rabbits, sheep, goats (Capra hircus) and cats (Tennyson and Millener 1994). For the next 70 years they were believed to be confined to 218 ha Rangatira (South East) Island (Fig. 1), although they may have persisted on the nearby and rarely visited Star Keys (10 ha). Although rodents and cats never established on Rangatira Island, the island was farmed from 1840 until the last stock were removed in 1961 (Nilsson et al. 1994); by the 1950s there was very little ground cover left, and snipe were rarely seen. Indeed, Bell (1955), and Dawson (1955) saw none during their visits in 1953 and 1954, respectively, and so the taxon came very close to extinction. Mangere Island has been free of terrestrial mammals since 1968, and 23 snipe were reintroduced from Rangatira Island by the New Zealand Wildlife Service in 1970 (Bell 1974); snipe are now common on both islands (Miskelly 1990).

Distributions of extant taxa of New Zealand snipe. Snipe on the Snares and Antipodes Islands occupy their entire historic ranges. Those on the three remaining island groups have passed through population size reductions of varying degrees, with shading showing their minimum distributions before partial range recovery (see text). We sampled all five taxa, and in the Auckland Islands obtained samples from Port Ross (Enderby and Rose Islands) and Adams Island, but not from Disappointment Island
The most extreme population bottleneck was experienced by the Campbell Island snipe, which was discovered on 19 ha Jacquemart Island (Fig. 1) only in 1997, 157 years after naturalists first visited the Campbell Island group (Miskelly 2000; Barker et al. 2005). It is now apparent that snipe were extirpated from 11,268 ha Campbell Island following the introduction of Norway rats about 1828 (Miskelly 2000), but survived fortuitously on sheer-sided Jacquemart Island, 1 km off the exposed south coast of the main island. Snipe from Jacquemart Island started to recolonise Campbell Island following the eradication of rats in 2001 (Miskelly and Fraser 2006a). The remarkable discovery of this previously unknown and critically endangered population prompted our investigation of the genetic relationships of Coenocorypha snipe.
Although the threats to these populations are more likely to be demographic ones in the shorter term, they also could be at risk from genetic erosion in the longer term. In particular we expect that genetic drift will be enhanced in small populations, and they likely experienced founder effects and both natural and human-induced population bottlenecks which led to loss of genetic variation, increased inbreeding and possibly lower fitness (Ramakrishnan et al. 2005; Woodworth et al. 2002). We therefore assayed variation in nine microsatellite loci and four mitochondrial genes from recently collected blood or feather samples to estimate levels of gene diversity, population structure and species limits in New Zealand snipes.
Methods
Sample collection
Samples from the six populations of New Zealand snipes were collected under permit between 2000 and 2007 (Table 1; Fig. 1). Because Mangere Island snipe are descended from 23 birds translocated from Rangatira Island in 1970, samples from both islands were pooled to comprise the Chatham Islands population. In the Auckland Islands the samples collected on Rose Island and adjacent Enderby Island were combined into the Port Ross samples (PROSS). Samples from the critically endangered Campbell Island population were obtained from the main island (Miskelly and Fraser 2006a, b). Most sampled birds were adults captured in hand nets, and either sampled for blood or feathers. Blood was preserved in 95% ethanol, and feathers were stored in a freezer once they reached the laboratory. The only isolated population not sampled was Disappointment Island within the Auckland Islands archipelago (Fig. 1).
Isolation and testing of microsatellite loci for polymorphism
Isolation of microsatellite loci was achieved using the enrichment protocol of Hamilton et al. (1999). About 5 mg of high molecular weight genomic DNA was isolated and digested using the restriction enzymes Hae III, Rsa I and Nhe I to create an appropriately sized genomic library (400–800 bp). The genomic library was then enriched for GT and CT repeats (using biotinylated oligonucleotides (CT)15 and (GT)15) and ligated into the vector pBluescript (Stratagene). Vectors were transformed into Escherichia coli XL1-Blue Electrocompetent (Stratagene) cells and grown on agar plates overnight. Positive clones were detected by hybridization with biotinylated oligonucleotides, using the Phototope Star Detection Kit (New England Biolabs). Colonies were picked randomly for subsequent sequencing. Plasmid DNA from positive clones were extracted using the Wizard Miniprep Kit (Promega, Madison, WI, USA), and sequenced on an ABI 3100 automated sequencer. Seventy-eight positive clones were sequenced of which 17 contained microsatellite repeats. Primers were designed for the sequences containing these microsatellites and an M13 tail was added to one primer (reverse or forward) of each primer pair to allow fluorescent labeling during amplification reactions (Schuelke 2000). Genomic DNA samples from 40 New Zealand snipes were used to test levels of polymorphism for each primer set. Amplification reaction volumes of 12.5 μl were used containing 100–200 ng of DNA, 1.25 μl of 10× EH buffer (Hagelberg 1994), 0.5 units of Taq DNA polymerase (Qiagen), 0.3 μl of 5 mM dNTPs, 2.5 pmoles of each primer and 2.5 pmoles of the appropriate fluorescent label. Amplification of all loci began with a 4 min denaturation at 94°C, followed by 36 cycles of 94°C for 15 s, 50°C for 25 s, 72°C for 25 s, with a final extension of 72°C for 4 min (Perkin Elmer 480 thermal cycler). Amplification products were then run on an ABI 3100 automated sequencer, and were scored using GeneMapper version 3.5 along with a LIZ size standard (Applied Biosystems). Nine polymorphic loci that amplified reliably and did not suffer from large allele dropouts or stuttering when checked with Micro-checker 2.2.3 (Van Oosterhout et al. 2004) were used to depict geographic patterns of population structure (Table 2). All loci were in linkage equilibrium, as tested with GENEPOP 3.4 (Raymond and Rousset 1995). Exemplars of alleles at each locus were sequenced to verify repeat motifs. Size homoplasy was detected only at the compound repeat locus SN19 in the 362 bp amplicon. We therefore sequenced all these amplicons from each individual to determine their repeat motifs and to score the alleles for the dinucleotide repeat region only. All but two birds in our study were sexed molecularly with primers that avoid the known length polymorphism in the Z product of shorebirds (AJB, unpublished data), and all loci were shown to be autosomal by heterozygous females.
Statistical analysis of microsatellite genotypes
Allele frequencies at each locus were calculated from genotype data of all individuals with amplifiable DNA using CONVERT 1.31 (Glaubitz 2004). Tests of H–W equilibrium and levels of genetic variation in population samples were calculated with POPGENE 1.31 (Yeh and Boyle 1997). To estimate levels of gene flow among the six populations we used a maximum likelihood method based on the coalescent as implemented in Migrate (Beerli and Felsenstein 1999, 2001). This analysis does not assume the equilibrium island model in which gene flow is symmetric among populations, but instead allows asymmetrical gene flow among populations. The following search parameters were used: 10 short chains sampling 100,000 trees and recording 5,000, followed by three long chains sampling 2,500,000 trees and recording 25,000. An adaptive heating scheme with four chains with start temperatures 1, 1.5, 3, and 1,000,000 was employed to more fully explore the likelihood surface. Initial estimates of population variation (θ) and gene flow were obtained using Fst (Beerli 2004). The analysis was run three times with different random numbers, and results were averaged because they gave similar estimates of the parameters.
We used the model-based clustering method implemented in STRUCTURE 2.2 (Pritchard et al. 2000) to test existing hypotheses of population structure based on the co-ancestry of alleles in the samples. The best-fitting model of population structure was chosen by inspection of the log-likelihoods of the data for K = 1 to 7 populations to find the highest likelihood of population clustering as suggested by Falush et al. (2003). Pairwise Fst values between populations were also computed in Arlequin v.2.000 (Schneider et al. 2000).
Mitochondrial DNA
We amplified and sequenced protein-coding genes because many silent sites at third positions in codons are free to vary, much like variable regions of the control region. Based on the rates of evolution in mtDNA genes in birds estimated by Pereira and Baker (2006) we chose four genes to sequence: ATPase6 and 8, COI, and COII.
Genomic DNA was extracted from blood samples using standard procedures (Sambrook et al. 1989). Samples were homogenized in a solution of STE buffer (100 mM NaCl; 10 mM Tris–HCl, pH 8.0; 1 mM EDTA, pH 8.0), 0.1% SDS, and 10 μg/ml proteinase K, and incubated overnight at 55°C. Dilutions of these crude DNA extractions were used as templates for amplification of the mtDNA genes via the Polymerase Chain Reaction (PCR) in a total reaction volume of 12 μl, consisting of 10 mM Tris–HCl, pH 8.3; 50 mM KCl; 2.5 mM MgCl2; 0.01% gelatine, 160 mg per ml BSA; 50 μM each dNTP; 0.4 μM each primer; and 1 U Taq DNA polymerase (Boehringer Mannheim). The following thermal cycle profile was carried out using a Perkin Elmer DNA 480 Thermal Cycler: 94°C for 45 s, 50°C for 45 s; and 72°C for 1 min 30 s, for 35 cycles. An initial denaturation step of 94°C for 5 min and a final extension step of 72°C for 7 min were used. Following amplification, the total volume of PCR product was subjected to electrophoresis through a 1.5% agarose gel in 1× TA buffer, and visualised using ethidium bromide and UV illumination. The band containing the PCR product was excised from the gel and purified via filter pipet-tip centrifugation (Dean and Greenwald, 1995). DNA sequencing reactions were cycle-sequenced as per manufacturer’s instructions and run on an ABI 3100 automated sequencer. DNA sequences from both the L- and H-strands were checked for ambiguities and the final consensus sequence was created for each gene fragment and aligned in ChromasPro ver. 1.41 (Technelysium Pty Limited, Tewantin, Australia), and then were concatenated in a matrix of 1,980 base pairs (bp). All sequences obtained in this study were deposited in GenBank (accession numbers GQ452385-GQ452696).
The program Modeltest 3.6 (Posada and Crandall 1998) was used to determine which model of sequence evolution best fitted the mtDNA sequences, which was the TrN (Tamura and Nei 1993) model + gamma-distributed rate variation among sites. The program Arlequin 2.000 (Schneider et al. 2000) was used to calculate haplotype diversity (Nei 1987). To show relationships among haplotypes, a median-joining haplotype network was constructed from the aligned sequence data using the program Network 4.1 (Bandelt et al. 1999).
To test whether the mtDNA sequences violated neutral expectations we used DNAsp (Rozas et al. 2003) to compute Tajima’s (1989) D-values, Fu and Li’s (1993) D* and F* test statistics, and Fu’s (1997) Fs statistic. Fs is more powerful for detecting an excess of young mutations (rare alleles) arising from the effects of population growth on neutral sequences or genetic hitchhiking on selected genes (Fu 1997), whereas D* and F* are best for detecting background selection against deleterious alleles (Fu 1997). Significance levels of the statistics were determined using coalescent simulations of 10,000 random samples based on the population parameter values of Fs and h to obtain critical values.
We conducted coalescent analyses to estimate population divergence times between populations using the program MDIV (Nielsen and Wakeley 2001). To correct for multiple substitutions at sites, the only available option of the HKY model of sequence evolution in MDIV was used. MDIV is based on a coalescent model that jointly estimates the divergence time and migration rates among pairs of populations using DNA sequence data. Four parameters can be estimated (θ, M, T and TMRCA), but we report only population divergence time T = t/N ef, where N ef is effective population size of females. Markov Chain Monte Carlo (MCMC) simulations generate posterior probability distributions whose modes represent the parameter estimates. Parameters were estimated using three runs of a 5,000,000 generation MCMC with an additional 2,000,000 generation burn-in period. To convert coalescent times to years before present (ybp), a mutation rate (μ) of 2.57 × 10−5/substitutions/locus/year was used for the protein-coding genes derived from Pereira and Baker (2006). This is based on 1.3 × 10−8 substitutions/site/year multiplied by the sequence length of 1,980 bp.
Results
Microsatellites
Allele frequencies
The six snipe populations exhibited striking patterns of variation in allele frequencies at the nine polymorphic loci (Fig. 2). The Chatham Islands population was fixed (100% frequency) for allele 2 at locus SN7 that was absent in all other populations; allele 5 at SN10 was also private. Similarly, the Snares population was fixed for private alleles 3 at locus SN8 and 1 at SN19, and had five additional private alleles. Of the nine microsatellite loci, seven were fixed (at SN2, SN6, SN7, SN8, SN11, SN19, SN20) and two (SN10, SN17) were almost fixed or had high frequencies for particular alleles in the Chatham Island population. Corresponding numbers in the Snares Islands population were six loci with fixed alleles (SN2, SN7, SN8, SN17, SN19, SN20) and two with almost fixed alleles (SN6, SN11). In the most endangered population (Campbell Island), four loci were fixed (SN2, SN8, SN17, SN20) and one was almost fixed (SN7). The fixed loci on Campbell Island were fixed for a common allele (allele 1 in SN20) in all other populations except Snares Islands (allele 3 in SN17, which was absent in the Snares Islands), or were common alleles in all of Port Ross, Adams and Antipodes Islands (allele 1 in SN2, allele 5 in SN8). The Antipodes Island population was fixed for allele 3 in SN6, and allele 3 in SN17, and had two private alleles (allele 1 in SN7, and allele 4 in SN19). The Adams Island population (within the Auckland Islands) was fixed for allele 3 in SN17, and had 11 private alleles. By contrast, the Port Ross population (also in the Auckland Islands) had no private alleles and only one fixed locus (SN17).

Allele frequency profiles for nine microsatellite loci in the six sampled island populations of New Zealand snipes. Allele frequencies are shown on the ordinate and loci and alleles on the abscissa
Genetic diversity
Despite the generally fast rate of mutation at microsatellite loci (~10−2 to 10−5 per locus/generation; Dallas 1992, Dib et al. 1996), levels of genetic diversity in New Zealand snipe populations ranged from low to extremely low (Table 3). In both the Chatham and Snares Island populations mean observed heterozygosity (H o = 0.019 and 0.064, respectively) approached zero due to the fixation of alleles at most loci, and the mean number of alleles per locus (N a = 1.44 and 1.33, respectively) was low. Higher values of these two measures of gene diversity were evident in the four other populations, although the recently discovered population on Campbell Island had low mean observed heterozygosity (H o = 0.185). The highest genetic diversity was detected in the two sampling sites from the Auckland Islands (Port Ross and Adams Island: H o = 0.346 and 0.533, respectively).
Population differentiation
The log-likelihood of the data in STRUCTURE was maximized for K = 6 populations, corresponding to the samples from each of the isolated island groups. Because of enhanced genetic drift and other population genetic effects in most of these populations, microsatellite alleles were not widespread geographically. Clusters of genotypes were largely confined to one population except in the Auckland Islands where admixture was apparent (Fig. 3). The proportion of membership of individuals in the inferred clusters for the Chatham Islands and Snares Island were high (>0.964), as expected from their low genetic diversity and fixed alleles at loci. Conversely, individuals from the two samples in the Auckland Islands had mixed ancestry primarily in two clusters, with Port Ross individuals having greater membership in one cluster (0.755) and Adams Island individuals with higher membership in another cluster (0.559). The proportion of membership was also high in the isolated Antipodes and Campbell Islands, but some individuals in the Auckland Islands have small to moderate membership coefficients with Antipodes and Campbell Island clusters. Additionally, pair-wise Fst values between the six populations were all significantly different from zero (P < 0.001) when Bonferroni adjustments for multiple comparisons were applied. The Antipodes and Campbell Island samples are well differentiated in microsatellites (Fst = 0.57), in contrast to mtDNA where they share the same haplotype.

Estimated population structure showing the proportion of ancestry of the 113 New Zealand snipes sampled in this study, based on microsatellite allele frequencies. Each individual is represented by a bar showing its estimated membership proportion in the six clusters. Three different runs of STRUCTURE produced almost identical results
The maximum likelihood estimates of historical gene flow (4Nm) among populations were computed in Migrate (Table 4). The Snares Island and Chatham Islands populations are clearly isolated from each other and from populations on the more southern islands in the Auckland, Antipodes, and Campbell archipelagos (all regarded as C. aucklandica, 4Nm ≤ 0.7). Gene flow estimates between the southern populations ranged to a high of 4Nm = 2.31 from Campbell Island into Adams Island, but were insufficient to homogenize their gene pools.
Mitochondrial DNA protein-coding genes
Haplotype frequency and genetic diversity
Only 18 variable sites were detected in the 1,980 bp of sequence from the four genes. The most variable gene was COII with eight segregating sites, followed by COI (six sites) and ATPase6 and ATPase8 with four sites. As with the microsatellites, most (14/16) substitutions were restricted to single populations and thus served as clean population-specific markers; the two remaining haplotypes were shared among the southern island groups (Auckland, Antipodes and Campbell Islands; Table 5). MtDNA gene diversity in each population was low except for Adams Island, as measured by the number of haplotypes they possessed, the average number of nucleotide differences between haplotypes, and haplotype diversity and nucleotide diversity (Table 6). The very small numbers of substitutions in the four genes were all A-G or C-T transitions, and jointly defined only 16 haplotypes across all six populations. The only population with noticeable genetic variation was on Adams Island, where nine haplotypes were found (Table 5). All the other populations were genetically depauperate. No genetic variation was detected in the Antipodes Island and the critically endangered Campbell Island populations, as all individuals were fixed for the same haplotype (which these two populations both shared with Adams Island; Table 5).
Population structure
The geographic pattern of population differentiation is displayed clearly in a median-joining network showing the mutational steps among haplotypes, and the genetic gap between the three groups of populations (Fig. 4). It also shows that genetic diversity is harbored principally in the Adams Island population, and that the network radiating from the common haplotype is not star-like but instead involves a number of mutational steps between different haplotypes. The network and a NJ tree of the 104 sequences (not shown) also demonstrated that the haplotypes are sorted into three reciprocally monophyletic clades corresponding to the Chathams Islands, Snares Island and the more southern populations, respectively. However, the haplotypes in the southern clade are admixed among the Auckland Islands, Antipodes Island and Campbell Island. Using an outgroup sequence of the four genes from the blackish oystercatcher (Haematopus ater) the root of the tree was located between the southern group and the Chathams plus Snares populations.

Median-joining network showing connections among New Zealand snipe mtDNA haplotypes. Haplotypes are identified as in Table 5, and mutations separating haplotypes on the network are shown by site position numbers in the sequences. Pies show the geographic distribution and proportion of the haplotypes in samples colour-coded to populations
Species limits
To help adjudicate species limits, we calculated population divergence times using the coalescent approach in the program MDIV (Nielsen and Wakeley 2001). Based on a mutation rate (μ) of 2.57 × 10−5 substitutions/locus/year estimated for the protein-coding genes in shorebirds, the split of the Chatham Islands plus Snares Island clade from the southern clade dates to about 96,000 ya. The divergence of the Chatham Islands and Snares populations was estimated at about 42,000 ya, and divergence within the southern populations dated to about 10,000 ya. These estimates are very approximate because of the confounding effect of enhanced genetic drift in small populations and the wide credibility intervals associated with effectively one mtDNA locus, but nevertheless are compatible with very recent population divergence in the southern group, following the last glacial maximum about 22,000 ya. This also explains why lineage sorting is incomplete in these populations.
Discussion
Low genetic diversity
One of the most striking findings of this study is that, with the exception of the largest population on Adams Island (Miskelly et al. 2006) in the Auckland Islands, these isolated populations of New Zealand snipes had very low variation in both nine microsatellite loci and four mtDNA protein-coding genes totaling 1,980 bp of sequence. Based on levels of microsatellite genetic diversity in 22 threatened bird species around the world listed in Table 1 of Jamieson et al. (2006), Chatham, Snares and Campbell Island snipes rank 2nd, 3rd and 4th after kakapo (Strigops habroptilus) in genetic impoverishment. The Antipodes Island snipe is 6th after the Mariana crow (Corvus kubaryi), the Port Ross population is 8th after the great bustard (Otis tarda), and the Adams Island population falls between the Taita Thrush (Turdus helleri) and the Eurasian Vulture (Gyps fulvus). Furthermore, microsatellite allele frequencies were either fixed or at high frequency at the majority of loci, and there was a relative dearth of intermediate frequency alleles. MtDNA haplotypes were also fixed in the Antipodes and Campbell Island populations, and both haplotype and nucleotide diversity were very low in all but the Adams Island population. Such low genetic diversity is the signature of enhanced genetic drift in small populations, recent population bottlenecks, or recent selective sweeps that have erased previous genetic variation. Selective sweeps would be expected to affect only some and possibly small regions of the nuclear genome, and so it seems unlikely that all the microsatellite loci would be affected as has occurred in New Zealand snipes.
Radiation of Coenocorypha snipe
Our analyses revealed a clear geographic pattern in which the Chatham Islands and Snares Island populations each have unique mtDNA haplotypes whereas the populations in the more southern islands share haplotypes. Microsatellites also recovered this pattern when analyzed according to membership coefficients in each population, indicating that populations in the southern group are genetically isolated from populations in the Chatham Islands and Snares Island. Even within the southern group (Auckland Islands, Antipodes Island and Campbell Island) gene flow is very low, indicating that they share ancestral polymorphisms which have not yet been completely sorted into monophyletic lineages. Based on our analyses, a prudent taxonomic approach would be to treat the three major populations in the southern group as one species C. aucklandica. Given the morphometric and plumage differences between these populations (Higgins and Davies 1996; Worthy et al. 2002; Miskelly and Fraser 2006b) which evolve more quickly under selection for local adaptation than do neutral genes, they probably warrant recognition as separate subspecies. Sample sizes from Campbell Island and Antipodes Island are small, and they share the common mtDNA haplotype probably due to genetic drift in these small populations. Nevertheless, they are clearly differentiated in microsatellite coancestry analyses. This result is predicted because experimentally manipulated population bottlenecks in Drosophila have been shown to have reduced heterozygosity rather than allelic diversity in highly polymorphic microsatellites, and also changed allele frequency distributions in different populations (England et al. 2003).
Despite their smaller body size and bill length compared to Auckland Island and Antipodes Island snipe (Miskelly and Fraser 2006b), snipes in the recently discovered population on Campbell Island clearly do not constitute a new species. Given the much older divergence of the reciprocally monophyletic assemblages on the Snares Islands and the Chatham Islands, their clear genetic separation in both microsatellite allele frequencies and mtDNA sequences, and the morphometric and plumage differences between them, they are strongly diagnosable. We therefore recommend that each be treated as separate phylogenetic species C. pusilla and C. huegeli, as suggested by Worthy et al. (2002).
Without genetic data from extinct mainland New Zealand species of Coenocorypha it is not possible to infer their phylogenetic relationships with the extant species. However, the split of the three extant species ~96,000 ya suggests one invasion of ancestral stock to the southernmost subantarctic islands, and a more recent split between C. huegeli and C. pusilla. Alternatively, the Snares Island snipe could possibly be closest genetically to the extinct snipes of the New Zealand mainland, as Worthy et al. (2002) could find no osteological differences between C. huegeli and C. iredalei, which were separated by a water gap of only 105 km. The much older split for C. aucklandica (about 96,000 ya) from the other two species and relatively high genetic diversity of the Adams Island population of C. aucklandica aucklandica indicate that snipe persisted on the Auckland Islands during the Pleistocene ice ages. This is contra Fleming (1982), who hypothesized “The specialized feeding behaviour of snipe cannot cope with frozen ground, which most northern species escape by migrating. Thus, snipe probably did not colonise the subantarctic islands until post-glacial times, in the past 10,000 years.” However, the radiation of C. aucklandica to Antipodes and Campbell Islands in the last 10,000 years is consistent with Fleming’s hypothesis.
Conservation management implications
Conservation management units are warranted for each of the five taxa sampled in this study. The short term conservation priority for Campbell, Chatham and Auckland Island snipes is to restore them to more (and in two cases, all) of their historic ranges and thereby greatly increase their population sizes (Roberts and Miskelly 2003). In the long term this is also the best strategy to prevent extinction from lower fitness associated with inbreeding and the accumulation of deleterious mutations. Irrespective of whether the erosion of genetic variation is a consequence of population bottlenecks or enhanced genetic drift in populations that have been small for a long time, the effect is potentially serious for these populations. Increased random drift can lead to inbreeding (Saccheri et al. 1999), loss of genetic variability, and possibly lowered individual fitness (Woodworth et al. 2002). This in turn has been associated with increased susceptibility to diseases, parasites and environmental stress (Lacy 1997; Reed et al. 2002). Population sizes need to become large enough so that beneficial mutations counteract this genetic load, but it usually requires a census population size of thousands rather than hundreds of individuals (Whitlock 2000).
The small samples we analyzed from different ends of the Auckland Islands archipelago may indicate that some genetic differentiation occurred when they were historically restricted to refugia in 57 ha Ewing Island and in the much larger Adams Island, respectively. This points to the possible importance of refugia in preserving ancestral genetic diversity, more of which might have survived in the small refugial population that survived on Disappointment Island. The New Zealand Department of Conservation is planning to eradicate cats and pigs on 50,990 ha Auckland Island, which will restore Auckland Island snipe to 100% of their historic range, and restore gene flow through the north-south axis of the archipelago. If this does not proceed soon, or is unsuccessful, we recommend translocation of Auckland Island snipe from Adams Island to Enderby Island to restore genetic diversity to the Port Ross snipe population.
References
Aikman H, Miskelly C (2004) Birds of the Chatham Islands. Department of Conservation, Wellington
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Barker D, Carroll J, Edmonds H, Fraser J, Miskelly CM (2005) Discovery of a previously unknown Coenocorypha snipe in the Campbell Island group, New Zealand subantarctic. Notornis 52:143–149
Beerli P (2004) Migrate: documentation and program, part of LAMARC version 2.0. Revised December 23, 2004. Distributed over the internet, http://evolution.gs.washington.edu/lamarc.html
Beerli P, Felsenstein J (1999) Maximum likelihood estimation of migration rates and population numbers of two populations using a coalescent approach. Genetics 152:763–773
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Nat Acad Sci USA 98:4563–4568
Bell LC (1955) Notes on the birds of the Chatham Islands. Notornis 6:65–68
Bell BD (1974) Mangere Island. Wildlife—a review, vol 5. New Zealand Wildlife Service, Department of Internal Affairs, Wellington, pp 31–34
Dallas JF (1992) Estimation of microsatellite mutation rates in recombinant inbred strains of mouse. Mamm Genome 3:452–456
Dawson EW (1955) The birds of the Chatham Islands 1954 Expedition. Notornis 6:78–82
Dean AD, Greenwald JE (1995) Use of filtered pipet tips to elute DNA from agarose gels. Biotechniques 18:980
Dib C, Faure S, Fizames C et al (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154
England PR, Graham HR, Osler GHR, Woodworth LM, Montgomery ME, David A, Briscoe DA, Frankham R (2003) Effects of intense versus diffuse population bottlenecks on microsatellite genetic diversity and evolutionary potential. Conserv Genet 4:595–604
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics 164:1567–1587
Fleming CA (1982) George Edward Lodge, the unpublished New Zealand bird paintings. Nova Pacifica, Wellington
Fu Y-X (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Fu Y-X, Li W-H (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Glaubitz JC (2004) CONVERT: a user-friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol Ecol Notes 4:309–310
Hagelberg EH (1994) Mitochondrial DNA from ancient bones. In: Herrman B, Hummel S (eds) Ancient DNA. Springer, New York, pp 195–205
Hamilton MB, Pincus EL, Di Fiore A, Fleischer RC (1999) Universal linker and ligation procedures for construction of genomic DNA libraries enriched for microsatellites. Biotechniques 27:501–507
Higgins PJ, Davies SJJF (1996) Handbook of Australian, New Zealand and Antarctic birds vol 3 Snipe to pigeons. Oxford University Press, Melbourne
Holdaway RN, Worthy TH, Tennyson AJD (2001) A working list of breeding bird species of the New Zealand region at first human contact. New Zealand J Zool 28:119–187
Jamieson IG, Wallis GP, Briskie JV (2006) Inbreeding and endangered species management: is New Zealand out of step with the rest of the world? Conserv Biol 20:38–47
Lacy RC (1997) Importance of genetic variation in the viability of mammalian populations. J Mammal 78:320–335
Millener PR (1999) The history of the Chatham Islands’ bird fauna of the last 7000 years, a chronicle of change and extinction. In: Proceedings of the 4th international meeting of the society of avian paleontology and evolution (Washington DC, June 1996) Smithsonian contributions to paleobiology, vol 89, pp 85–109
Miskelly CM (1987) The identity of the hakawai. Notornis 34:95–116
Miskelly CM (1988) The Little Barrier Island snipe. Notornis 35:273–281
Miskelly CM (1990) Breeding systems of New Zealand snipe Coenocorypha aucklandica and Chatham Island snipe C. pusilla; are they food limited? Ibis 132:366–379
Miskelly CM (2000) Historical records of snipe from Campbell Island, New Zealand. Notornis 47:131–140
Miskelly CM, Fraser JR (2006a) Campbell Island snipe (Coenocorypha undescribed sp) recolonise subantarctic Campbell Island following rat eradication. Notornis 53:353–359
Miskelly C, Fraser J (2006b) Campbell Island snipe survey, January 2006. Department of Conservation, Wellington
Miskelly CM, Sagar PM, Tennyson AJD, Scofield RP (2001) Birds of the Snares Islands, New Zealand. Notornis 48:1–40
Miskelly CM, Walker KJ, Elliott GP (2006) Breeding ecology of three subantarctic snipes (genus Coenocorypha). Notornis 53:361–374
Miskelly CM, Dowding JE, Elliott GP, Powlesland RG, Robertson HA, Sagar PM, Scofield RP, Taylor GA (2008) Conservation status of New Zealand birds, 2008. Notornis 55:117–135
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nielsen R, Wakeley J (2001) Distinguishing migration from isolation: a Markov Chain Monte Carlo approach. Genetics 158:885–896
Nilsson RJ, Kennedy ES, West JA (1994) The birdlife of South East Island (Rangatira), Chatham Islands, New Zealand. Notornis 41(supplement):109–125
Pereira SL, Baker AJ (2006) A mitogenomics timescale for birds detects variable phylogenetic rates of molecular evolution and refutes the standard molecular clock. Mol Bio Evol 23:1731–1740
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Ramakrishnan U, Hadly EA, Mountain JL (2005) Detecting past population bottlenecks using temporal genetic data. Mol Ecol 14:2915–2922
Raymond M, Rousset F (1995) GENEPOP: population genetics software for exact tests and ecumenicism. J Heredity 86:248–249
Reed DH, Briscoe DA, Frankham R (2002) Inbreeding and extinction: the effect of environmental stress and lineage. Conserv Genet 3:301–307
Roberts A, Miskelly C (2003) Recovery plan for the snipe species of New Zealand and the Chatham Islands (Coenocorypha spp) tutukiwi, 2003–2015. Department of Conservation, Wellington
Rozas J, Sanchez-DelBarrio JC, Messequer X, Rozas R (2003) DNAsp, DNA polymorphism analysis by the coalescent and other methods. Bioinformatics 19:2496–2497
Saccheri IJ, Wilson IJ, Nichols RA, Bruford MW, Brakefield PM (1999) Inbreeding of bottlenecked butterfly populations: estimation using the likelihood of changes in marker allele frequencies. Genetics 151:1053–1063
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning. Cold Spring Harbor Laboratory, Cold Spring Harbor
Schneider S, Roessli D, Excoffier L (2000) Arlequin ver 2000: a software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Tennyson AJD, Millener PR (1994) Bird extinctions and fossil bones from Mangere Island, Chatham Islands. Notornis 41(supplement):165–178
Turbott EG (1990) Checklist of the birds of New Zealand and the Ross Dependency, Antarctica. Random Century and Ornithological Society of New Zealand, Auckland
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Whitlock MC (2000) Fixation of new alleles and the extinction of small populations: drift load, beneficial alleles, and sexual selection. Evolution Int J org Evolution 54:1855–1861
Woodworth LM, Montgomery ME, Briscoe DA, Frankham R (2002) Rapid genetic deterioration in captive populations: causes and conservation implications. Conserv Genet 3:277–288
Worthy TH (2003) A new extinct species of snipe Coenocorypha from Vitilevu, Fiji. Bulletin of the British Ornithologists’ Club 123:90–103
Worthy TH, Holdaway RN (2002) The lost world of the moa, prehistoric life of New Zealand. Canterbury University Press, Christchurch
Worthy TH, Miskelly CM, Ching BA (2002) Taxonomy of North and South Island snipes (Aves: Scolopacidae: Coenocorypha) with analysis of a remarkable collection of snipe bones from Greymouth, New Zealand. New Zealand J Zool 29:231–244
Yeh FC, Boyle TJB (1997) Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belg J Bot 129:157
Acknowledgments
We thank BDG Synthesis, the Natural Sciences and Engineering Research Council of Canada (grant A 200 to AJB), and the New Zealand Department of Conservation (DOC) for financial support for fieldwork and labwork. Snipe blood and feather samples were collected with the assistance of Dave Barker, Karen Barlow, Jeremy Carroll, Graeme Elliott, James Fraser, Sheryl Hamilton, Christine Reed, Kath Walker, Murray Willans, and Al Wiltshire. Access to nature reserves and collection of samples was authorised by the DOC Southland and Wellington Conservancies. Figure 1 was prepared by Chris Edkins, DOC.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Baker, A.J., Miskelly, C.M. & Haddrath, O. Species limits and population differentiation in New Zealand snipes (Scolopacidae: Coenocorypha). Conserv Genet 11, 1363–1374 (2010). https://doi.org/10.1007/s10592-009-9965-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-009-9965-2