
Abstract
Freshwater pearl mussels (Margaritifera margaritifera) are among the most critically threatened bivalve molluscs worldwide. An understanding of spatial patterns of genetic diversity is crucial for the development of integrative conservation strategies. We used microsatellites to study the genetic diversity and differentiation of 14 populations of M. margaritifera in central Sweden, an area which was described as a major secondary contact zone in postglacial colonisation for other species. Genetic diversity of Swedish pearl mussel populations was much greater than in central and southern Europe but similar to the genetic diversity observed in the northeastern portion of their European range. Genetic differentiation among populations was pronounced but to a large extent independent from present-day drainage systems. The complex patterns of genetic diversity and differentiation in pearl mussel seem to be strongly influenced by the species’ high degree of specialisation and extraordinary life history strategy which involves facultative hermaphrodism and an obligatory encystment stage on a host fish. Genetic drift effects and anthropogenic disturbances resulting in reduction of population size and loss of connectivity are less pronounced in northern pearl mussel populations compared to those in central and southern Europe.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Freshwater bivalve molluscs (order Unionoida, also known as naiads) are often considered the most endangered group of animals in the world (Bogan 1993; Neves et al. 1997; Lydeard et al. 2004; Strayer et al. 2004). The complex life cycle of unionoids typically involves an encystment phase on a host fish. Their specific water quality and substratum requirements during different stages of development make them good indicator species for aquatic ecosystem health. Given the important roles of freshwater bivalves in particle filtration and processing, nutrient release and sediment mixing, the decline or extinction of freshwater mussels can profoundly affect ecosystem processes in aquatic habitats (Vaughn and Hakenkamp 2001; Geist and Auerswald 2007).
One of the most critically threatened naiads in Europe is the freshwater pearl mussel (Margaritifera margaritifera). M. margaritifera was a formerly widespread and abundant species, distributed from the Arctic and temperate regions of Western Russia through Europe to the north-eastern seaboard of Northern America (Jungbluth et al. 1985). Several studies have revealed dramatic declines throughout its range (e.g. Bauer 1988), and the species is at present under a serious threat of extinction in Europe with only a small number of successfully recruiting populations remaining (Ziuganov et al. 1994; Young et al. 2001; Geist 2005; Geist and Auerswald 2007). Excessive pearl fishing, habitat destruction by water pollution, eutrophication, acidification, river engineering and the local decline of host fish populations have all contributed to the decline of freshwater pearl mussels (Young et al. 2001; Geist 2005). The great reproductive potential in concert with the longevity of pearl mussels, and the robust status of host fish stocks in many European watersheds (Geist et al. 2006) suggest that the species has the potential for recovery if immediate action is taken. Studies into the life-history traits of freshwater pearl mussel populations along a latitudinal gradient (from Spain to the polar circle) revealed a strong variation in life-history traits such as life span and reproductive success (Bauer 1992). These differences may also be reflected in the genetic structure of populations in northern Europe versus populations in the southern portion of the species’ range. The recognition of these regional genetic patterns may be crucial for the development of effective conservation strategies for pearl mussels.
Recent studies have demonstrated that knowledge of the genetic structure of freshwater pearl mussel populations can be extremely useful for their conservation (Geist et al. 2003, 2008; Marchordom et al. 2003; Geist and Kuehn 2005, 2008; Bouza et al. 2007). The lowest genetic diversity of pearl mussels and a strong genetic differentiation of populations were observed in the southwestern portion of their European range (Bouza et al. 2007) despite the fact that this area was an important refugium for many species during the proposed glacial maximum during the pleistocene (Taberlet et al. 1998; Hewitt 2000). In contrast, higher genetic diversity was found in eastern and north-eastern Europe (Geist and Kuehn 2005, 2008), but no study has yet investigated the genetic structure of pearl mussel populations in north-western Scandinavia. A geographical zone in central Sweden has been described as a major European contact zone for genetically divergent evolutionary lineages in many species (reviewed in Taberlet et al. 1998; Hewitt 2000), including aquatic taxa and fish species (e.g. Gum et al. 2009). This region was thus selected as a candidate area for studying the genetic structuring of pearl mussels and for comparing it with results on the genetic variation within and between populations from central, southern and northeastern Europe.
The objective of this study was to analyse the spatial pattern of genetic diversity and differentiation of freshwater pearl mussels from three major drainage systems in the northern portion of their European range in the context of the species life history strategy and the known contact zone.
Materials and methods
Sampling strategy
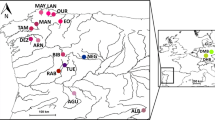
A total of 250 individuals from 14 pearl mussel populations were sampled from three major drainage systems of Ångermanälven (four populations), Gideälven (three populations) and Ljungan (seven populations) in county Västernorrland in central Sweden during August 2008. The selection of sampling sites was based on a representative distribution of populations from three major drainage systems in the area, from populations of good versus bad prior ecological population assessment by the district of Västernorrland (Söderberg et al. 2008; H. Söderberg, personal communication), as well as from downstream populations close to the sea versus upstream populations far from the sea (in order to include possible differences in the host fish spectrum, Atlantic salmon, Salmo salar and brown trout, Salmo trutta). Special attention was attributed to representative sampling of mussels over the distribution stretch in the respective rivers and to representative sampling of all age classes except for mussels <4.5 cm maximum shell length. Smaller mussels were excluded since they live buried in the stream substratum and could have been harmed during sampling. In one population (SSB) only six mussels were recovered during intensive search. This number appears to be close to the remaining total number of mussels in this river. Descriptions of the sampling locations are provided in Table 1 and Fig. 1. Haemolymph sampling as described in Geist and Kuehn (2005) was carried out in order to prevent any harm to the mussels. All mussels were returned to the original sites after the sampling.

Sampling locations (black circles) of freshwater pearl mussel (Margaritifera margaritifera) populations in Sweden and magnification of the sampling sites from three major drainage systems Ångermanälven, Gideälven and Ljungan; sample codes according to Table 1
DNA isolation and microsatellite analyses
Haemolymph samples were transferred into 1.7 ml Eppendorf vials, cooled at 2°C during the sampling trip and then immediately processed in the laboratories of Technische Universität München, Germany. After centrifugation at 14,000g for 5 min the supernatant was discarded and DNA was isolated from the remaining cellular pellet with the NucleoSpin Tissue Kit (Machery-Nagel) following the manufacturer’s instructions for preparation of tissue material. In order to allow comparability of results with other genetic studies on pearl mussels, all genetic analyses were conducted based on genotyping of nine species-specific standard microsatellite markers for M. margaritifera as described in Geist et al. (2003), Geist and Kuehn (2005, 2008). Polymerase chain reactions (PCRs) were performed in a total volume of 12.5 μl with the following components: 25–50 ng of genomic DNA, 200 nM of each primer, 0.2 mM of each dNTP, 3 mM MgCl2 (2 mM MgCl2 for locus MarMa5280), 1× PCR buffer (10 mM Tris–HCl, 50 mM KCl, 0.08% Nonidet P40), and 0.25 U Taq DNA Polymerase (Qbiogene). PCR products were separated on 5% denaturing 19:1 acrylamide:bisacrylamide gels on ALFexpressII DNA analyser and scored with Allelelinks 1.02 software (Amersham Pharmacia Biotech). Electrophoresis was carried out with two internal standards (70, 300 bp) in each lane. Additionally, an external standard (50–500 bp ladder) and a previously sequenced reference sample were included on each gel in order to ensure exact scoring and to facilitate cross-referencing among gels.
Statistical and population genetic analyses
Allele frequencies, average allele numbers per locus (A), and expected and observed heterozygosities (H e , H o) were calculated with GENEPOP v. 3.4 (Raymond and Rousset 1995a). GENEPOP v. 3.4 was also used to test the genotypic distribution within populations for conformance with Hardy–Weinberg (HW) expectations (Hardy–Weinberg exact test). Values of F IS and allelic richness (A R) as a standardized measure of the number of alleles per locus corrected by the sample size were calculated with the FSTAT v. 2.9.3 program package (Goudet 1995). Alleles were considered private if they showed a frequency higher than 5% in one population and did not occur in any other population.
Genetic differentiation between pairs of populations was estimated by F ST (Weir and Cockerham 1984). Tests for significant population differentiation among all pairs of populations were performed with GENEPOP using 100,000 iterations and 1,000 de-memorisation steps (Raymond and Rousset 1995b). Nei D A genetic distance (Nei et al. 1983) was calculated using the DISPAN program (Ota 1993). ARLEQUIN 3.0 software (Excoffier et al. 2005) was used to hierarchically quantify genetic population structure by analysis of molecular variance (AMOVA; Excoffier et al. 1992), and to incorporate molecular information based on allelic frequencies. All probability tests were performed applying the Markov Chain algorithm (Guo and Thompson 1992; Raymond and Rousset 1995b). Sequential Bonferroni adjustments (Rice 1989) were used to correct for multiple tests. The Bayesian approach of population assignment test (Cornuet et al. 1999; ‘as it is’ option) implemented in the GENECLASS 1.0.02 program (Piry and Cornuet 1999) was used to estimate the likelihood of an individual’s multilocus genotype to be assigned to the population from which it was sampled. Relatedness between individuals was estimated based on the F-value from the 2MOD program (Ciofi and Bruford 1999) which refers to the probability that two genes share a common ancestor within a population and correlates with effective population sizes. The 2MOD program was also used to investigate the population history of the freshwater pearl mussel populations based on the coalescent theory. The method uses the comparison of the relative likelihoods of a model of immigration-drift equilibrium (gene flow model) versus drift. A Markov Chain Monte Carlo simulation (100,000 iterations) was computed, and the first 10% of the output were discarded in order to avoid bias due to the starting conditions.
In order to determine the number of genetic clusters (K) and to probabilistically assign individuals to these clusters, the Bayesian clustering method as proposed by Pritchard et al. (2000) was carried out using STRUCTURE 2.2 software (Pritchard et al. 2000). This model-based Bayesian approach was selected since it excludes prior information on the origin of individuals. We tested K from one to 14 with ten iterations (20,000 burn-in, 200,000 Markov chain Monte Carlo replicates in each run) to assess convergence of ln Pr(X|K). The number of clusters present was then determined from posterior probabilities of K and additionally by an ad hoc statistic ΔK based on the rate of change in the log probability of data (Evanno et al. 2005). For the determined values of K, we assessed the average proportion of membership (admixture coefficient, q) of the samples to the inferred clusters using CLUMPP (Jakobsson and Rosenberg 2007), applying the LargeKGreedy algorithm.
In order to combine geographical and genetic data and to identify zones of reduced gene flow between populations, we generated a synthesis map with the first principal component (PC) scores obtained from a PC analysis using allele frequencies of each locus and each population as variables according to Piertney et al. (1998). PC1 scores were interpolated within space by the Kriging interpolation procedure (Journel and Huijbregts 1978). The interpolated lines in the resulting diagram represent zones of equal genetic divergence (isogenes). The number of isogenes between two sampling sites shows the degree of their genetic differentiation. The colour shows the value the first principal component. The analyses were carried out with STATISTICA 6.0 and SURGE 1.4.0. In addition, a Mantel test with 1,000 iterations implemented in GENALEX 6 (Peakall and Smouse 2006) was used to test for the correlation of geographic distance among population pairs (measured by the shortest waterway distance between populations on 1:25,000 and 1:65,000 scale maps, Norrgrann 2001) with their genetic differentiation [measured by (F ST/(1−F ST); Rousset 1997].
Results
Genetic diversity
Genetic diversity of freshwater pearl mussels in their northern European range was higher than previously described for central and southern European populations. A mean number of 11 alleles per locus were observed for the nine microsatellite loci used in this study. The number of alleles per locus ranged from one at locus MarMa5280 to 21 at locus MarMa3621. Identical multilocus genotypes were only found in the genetically least diverse population SMT, where six redundant genotypes occurred. Allelic variation, expressed by the average number of alleles per locus (A) and allelic richness (A R) varied strongly between populations and averaged 4.4 and 3.0 over all loci and populations, respectively. A summary of the microsatellite diversity indices is provided in Table 2. Both the highest (A = 6.7, A R = 3.6) and the lowest genetic diversity (A = 2.2, A R = 1.6) were observed within the Gideälven drainage system. Only one population, SMT, showed a distinctively low genetic diversity compared to all other populations. Expected and observed heterozygosities (H e and H o) averaged 0.496 and 0.444, respectively, and were also exceptionally low for population SMT (Table 2). The inbreeding coefficient F IS was highest in population SMT as well. The proportion of common ancestors within each population inferred from the F-values of the 2MOD program covered a wide range, from F = 0.028 in the genetically diverse population SGI to F = 0.592 in population SMT. Private alleles occurred at six different loci and in six populations from all three drainage systems. They consistently occurred at frequencies of 10% or less. The maximum of private alleles (3) was found in population SGI where the maximum values of H e and H o (0.580 and 0.528, respectively) were found. Six out of the fourteen populations (42%) significantly deviated from expected Hardy–Weinberg proportions at single loci after Bonferroni correction (Table 2).
AMOVA analyses of hierarchical gene diversity revealed that 76.9% of the genetic variation was accounted within individuals, 6.8% was due to differences among individuals within populations and 16.3% was due to differences among populations within drainage systems. Only 0.01% of the variation was due to differences among drainage systems. The global fixation indices were 0.082, 0.163, 0.231 for F IS, F ST and F IT, respectively. F SC and F CT were 0.163 and <0.001.
Genetic differentiation
Pearl mussel populations in county Västernorrland, Sweden showed a drainage-independent, strongly structured genetic differentiation pattern, with a mean F ST of 0.163. Genetic distance was very great between some population pairs with F ST values as high as 0.500 between SMT and SGA and a maximum Nei D A of 0.405 (Table 3). In contrast, populations in close geographical vicinity had lowest genetic differentiation, as evident for the four geographically adjacent populations SNB, SRA, SVA, SVB for which pairwise F ST values were consistently ≤0.070. The mantel test found no significant correlation between geographic (measured along waterways) and genetic differentiation of populations (r 2 = 0.028, P = 0.144). The results of the distance matrices (Table 3) and the combined incorporation of geographical and genetic data in the synthesis map (Fig. 2) clearly show that the observed genetic differentiation pattern is largely independent from the three major drainage systems in the area. For instance, populations SHE (from the northernmost Gideälven drainge) and SGR and SHA (both from the southernmost Ljungan drainage) were only moderately differentiated as evident from similar z-values in the PCA (Fig. 2) and pairwise F ST values of 0.090 and 0.102, respectively (Table 3). In contrast, genetic differentiation within the Gideälven drainage system was much more pronounced with the geographically adjacent populations SHE and SMT showing distinct z-values and a pairwise F ST value of 0.436. The drainage-independent genetic population structure is also supported by the results of the assignment test, by the AMOVA analyses of hierarchical gene diversity, and by the genetic cluster analyses. Individual multilocus genotype based STRUCTURE analyses identified two hierarchical levels of structure with three or six genetic clusters of pearl mussel populations using ΔK and ln Pr(X|K) analyses (Fig. 3). None of the drainage systems is exclusively assigned to one single genetic cluster, but populations with geographical proximity within drainage systems (SVB, SVA, SRA, SNB) were consistently assigned to the same genetic cluster for K = 3 and for K = 6 (Table 4). The high genetic variability and the missing link between genetic differentiation and drainage systems is also reflected in the weak assignment of individuals to their source populations and their drainage systems: Only 125 out of the 250 individuals (50%) were correctly assigned to their sampling site and 163 out of 250 (63%) were correctly assigned to their drainage of origin (Table 5). One exceptionally high rate of correct self-assignment was evident for population SMT (100% assignment to population of origin and to drainage system). The strong differentiation of this population to other populations is likely to be influenced by strong genetic drift effects since SMT was simultaneously found to have lowest genetic diversity of all investigated populations. In contrast, none of the specimens from population SGI was assigned to their sampling site and only 10% were correctly assigned to the drainage of origin. At the same time, population SGI had the lowest probability of common ancestors and was one of the genetically most variable populations (Table 2).

Synthesis map combining geographical and genetic data using principal component analysis (PCA) with the x and y axis representing the geographical latitude and longitude of the sampled populations and colours and topography representing the first principal component scores (z-values) obtained from allele frequencies of each locus and population as variables. The interpolated lines in the resulting diagram represent zones of equal genetic divergence (isogenes)

Mean (±SD) of log likelihood values (14 independent runs), ln(X|K), and rate of change in the log probability of data between successive values of K, ΔK, for the Margaritifera margaritifera dataset (n = 250)
Based on the results of the 2MOD program (Ciofi and Bruford 1999), the relative likelihood of the model of gene flow-drift equilibrium versus drift revealed a gene-flow model (P = 1.0) for the Swedish pearl mussel populations.
Discussion
Genetic diversity
This study represents the first analyses of the genetic diversity of endangered freshwater pearl mussel populations in the northwestern portion of their Scandinavian distribution range using highly variable microsatellite markers. In congruence with genetic studies in southern (Bouza et al. 2007), central, and eastern European freshwater pearl mussel populations using the same panel of microsatellites (Geist and Kuehn 2005, 2008), a substantial degree of genetic structure within small geographical scales was observed. Genetic diversity of Swedish pearl mussels as measured by mean number of alleles, allelic richness, observed and expected heterozygosities, F IS and F-values was on average higher than in populations from central Europe and from the Iberian peninsula but similar to those in the north-eastern distribution range. For instance, allelic richness was consistently below 2.0 for all investigated central European populations (Geist and Kuehn 2005) and averaged 2.1 for all Iberian populations (Bouza et al. 2007). In thirteen out of the 14 Swedish populations investigated in this study, allelic richness was higher than 2.7 but did not exceed values >4.0 which were recorded in populations from Finish Lapland (Geist and Kuehn 2008). Observed and expected heterozygosities in some Swedish populations even exceeded the values from Finish Lapland and mark the highest values ever described in the species. This pattern is also reflected in the probability of common ancestors (2MOD), which was much lower in Swedish populations compared to those from other areas. Consequently, the most inclusive measure of all inbreeding (F IT) in Swedish populations was only 50% of the F IT value observed in central European populations (0.231 and 0.423, respectively). A number of reasons may contribute to the high genetic diversity observed in the north of the pearl mussel distribution range: several studies have suggested that central Sweden is a major European contact zone where distinct evolutionary lineages have met in diverse taxonomic groups, resulting in an overall increased genetic diversity (Taberlet et al. 1998; Hewitt 2000). These patterns have also been observed in aquatic species such as grayling (Thymallus thymallus; Gum et al. 2009), a fish which frequently co-occurs with freshwater pearl mussels (Geist et al. 2006). The validity of the contact zone in pearl mussel could explain the high genetic diversity found in this area. Due to the complex life cycle of pearl mussels which involves an encystment phase on the gills of a host fish, a greater spectrum of host fish stocks may also render a greater genetic diversity of the pearl mussel. Whereas brown trout (Salmo trutta) is currently the only available host fish for pearl mussels in central Europe (Wächtler et al. 2001; Geist et al. 2006), central Sweden is also a major habitat for Atlantic salmon (Salmo salar) and for migratory forms of the Salmo trutta complex which are all suitable host fishes for pearl mussel, too (Ranke et al. 1999). Within the study area, the Gideälven is the most important salmon stream in which two forms of the Salmo trutta complex, the migratory sea trout and the brown trout also occur (H. Söderberg, personal communication). The most downstream population in the Gideälven mainstream (SGI) was found to have highest genetic diversity, lowest probability of common ancestors and the smallest rate of correct assignment of all investigated populations, indicating that a link between diversity of host fish species and genetic diversity in pearl mussel may exist.
One of the most important factors on a larger geographical scale is that genetic diversity of pearl mussels seems to be closely linked to latitudinal differences in the ecology and life-history strategy of the species (Bauer 1992; Geist and Kuehn 2008). Margaritifera margaritifera has maintained highest population densities in optimal northern habitats and longevity in northern populations can exceed the reproductive life span of central and southern European populations by more than 100 years, which increases the chances of contribution of individual parent mussels to the next generation. Both factors contribute to reduced genetic drift over time and may also explain the high genetic diversity of pearl mussel in the northern portion of its range (Geist and Kuehn 2008). This finding may be attributable to the specialisation and exceptional life history of pearl mussel since the observations of highest genetic diversity in post-glacially colonised areas are inconsistent with the theoretical expectation that genetic diversity should decrease with distance from glacial refugia (Hewitt 1996) and also with results from North American mussels where genetic diversity was found to decrease in northern mussel populations relative to southern populations (Elderkin et al. 2008). The strong effect of life history strategy and ecological niche on the genetic pattern observed in pearl mussel is also supported by the negative correlation between genetic diversity of pearl mussel and brown trout (Geist and Kuehn 2008). Northern pearl mussel populations are located in areas with a low density of humans and seem to be less influenced by anthropogenic disturbances such as habitat loss, eutrophication, siltation and habitat fragmentation compared to populations in central and southern Europe (Geist and Auerswald 2007), thus increasing the chances of genetically diverse juvenile recruitment at low rates of genetic drift. Pronounced genetic drift effects resulting in reduction of genetic diversity and overestimation of genetic differentiation have mostly been reported for peripheral and disturbed populations in southern and central Europe (Bouza et al. 2007; Geist and Kuehn 2005). This effect appears to significantly influence the overall population structuring of pearl mussels and has also been described to increase the distinctiveness of some North American mussel populations (e.g. Grobler et al. 2006).
Genetic differentiation
The observed pattern of pearl mussel population differentiation in central Sweden was very structured but largely independent from present-day drainage systems with only 0.01% of the genetic variation being due to differences among drainage systems. As evident from the Mantel test, an isolation-by-distance model does not explain the genetic differentiation patterns observed. At smaller geographical scales within drainages, however, a closer relatedness of populations was observed, as evident for the clustering of populations SVA, SVB, SRA and SNB. The lack of structure among drainages could theoretically be due to movements of host fish (historically or recent), either due to naturally large ranges (as evident in salmon and migratory brown trout) or anthropogenic movements of fish. Indeed, these hypotheses are supported by the gene-flow model for the Swedish pearl mussel populations. However, the genetic differentiation among populations based on estimates of F ST and Nei D A, as well as the results of the structure analyses, the synthesis map and the occurrence of unique private alleles in six populations all indicate a pronounced genetic structure of Swedish pearl mussel populations. Strong population differentiation within single tributaries has also previously been described in pearl mussel from central European areas (Geist and Kuehn 2005) and in North American mussel species (e.g. Berg et al. 2007) but may be species specific even for mussels that occur in the same ‘bed’ (Elderkin et al. 2008).
The weak rates of correct assignment to the populations of origin are in sharp contrast to central and southern European populations, where 92% (Iberian Peninsula; Bouza et al. 2007) and 79% (central Europe; Geist and Kuehn 2005) of the individuals were correctly assigned to their sampling sites. These differences can largely be explained by the greater genetic variability and by higher gene flow in Swedish pearl mussel populations compared to those in more southern areas. The genetically most variable Swedish populations (e.g. SGI, SHE) had very low rates of correct assignment to their sampling site (SGI = 0%, SHE = 5%) and drainage of origin (SGI = 10%, SHE = 25%), whereas the genetically least variable population SMT which lies in the same drainage and between the two most variable ones had 100% correct assignment of specimens to their original stream from which they were sampled.
The degree of genetic differentiation among pearl mussel populations is generally influenced by the colonisation history of populations, by local adaptations due to mutation and selection, as well as by drift and migration, and ultimately by the life history strategy and demography of populations. Colonisation of pearl mussel populations in Sweden must have happened after the last ice age, probably not earlier than about 10,000 years ago (Hamilton et al. 1989). The pearl mussel colonisation history is probably closely linked to the colonisation history of the fish host (Geist and Kuehn 2008) but the roles of brown trout (Salmo trutta), salmon (Salmo salar) and arctic charr (Salvelinus alpinus) in this process remain unresolved. Among freshwater invertebrates, population structure has been negatively correlated with degree of isolation (Miller et al. 2002) and dispersal ability (Bilton et al. 2001). The strong dependence of pearl mussel migration on passive glochidial transport via host fish and the high degree of specialisation of pearl mussels to extremely oligotrophic conditions probably both contribute to stronger mechanisms of isolation compared to other co-occurring aquatic species, even within adjacent populations in the same drainage system (Geist and Kuehn 2008). In addition, female mussels can switch to facultative hermaphrodites and periodically produce enormous numbers of offspring (Bauer 1987). This extraordinary reproductive strategy is likely to increase genetic founder/drift effects and to result in elevated levels of inter-population differentiation. In addition, anthropogenic factors which directly reduce the effective population sizes and which influence habitat quality and fragmentation can enhance genetic drift effects and induce genetic bottlenecks (Geist and Kuehn 2005). This effect is strongest in central and southern European populations, but also partially occurs in the dataset from Swedish populations, where the strong differentiation of the small population SMT to other populations may be overestimated due to the extremely low genetic diversity in this population. Population SMT only comprised <100 individuals of similar size (and age) and was the only population where redundant genotypes were found, indicating that this population may have reached a severe genetic bottleneck.
Adaptation to specific habitat conditions may also contribute to the genetic differentiation patterns observed yet remains untested. Culturing and breeding experiments indicate that genetically differentiated pearl mussel populations may have different survival rates and dietary preferences (Lange, personal communication).
Conclusions
The results of this study in concert with previous results from other geographical regions (Bouza et al. 2007; Geist and Kuehn 2005, 2008) clearly suggest that the spatial genetic structuring of pearl mussels is more complex than that in other species. Whereas drainage-specific genetic structuring is a common observation in many fish species (e.g. Hänfling and Brandl 1998; Gross et al. 2001), patterns of genetic diversity and differentiation of pearl mussels and of co-occuring species in the same habitats, including their host fish, seem to differ in many cases (Geist and Kuehn 2008). The high genetic diversity of pearl mussel in the north of its European range is inconsistent with the expectation that genetic diversity would be lowest in post-glacially colonised areas. The life history strategy and the narrow ecological niche of the species, as well as colonisation history and anthropogenic habitat modifications all shape the degree of genetic diversity and differentiation of M. margaritifera. The cryptic spatial genetic population structure of pearl mussels thus requires detailed genetic studies in order to be able to fully reveal these patterns and to be able to develop sound conservation strategies which conserve a maximum of the species’ evolutionary potential. The recognition of genetic clusters and the identification of priority clusters for conservation can be a useful conservation tool, as described for migratory fish species (Geist et al. 2009). Studies which link genetic patterns of diversity and differentiation with ecological habitat features and with the life history of species may increase our understanding of the cryptic genetic structure in freshwater pearl mussels and other aquatic mollusc species.
References
Bauer G (1987) Reproductive strategy of the freshwater pearl mussel Margaritifera margaritifera. J Anim Ecol 56:691–704. doi:10.2307/5077
Bauer G (1988) Threats to the freshwater pearl mussel Margaritifera margaritifera in central Europe. Biol Conserv 45:239–253. doi:10.1016/0006-3207(88)90056-0
Bauer G (1992) Variation in the life span and size of the freshwater pearl mussel. J Anim Ecol 61:425–436. doi:10.2307/5333
Berg DJ, Christian AD, Guttman SI (2007) Population genetic structure of three freshwater mussels (Unionidae) species within a small stream system: significant variation at local spatial scales. Freshw Biol 52:1427–1439. doi:10.1111/j.1365-2427.2007.01756.x
Bilton DT, Freeland JR, Okamura B (2001) Dispersal in freshwater invetebrates. Annu Rev Ecol Syst 32:159–181. doi:10.1146/annurev.ecolsys.32.081501.114016
Bogan AE (1993) Freshwater bivalve extinctions (Mollusca: Unionoida): a search for causes. Am Zool 33:599–609
Bouza C, Castro J, Martínez P, Amaro R, Fernández C, Ondina P, Outeiro A, San Miguel E (2007) Threatened freshwater pearl mussel Margaritifera margaritifera L. in NW Spain: low and very structured genetic variation in southern peripheral populations assessed using microsatellite markers. Conserv Genet 8:937–948. doi:10.1007/s10592-006-9248-0
Ciofi C, Bruford MW (1999) Genetic structure and gene flow among komodo dragon populations inferred by microsatellite loci analysis. Mol Ecol 8:17–30. doi:10.1046/j.1365-294X.1999.00734.x
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999) New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153:1989–2000
Elderkin CL, Christian AD, Metcalde-Smith JL, Berg DJ (2008) Population genetics and phylogeography of freshwater mussels in North America, Elliptio dilatata and Actinonaias ligamentina (Bivalvia: Unionidae). Mol Ecol 17:2149–2163. doi:10.1111/j.1365-294X.2008.03745.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Geist J (2005) Conservation genetics and ecology of european freshwater pearl mussels (Margaritifera margaritifera L.) Ph.D. thesis, Technische Universität München, Germany. Available at: http://tumb1.biblio.tu-muenchen.de/publ/diss/ww/2005/geist.pdf. Accessed 12 May 2009
Geist J, Auerswald K (2007) Physicochemical stream bed characteristics and recruitment of the freshwater pearl mussel (Margaritifera margaritifera). Freshw Biol 55:2299–2316. doi:10.1111/j.1365-2427.2007.01812.x
Geist J, Kuehn R (2005) Genetic diversity and differentiation of central European freshwater pearl mussel (Margaritifera margaritifera L.) populations: implications for conservation and management. Mol Ecol 14:425–439. doi:10.1111/j.1365-294X.2004.02420.x
Geist J, Kuehn R (2008) Host-parasite interactions in oligotrophic stream ecosystems: the roles of life history strategy and ecological niche. Mol Ecol 17:997–1008. doi:10.1111/j.1365-294X.2007.03636.x
Geist J, Rottmann O, Schröder W, Kühn R (2003) Development of microsatellite markers for the endangered freshwater pearl mussel Margaritifera margaritifera L. (Bivalvia: Unionoidea). Mol Ecol Notes 3:444–446. doi:10.1046/j.1471-8286.2003.00476.x
Geist J, Porkka M, Kuehn R (2006) The status of host fish populations and fish species richness in European freshwater pearl mussel (Margaritifera margaritifera) streams. Aquat Conserv 16:251–266. doi:10.1002/aqc.721
Geist J, Wunderlich H, Kuehn R (2008) Use of mollusc shells for DNA-based molecular analyses. J Molluscan Stud 74:337–343. doi:10.1093/mollus/eyn025
Geist J, Kolahsa M, Gum B, Kuehn R (2009) The importance of genetic cluster recognition for the conservation of migratory fish species: the example of the endangered European Huchen (Hucho hucho L.). J Fish Biol (in press)
Goudet J (1995) F-STAT version 1.2: a computer program to calculate F-statistics. J Hered 86:485–486
Grobler PJ, Jones JW, Johnson NA, Beaty B, Struthers J, Neves RJ, Hallerman EM (2006) Patterns of genetic differentiation and conservation of the slabside pearlymussel, Lexingtonia dolabelloides (Lea, 1840) in the Tennessee river drainage. J Molluscan Stud 72:65–72. doi:10.1093/mollus/eyi055
Gross R, Kühn R, Baars M, Schröder W, Stein H, Rottmann O (2001) Genetic differentiation of European grayling populations across the Main, Danube and Elbe drainages in Bavaria. J Fish Biol 58:264–280. doi:10.1111/j.1095-8649.2001.tb00513.x
Gum B, Gross R, Geist J (2009) Conservation genetics and management implications for European grayling, Thymallus thymallus: synthesis of phylogeography and population genetics. Fish Manag Ecol 16:37–51. doi:10.1111/j.1365-2400.2008.00641.x
Guo SW, Thompson EA (1992) Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometric 48:361–372. doi:10.2307/2532296
Hamilton KE, Ferguson A, Taggart JB, Tomasson T, Walker A, Fahy E (1989) Post-glacial colonisation of brown trout, Salmo trutta L.: Ldh-5 as a phylogeographic marker locus. J Fish Biol 35:651–664. doi:10.1111/j.1095-8649.1989.tb03017.x
Hänfling B, Brandl R (1998) Genetic variability, population size and isolation in distinct populations in the freshwater fish Cottus gobio L. Mol Ecol 7:1625–1632. doi:10.1046/j.1365-294x.1998.00465.x
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc Lond 58:247–276
Hewitt G (2000) The genetic legacy of the quaternary ice ages. Nature 405:907–913. doi:10.1038/35016000
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. doi:10.1093/bioinformatics/btm233
Journel A, Huijbregts C (1978) Mining geostatistics. Academic Press, London, UK
Jungbluth JR, Coomans HE, Groh H (1985) Bibliographie der Flussperlmuschel Margaritifera margaritifera (Linn. 1785). Verslagenen Technische Gegevens No. 41 Institut voor Taxonomische Zoologie, Universiteit Amsterdam
Lydeard C, Cowie RH, Ponder WF et al (2004) The global decline of nonmarine molluscs. Bioscience 54:321–330. doi:10.1641/0006-3568(2004)054[0321:TGDONM]2.0.CO;2
Marchordom A, Araujo R, Erpenbeck D, Ramos MA (2003) Phylogeography and conservation genetics of the endangered European Margaritiferidae (Bivalvia: Unionoidea). Biol J Linn Soc Lond 78:235–252. doi:10.1046/j.1095-8312.2003.00158.x
Miller MP, Blinn DW, Keim P (2002) Correlations between observed dispersal capabilities and patterns of genetic differentiation in populations of four aquatic insect species from the Arizona white mountains, USA. Freshw Biol 47:1660–1673. doi:10.1046/j.1365-2427.2002.00911.x
Nei M, Tajima F, Tateno Y (1983) Accuracy of genetic distances and phylogenetic trees from molecular data. J Mol Evol 19:153–170. doi:10.1007/BF02300753
Neves RJ, Bogan AE, Williams JD, Ahlstedt SA, Hartfield PW (1997) Status of aquatic molluscs in the southeastern United States: a downward spiral of diversity. In: Benz GW, Collins DE (eds) Aquatic fauna in peril: the southeastern perspective. Southeast Aquatic Research Institute, Lenz Design and Communications, Decatur Special Publication 1
Norrgrann O (2001) Spridningsavstånd mellan och fragmentering av bestånd av flodpärlmussla (Margaritifera margaritifera) i Västernorrlands län. Självständigt arbete Institutionen för Naturvetenskap och Miljö. Stencil. English abstract. Mitthögskolan, Härnösand. 21 pp
Ota T (1993) Dispan: genetic distance and phylogenetic analyses software. Pennsylvania State University, Pennsylvania
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–855. doi:10.1111/j.1471-8286.2005.01155.x
Piertney SB, MacColl ADC, Bacon PJ, Dallas JF (1998) Local genetic structure in red grouse (Lagopus lagopus scoticus): evidence from microsatellite DNA markers. Mol Ecol 7:1645–1654. doi:10.1046/j.1365-294x.1998.00493.x
Piry S, Cornuet JM (1999) GENECLASS: a program for assignation and exclusion using molecular markers. URLB/INRA, France
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:845–859
Ranke W, Rappe C, Soler T, Funegård P, Karlsson L, Thorell L (1999) Baltic salmon rivers—status in the late 1990 s as reported by the countries in the Baltic region. The Swedish Environmental Protection Agency, Gothenburg, Sweden, 69 pp
Raymond M, Rousset F (1995a) GENEPOP version 3.4: population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Raymond M, Rousset F (1995b) An exact test for population differentiation. Evolution 49:1280–1283. doi:10.2307/2410454
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225. doi:10.2307/2409177
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Söderberg H, Karlberg A, Norrgrann O (2008) Status, trender och skydd för flodpärlmusslan I Sverige. ISBN: 1403-624X
Strayer DL, Downing JA, Haag WR et al (2004) Changing perspectives on pearly mussels, North America’s most imperilled animals. Bioscience 54:429–439. doi:10.1641/0006-3568(2004)054[0429:CPOPMN]2.0.CO;2
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464. doi:10.1046/j.1365-294x.1998.00289.x
Vaughn CC, Hakenkamp CC (2001) The functional role of burrowing bivalves in freshwater ecosystems. Freshw Biol 46:1431–1446. doi:10.1046/j.1365-2427.2001.00771.x
Wächtler K, Dreher-Mansur MC, Richter T (2001) Larval types and early postlarval biology in naiads (Unionoida). In: Bauer G, Wächtler K (eds) Ecology and evolution of the freshwater mussels Unionoidea. Springer, Heidelberg. Ecol Stud 145:93–125
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. doi:10.2307/2408641
Young MR, Cosgrove PJ, Hastie LC (2001) The extent of, and causes for, the decline of a highly threatened naiad: Margaritifera margaritifera. In: Bauer G, Wächtler K (eds) Ecology and evolution of the freshwater mussels Unionoidea. Springer, Heidelberg. Ecol Stud 145:337–357
Ziuganov V, Zotin A, Nezlin L, Tretiakov V (1994) The freshwater pearl mussels and their relationships with salmonid fish. VNIRO, Russian Federal Institute of Fisheries and Oceanography, Moscow, 104 pp
Acknowledgments
This work was initiated and financially supported by the Nature Conservation Office at the County Administrative Board of Västernorrland.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Geist, J., Söderberg, H., Karlberg, A. et al. Drainage-independent genetic structure and high genetic diversity of endangered freshwater pearl mussels (Margaritifera margaritifera) in northern Europe. Conserv Genet 11, 1339–1350 (2010). https://doi.org/10.1007/s10592-009-9963-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-009-9963-4