
Abstract
A genetic analysis of freshwater pearl mussel Margaritifera margaritifera populations from NW Spain, a peripheral area of its European distribution, was carried out using microsatellite markers. These populations were formerly reported as genetically differentiated on the basis of growth and longevity studies. Ten loci previously characterized in populations from central Europe were used to comparatively analyze the genetic variability at the southern edge of the species’ range. Iberian pearl mussel populations showed very low genetic variability and significant high genetic differentiation. Half of the total genetic diversity observed appeared to be distributed between populations, which suggested a highly structured adaptive potential in pearl mussel at the southern peripheral distribution of the species. Population distinctiveness was evidenced by assignment tests, which revealed a high accuracy of individual assignments to their population of origin. All data suggested low effective population size and major effects of genetic drift on population genetic structure. In order to avoid further loss of genetic variation in biologically distinctive populations from NW Spain, prioritization of genetic resources of this species is required for conservation and management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Freshwater mussels or naiads (Bivalvia, Order Unionoida) are among the highest endangered group of invertebrates due to habitat destruction and other anthropogenic perturbations (Lydeard et al. 2000; Roe and Hoeh 2003). The long-lived naiad Margaritifera margaritifera (Family Margaritiferidae) is listed as “threatened” by IUCN (2004), and recently appears as one of the most critically threatened naiad in Europe due to the catastrophic declines and local extinctions throughout most of its range throughout the 20th century (Bauer 1983, 1986, 1988; Beasley and Roberts 1996; Ziuganov et al. 1998; Young et al. 2001; Velasco et al. 2002; Reis 2003). The freshwater pearl mussel is distributed from the artic and temperate regions of western Russia, westwards through Europe to the northeastern seaboard of North America (Young et al. 2001). Bauer (1992) studied 48 populations of this species across Europe and found a latitudinal variation (north Spain to polar circle) among several life-history traits. Their study showed that reproductive success was positively correlated with both maximum life span and maximum shell length, but negatively with growth rate. Iberian populations exhibit the highest growth rates, the shortest life span and the smallest maximum shell length known for M. margaritifera (San Miguel et al. 2004). Furthermore, adult specimens in these populations likely produce fewer offspring (Ziuganov et al. 2000; San Miguel et al. 2004). It has long been recognized that in a wide variety of organisms, mortality increases with growth rate but decreases with body size (Gunderson 1980; Roff 1984, 1992, 2001). Iberian populations can be considered peripheral because they are lying at the southern edge of the species’ range and appear to be close to their physiological (and reproductive) limits (San Miguel et al. 2004). Geographical peripheral populations, separated from central ones by spatial distance, are more likely to be imperilled than central ones, as has been demonstrated in different animal and plant species (Lesica and Allendorf 1995; Durka 1999; Pedersen and Loeschcke 2001; Guo et al. 2005). In comparison with central populations, peripheral populations may show less genetic variation and substantial genetic divergence caused by genetic drift and selection (Lesica and Allendorf 1995; Vucetich and Waite 2003; Garner et al. 2004). It is argued that divergence associated with geographic isolation will be more noticeable in quantitative characters (Schwaegerie et al. 1986). In plants, it has been shown that self-fertilizing populations occur at the geographic periphery of outcrossing species or species complexes (Solbrig and Rollins 1977), which probably has a strong impact on the population genetic structure of peripheral populations. Pearl mussels are normally dioecious (Bauer 1987; Ziuganov et al. 1994), but females have the ability to switch over to hermaphroditism under certain conditions (e.g., low density). There is some evidence for a high percentage of hermaphrodite individuals in Iberian populations (Grande et al. 2001; Reis 2003), suggesting a high prevalence of selfing related to their peripheral habitat and low densities. Selfing might genetically impoverish these populations, increasing their extinction risk. However, whether or not peripheral populations of freshwater pearl mussel show less and highly structured genetic variation remains uncertain.
Although over the last decade there has been an increasing interest in all aspects of unionoid mussel biology, including genetics (Roe and Hoeh 2003), there is little genetic information available on M. margaritifera. Allozyme studies of Margaritifera species, including M. margaritifera from Spain, revealed very low genetic variability (Davis and Fuller 1981; Machordom et al. 2003; Curole et al. 2004); these latter authors warned about the need for specific management of populations, and protection of species. However, the very low allozymic variation, particularly in M. margaritifera, limit more detailed analysis of population genetic structure. Therefore, the need for additional studies using hypervariable genetic markers has been proposed (Curole et al. 2004). Recently, microsatellite loci has demonstrated appropriate resolution to reveal the genetic structure for central European populations of freshwater mussel, providing useful information for conservation and management programs (Geist and Kuehn 2005). However, nothing is known about microsatellite variation in peripheral populations of this species.
Local wildlife authorities and ecological organizations in NW Spain have recently become concerned about the situation of the last pearl mussel populations in this area. In the present study, ten microsatellite markers (Geist et al. 2003) were used to analyze genetic diversity and differentiation among M. margaritifera populations from the main river basins in NW Iberian Peninsula. We compared levels and patterns of genetic variation of peripheral populations from southern European areas with previous published data from central Europe. The results obtained revealed very low variability and high genetic differentiation in freshwater pearl mussel populations from NW Spain. This information will be essential for conservation and management of genetic resources of this species at the southern border of its European distribution.
Material and methods
Biological samples and DNA extraction
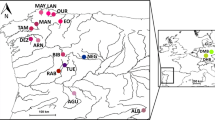
Samples of M. margaritifera were collected in 2002 and 2003 from seven sites from six river drainages in north-western Spain (Fig. 1). The sampling area from north to south covered the following river basins: the Eo river flowing into the Cantabric Sea (EO), the Mandeo river into the Artabric gulf (MD), and the rivers Tambre (TA), Ulla (AG, Arnego tributary), Miño (TR and MR, Trimaz and Mera tributaries) and Limia (SA, Salas tributary) river basins into the Atlantic Ocean.

Sampling site locations of Margaritifera margaritifera populations from six river basins in NW Spain (EO, MD, TA, AG and SA, were from the Eo, Mandeo, Tambre, Ulla and Limia river basins, respectively; the other two sites were MR and TR, from the Miño, the main river basin in the area under study). Coordinates of the map: upper left border: 43°16′ N/7°15′ W; lower right border: 40°4′ N/1°35′ W
Total DNA was extracted from foot muscle following the CTAB (hexadecyltrimethylammoniumbromide) procedure described in Winnepennincks et al. (1993). The DNA obtained was resuspended in TE buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA) to a final concentration of about 100 ng/μl, and used as template in polymerase chain reactions (PCR).
Microsatellite analysis
Ten microsatellite loci from M. margaritifera (MarMa1632, MarMa3050, MarMa3621, MarMa4143, MarMa4277, MarMa4315, MarMa4322, MarMa4726, MarMa4859 and MarMa5167) were amplified using the oligonucleotide primers reported by Geist et al. (2003). PCR reactions were performed using 15 μl of mixture reaction containing 40 ng of template DNA, 100 μM of each dNTP, 15 pmol of each primer (the forward was fluorescently labelled with Cy5), 1XPCR Gold Buffer (15 mM Tris–HCl, pH 8.0, 50 mM KCl), 1.5 mM MgCl2 and 0.5 U Amplitaq Gold DNA polymerase (Applied Biosystems). Amplifications were carried out in a MJ Research PTC-100 thermocycler under the following conditions: an initial denaturation step at 95°C for 5 min, followed by 35 cycles at 94°C for 45 s, 50 s at the specific annealing temperature (as in Geist and Kuehn 2005, with slight modifications) and 72°C for 50 s, and a final extension step at 72°C for 10 min. Alleles were scored on High Resolution acrylamide gels run in an ALFexpress II automatic sequencer using Alfwin Fragment Analyser 2.1 software (Amersham Biosciences). A sample of 25 individuals from the Elbe drainage (ZI) was analyzed for the same microsatellite set of this study as a reference for genetic comparison with previous data on central European populations of this species (Geist and Kuehn 2005). Allelic nomenclature was established by mutual comparison between individual genotypes at the six loci common to both studies (J. Geist, personal communication).
Data analysis
Unbiased expected heterozygosity (H e), mean number of alleles per locus (A) and proportion of polymorphic loci (P99 and P95) were computed to estimate genetic diversity (Nei 1987). Allelic richness (Â) per locus was obtained using the rarefaction method of fstat 2.9.3 (Goudet 1995) for normalizing sample size. Departure from Hardy–Weinberg equilibrium (H-W) per locus and genotypic linkage equilibrium between all pairs of loci were tested by exact tests using genepop 3.1 (Raymond and Rousset 1995). The F IS estimate was computed according to Weir and Cockerham (1984) using genepop 3.1. Assuming that departure from H-W equilibrium is only due to inbreeding produced by self-fertilization, the selfing rate S was estimated using the classical relationship S = 2 F IS/(1+ F IS) (Hedrick 2005). The program micro-checker (van Oosterhout et al., 2004) was used to check for genotyping accuracy and to detect evidences of null alleles or large allele dropout.
In order to detect if the populations have experienced a reduction in effective size, two different methods were applied. First, using the program bottleneck 1.2.02 (Piry et al. 1999), populations were tested under different mutational models (IAM, SMM and the intermediate two-phased model, TPM) for heterozygosity excess given the number of alleles observed, as expected following a recent bottleneck (Cournet and Luikart 1996). Significance of heterozygosity excess was checked using Wilcoxon signed-rank tests. Second, as bottlenecks also lead to the loss of rare alleles, the ratio of the number of alleles to the range of allele size (Garza and Williamson 2001) was calculated for each population using the program agarst (Harley 2001).
Homogeneity tests were performed between pairs of samples by exact probability tests using genepop 3.1. The program fstat v 2.9.3 was used to calculate global estimates of genetic differentiation using G ST (Nei 1987) and R ST (Slatkin 1993) statistics based on the infinite alleles (IAM) and the stepwise (SMM) mutation models, respectively. To estimate the genetic differentiation between all pairs of populations, pairwise F ST based on polymorphic loci (Weir and Cockerham 1984) were calculated using fstat v 2.9.3. The use of absolute measures of divergence has been recommended, since relative measures of differentiation between populations are dependent on the extent of within-population diversity. In this study, we have tried to standardise FST values according to levels of variability using Nei’s estimator of minimum genetic differentiation D m (Nei 1987), as suggested by Hedrick (2005). Absolute genetic distances between pairs of samples were also obtained by using the estimator D A over all loci (Nei 1987). The application of D A in our study will permit to establish direct comparisons with D A genetic distances between pairs of central European populations reported by Geist and Kuehn (2005). Genetic relationships among populations were obtained from D A genetic distance matrix by generating a multidimensional scaling method (MDS) and a minimum spanning tree (MST; ntsys statistical package; Rohlf 1992). Isolation by distance was tested using the isolde program in genepop 3.1. The correlation between geographic distances (in km, measured directly following the river contours) and genetic distance estimated by D A and F ST/(1–F ST) was tested (Slatkin 1993; Rousset 1997) using the isolde program in genepop 3.1. The statistical significance of the correlation was tested using a Mantel randomization test and 1,000 permutations. The program geneclass2 (Piry et al. 2004) was used to estimate the likelihood of an individual’s multilocus genotype occurring in a given population, and indirectly the level of population genetic differentiation. As in Geist and Kuehn (2005), population history was investigated with coalescent models by using the 2mod program (Ciofi et al. 1999). The method is based on the comparison between the likelihoods for a gene flow-drift equilibrium model and a pure drift model, assuming negligible mutation effects (Goodman et al. 2001). The probability that two genes shared a common ancestor within a population was also estimated for each population (F 2mod value). A simulation (100,000 iterations) was computed and the first 10% of the output was discarded in order to avoid bias to the starting conditions. The sequential Bonferroni correction was employed when multiple tests were carried out (Rice 1989).
Results
Genetic diversity within populations
The range of allele size and frequency distribution at the ten microsatellite loci analyzed in a central European pearl mussel population (ZI, Geist and Kuehn 2005) were highly concordant with the results obtained in the Iberian populations studied. This agreement supports the accuracy of the allelic scoring process in both studies and allowed comparison between them. The main discrepancy was related to the allelic nomenclature used in both studies, since a slippage of the allelic series by two and three bp was observed at MarMar4143 and MarMar5167 loci, respectively.
Two out of 10 microsatellite loci studied were monomorphic in all NW Spanish populations (MarMa4315 and MarMa4322; Appendix A; Table 1). The loci MarMa4726 and MarMa1632 were weakly polymorphic and only showed variation in the TR and/or MR samples. MarMa4859 and Marma4277 were the most polymorphic loci. However, the eight polymorphic loci showed low variability (Table 1), yielding very low average estimates of genetic variability within population (Table 2). The average number of alleles per locus ranged from 1.2 in AG to 2.5 in EO and the expected heterozygosity from 0.010 in AG to 0.279 in TR. The lowest values for genetic variability were found in the AG population from the Ulla basin, in which no loci were polymorphic using the 95% criterion (Fig. 1; Table 2). Maximum values of genetic diversity were observed in the TR population (A = A R = 2.4; H e = 0.279), one of two samples from the Miño basin (Fig.1; Table 2). In contrast, higher variability was observed in the ZI population from central Europe for the same set of 10 loci, all of which were polymorphic at the 95% criterion (A = 3.8; A R = 3.6; H e = 0.498; Table 2).
A total of 15 out of 27 tests showed significant deviations from Hardy–Weinberg expectations. Eight of these remained statistically significant after sequential Bonferroni correction (29.5%; Table 1). All significant deviations were due to heterozygote deficiency (mean F IS: 0.627 ± 0.041), mostly restricted to MarMa4859 and MarMa4277 loci in most populations (mean F IS: 0.546 ± 0.091 and 0.361 ± 0.100, respectively; Table 1). Statistically significant deviations were not systematic, occurring at different loci in different populations (Table 1). The application of micro-checker only revealed deviations from the expected homozygote and heterozygote distributions attributable to the presence of null alleles over populations at locus MarMa4859. In the EO population, all polymorphic loci showed heterozygote deficiency (mean F IS: 0.534 ± 0.018). These deficits were significant for three loci after sequential Bonferroni correction. A high average selfing rate of S = 0.62 ± 0.04 was observed, ranging from 0.48 in TA to 0.70 in EO and MR (Table 1). The proportion of common ancestors within each population as inferred from the F 2mod values ranged from 0.28 in TA to 0.84 in AG, with a global average of 0.493 ± 0.084 for all populations under study (Table 1). Exact probability tests for genotypic disequilibrium between all pair of loci across all populations revealed a single significant value among 28 comparisons. This value was not significant after sequential Bonferroni correction. When each population was tested separately, genotypic equilibrium between all pairs of loci was generally observed. Only a single pair of loci showed genotypic association after Bonferroni correction in the EO population (MarMa3621–MarMa4859).
Bottleneck tests based on allele frequency distributions using different mutation models (either IAM, SMM or TPM) were not significant for any of the four populations studied (EO, TA, MR and TR). The remaining samples could not be analyzed using this method because of low genetic variation (MD, AG and SA, less than four polymorphic loci; Cournet and Luikart 1996). The ratio of the number of alleles over the allele size distribution provided high M values for all populations excluding AG, which was 0.4 (Table 1). This value is considerably lower than the 0.68 value reported by Garza and Williamson (2001) as a strong indication for past reduction in population size.
Genetic differentiation between populations
Overall G ST and R ST estimates of differentiation among populations were high and significant (0.506 and 0.449, respectively; Table 3). Homogeneity test performed for each locus across populations were also highly significant. Pairwise F ST values ranged from 0.194 (TR-MR) to 0.856 (AG-MD), and all were statistically significant after correction for multiple tests (Table 4). Interestingly, two pairs of Spanish populations showed higher F ST values (>0.8) than any pair involving ZI, the reference central European population (Table 4). Because F ST is highly dependent on the intrapopulation diversity some values were clearly overestimates, as confirmed by using minimum genetic distances to standardize F ST values (D m; Table 3). By contrast to FST values, the absolute measures of genetic divergence were higher for the ZI population (D m > 0.44; D A > 0.58, between ZI-TR; mean D m and D A = 0.486 and 0.645, respectively) than for any pair involving Spanish populations (mean D m and D A = 0.183 and 0.231, respectively). The D A genetic distances between all pairs of Iberian populations ranged from a minimum between TR and MR (D A = 0.112), the two populations from Miño basin, to maximum figures for SA-AG and SA-MR pairs (D A = 0.389 and 0.328, respectively; Table 4). The distinctness of populations was also supported by the presence of private alleles and the assignment tests. A total of 92.2% of the individuals was correctly assigned to the population of origin (Table 5). Eleven private alleles occurred at four loci in five populations, some of them at high frequencies. The highest number was five in the EO population (Table 2; Appendix A). The samples from Miño basin were the most closely related, and were grouped with the Atlantic sample TA (Fig. 2). The remaining Atlantic and Cantabric populations (EO, MD, AG) were genetically very divergent and did not show an obvious geographical pattern (Fig. 2). Very low and nonsignificant correlations were observed between pairwise estimates of genetic differentiation and geographical distances.

Genetic relationships among Iberian Margaritifera margaritifera populations obtained by three dimensional multidimensional scaling (MDS) with superimposed minimum spanning tree (MST) based on the D A genetic distance matrix between all pairs of populations, including a central European reference sample (ZI). The stress value (0.01) indicates a good fit of the MDS to genetic distance matrix. Population codes are presented in Fig. 1
Discussion
Genetic variability within population
Genetic variability of the Iberian M. margatifera populations was very low. All showed 30% to 70% decrease in allelic richness, and up to 98% in heterozygosity with respect to the central European population (ZI) genotyped in our study (Table 2). In comparison with the 24 central European populations previously studied by Geist and Kuehn (2005), the average variability was significantly lower in the Iberian populations studied (H e: 0.173 vs 0.323; P < 0.05). In addition, the proportion of populations with a low number of polymorphic loci (less than four variable markers in 43% of the populations) was much higher than previously reported in this species (20%). Given the common subset of loci used in both studies, these data indicate reduced genetic variation in southern European populations. The results are in agreement with the general prediction for geographically peripheral populations i.e. reduced genetic variability as a result of small population size and spatial isolation (Lesica and Allendorf 1995).
Low genetic variation in pearl mussel populations may be explained by different factors. Anthropogenic influence has lead to fragmentation and deterioration of the sensitive habitat of this specialized mussel. In addition, a complex life cycle involving a parasitic phase probably explains the presence of small populations interconnected by low gene flow. Among the available host in NW Spain are Atlantic salmon (Salmo salar) and principally the brown trout with both resident and anadromous ecotypes (Salmo trutta). Despite extremely high mussel fecundities only a small percentage of larvae survive past metamorphosis to the juvenile stage (Curole et al. 2004). The combination of high reproductive rates and the variance in the reproductive success of few parents can increase the variance in family size, and so decreases the ratio Ne/N (Hedgecock 1994). This effect in combination with the habitat perturbation may dramatically reduce effective population size. Therefore, pearl mussel populations would be subjected to strong genetic drift effects resulting in low levels of genetic variability within populations, and high genetic differentiation between populations. The predominant effect of drift on population genetic composition was supported by the results of the 2mod analysis with a higher probability of supporting a gene flow-drift disequilibrium model (P = 0.997), as well as by high F ST values (see below). However, the test by Cornuet and Luikart (1996) did not provide evidence for a recent bottleneck in any population studied. These results might be explained by low statistical power because of the limited number of loci and their low level of polymorphism in some populations. In addition, this test may fail to detect recent reductions in effective population size if it has been low for several generations (Luikart et al. 1998; Hansen et al. 2000). The approach by Garza and Williamson (2001) suggested past reduction in population size for AG (M < 0.7; Table 1). Although caution should be observed when interpreting M estimates calculated with a limited number of loci (8 out of 10 loci were monomorphic in AG), even a pair of loci have provided indirect evidence of genetic bottlenecks using this method (Gagnon and Angers 2006).
Biological characteristics of the pearl mussel and its anadromous hosts, sea trout and Atlantic salmon may be under stress in these species at the southern periphery of their European distribution (Bouza et al. 1999; Morán et al. 2005). More stressful habitats are usually expected for marginal populations at the periphery of a species distribution area (Pedersen and Loeschcke 2001). In addition, the southern peripheral populations of M. margaritifera could be genetically less variable than populations from central Europe because a higher prevalence of selfing in the long term (Grande et al. 2001; Reis 2003; Charlesworth 2003). Assuming that the heterozygote deficiencies detected in our study were mainly caused by non-random mating, the estimated selfing rates were very high (Table 1). The highest inbreeding values inferred by F2mod were observed in the less variable populations, the maximum in AG (F > 80%) with extremely low levels of genetic variability. This could be a general phenomenon for genetically eroded populations of the species (Geist and Kuehn 2005), but more acutely present in the southern peripheral areas as shown in this study. Inbreeding should affect all loci, generating uniform heterozygote deficiencies across polymorphic loci, as occurs in the EO population. The observed variability in heterozygote deficits across loci in the remaining populations (Table 1) requires other explanations. For example, sampling error increasing the variance of F IS among loci cannot be excluded. Another explanation could be the presence of null alleles (Brookfield 1996), particularly for MarMa4859 and MarMa4277 loci which show high F IS across loci in most populations. The application of micro-checker only revealed the presence of null alleles at locus MarMa4859 over most populations. By contrast, in the EO population, all polymorphic loci showed a general excess of homozygotes for most allele size classes, attributable to the presence of null alleles according to micro-checker. It is unlikely that all loci show null alleles at high frequencies in this single population, inbreeding appearing as a more likely explanation of the deviations of HW equilibrium detected over all loci in this case. Additional factors such as the Wahlund effect, caused by sampling a mixture of genetically differentiated populations across river basins mediated by anadromous hosts should also be considered. This could be particularly likely in the Eo river basin where different freshwater mussel populations exist along the fluvial course, despite no data on the genetic differentiation among them exist to date (San Miguel et al. 2004). To confirm this aspect, further studies on pearl mussel genetic structure within EO basin would be required. Fine-scale genetic analysis involving relatedness estimation in relation with spatial distances between individuals would be necessary to disentangle selfing, mating among relatives, null alleles, and the Wahlund effect as causes of the HW deviations detected (Curole et al. 2004; Vekemans and Hardy 2004).
Genetic variability among populations
Half of the total genetic diversity observed was located between populations (Table 3). Population differentiation was also demonstrated by assignment tests. Because no significant correlation between geographical and genetic distances could be found, isolation by distance seems unlikely. Patterns of microsatellite genetic differentiation of Iberian pearl mussel populations studied could be due to different factors. First, the microhabitat availability within each river basin because the strong physical habitat preferences of the species (substratum descriptors, aquatic vegetation cover, mean water depth and current velocity etc.; Zyuganov 1994; San Miguel et al. unpublished data). This is particularly important in peripheral areas where less favourable habitats are expected. Second, the existence of a parasitic stage in the life cycle is another important factor shaping the genetic structure of populations of this species. The mobility of infected hosts would be one of the major factors influencing gene flow because of the limited dispersal of the glochidial larval stage. While gene flow among river basins has not been proven to date, it can not be ruled out, at least during past flood events along evolutionary history (Machordom et al. 2003). Host population genetic structure probably influences the distribution of genetic variability of the mussel. On one hand, genetically eroded populations of the host with small population sizes might reduce the infestation success of glochidia. However, extremely low genetic variation in some populations (e.g. AG) was not associated with low variation in brown trout samples from the same sampling area (Bouza et al. 1999; unpublished data). On the other hand, high genetic differentiation among brown trout populations has been described in the area under study, with restricted gene flow among populations even in proximity (Bouza et al. 1999; unpublished data). The highest genetic similarity observed in this study between MR and TR pearl mussel populations (Fig. 2) was in agreement with high gene flow among brown trout populations in tributaries of the Miño basin’s upper course. However, the scattered pattern of genetic variation observed in the remaining pearl mussel populations (Fig. 2) was not strictly related to the genetic differentiation observed among brown trout populations from Cantabric, Artabric and Atlantic basins (Bouza et al. 1999; unpublished data). These comparisons of genetic structure between host and mussel should be taken with caution since the reduced number of pearl mussel populations studied. The preference for resident brown trout in pearl mussel central European populations (Geist and Kuehn 2005) should probably be the case for some populations in this study (MR, TR and SA). These samples were from Miño and Limia rivers basins located below the current limit of distribution of the anadromous brown trout (Bouza et al. 1999). Both resident and anadromous brown trout as well as Atlantic salmon are present in the remaining river basins studied, especially in the Cantabric and Artabric areas. The possibilities of recruitment of the glochidium stage could be probably increased in these rivers (Fernández et al. 2006). Finally, habitat fragmentation together with small effective sizes population has resulted in high differentiation among NW Spanish populations. Strong genetic drift on populations studied was supported by high global and pairwise F ST figures, as well as by the presence of private alleles, even between close populations. The importance of stochastic phenomena influencing the population genetic structure of freshwater pearl mussels was also pointed by Geist and Kuhen (2005).
As expected, the central European population used as reference in this study (ZI) was highly differentiated with respect to the NW Spanish populations (F ST = 0.585; D A = 0.635). This is in agreement with both previous studies on M. margaritifera populations using microsatellites (Geist and Kuehn 2005; global F ST = 0.374 Central Europe) and low polymorphic allozymes (Machordom et al. 2003; global F ST = 0.320 including northern and southern European populations). However, the peripheral southern populations studied revealed the higher level of genetic structure (G ST = 0.506). The average genetic distance observed in the area under study (D A = 0.231) was much higher than the average between populations from Elbe and Danube river basins flowing into Atlantic and Black Sea drainages (D A = 0.164 recalculated from Geist and Kuehn 2005). These results suggested very low gene flow and a high degree of genetic variation among populations.
Management implications
The present study represents the first population genetic analysis of M. margaritifera from NW Spain using microsatellite loci. These populations are exceptional, based on growth and longevity data (San Miguel et al. 2004). Our results showed very low and structured genetic variation at the southern peripheral range of the species, suggesting a high risk of extinction for many of these biologically distinctive populations. The combination of data from neutral markers and adaptive characters support the priority for conservation of endangered Iberian populations. More detailed knowledge of the genetic diversity within and among populations will be essential to develop strategies for conservation and management, including future plans for captive breeding and reintroduction. In addition, the conservation of mussel habitat should be considered a priority in the protection of this threatened species.
References
Bauer G (1983) Age structure, age specific mortality rates and population trend of the freshwater pearl mussel (Margaritifera margaritifera) in North Bavaria. Arch Hydrobiol 98:523–532
Bauer G (1986) The status of the freshwater pearl mussel Margartifera margaritifera L. in the South of its european range. Biol Conserv 38:1–9
Bauer G (1987) Reproductive strategy of the freshwater pearl mussel Margaritifera margaritifera. J Animal Ecol 56:691–704
Bauer G (1988) Threats to the freshwater pearl mussel Margaritifera margaritifera L. in central Europe. Biol Conserv 45:239–253
Bauer G (1992) Variation in the life span and size of the freshwater pearl mussel. J Anim Ecol 61:425–436
Beasley CR, Roberts D (1996) The current distribution and status of the freshwater pearl mussel Margaritifera margaritifera L. 1758 in north-west Ireland. Aquatic Conservation. Mar Freshwater Ecosyst 6:169–77
Bouza C, Arias J, Castro J, Sanchez L, Martinez P (1999) Genetic structure of brown trout, Salmo trutta L. at the southern limit of the distribution range of the anadromous form. Mol Ecol 8:1991–2001
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5:453–455
Charlesworth D (2003) Effects of inbreeding on the genetic diversity of populations. Philos T Roy Soc B 358:1051–1070
Ciofi C, Beaumont MA, Swingland IR, Bruford MW (1999) Genetic divergence and units for conservation in the Komodo Dragon Varanus komodoensis. Proc R So Lond Ser B 266:2269–2274
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–20014
Curole JP, Foltz DW, Brown KM (2004) Extensive allozyme monomorphism in a threatened species of freshwater mussel, Margaritifera hembeli Conrad (Bivalvia: Margaritiferidae). Conserv Genet 5:271–278
Davis GM, Fuller SLH (1981) Genetic relationships among recent Unionacea (Bivalvia) of North America. Malacologia 20:217–253
Durka W (1999) Genetic diversity in peripheral and subcentral populations of Corrigiola litoralis L. (Illecebraceae). Heredity 83:476–484
Fernández C, Outeiro A, Ondina P, Amaro R and San Miguel E (2006) The freshwater mussel Margaritifera margaritifera (L.) in Galicia (NW of Spain): Preliminary study in Eo and Masma Rivers. In: Abstracts of the international congress on Bivalvia, Universitat Autònoma de Barcelona, Barcelona, July 2006
Gagnon MC, Angers B (2006) The determinant role of temporary preglacial drainages on the genetic structure of fishes. Mol Ecol 15:1051–1065
Garner TWJ, Pearman PB, Angelone S (2004) Genetic diversity across a vertebrate species’ range: a test of the central–peripheral hypothesis. Mol Ecol 13:1047–1053
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Geist J, Kuehn R (2005) Genetic diversity and differentiation of central European freshwater pearl mussel (Margaritifera margaritifera L.) populations: implications for conservation and management. Mol Ecol 14:425–439
Geist J, Rottmann O, Schröder W, Kühn R (2003) Development of microsatellite markers for the endangered freshwater pearl mussel Margaritifera margaritifera L. (Bivalvia: Unionoidea). Mol Ecol Notes 3:444–446
Goodman SJ, Tamate HB, Wilson R, Nagata J, Tatsuzawa S, Swanson GM, Pemberton JM, McCullough DR (2001) Bottlenecks, drift and differentiation: the population structure and demographic history of sika deer (Cervus nippon) in the Japanese archipelago. Mol Ecol 10:1357–1370
Goudet J (1995) Fstat (Version 1.2): a computer program to calculate F-statistics. J Heredity 86:485–486
Grande C, Araujo R, Ramos MA (2001) The gonads of Margaritifera auricularia (Spengler, 1793) and M. margaritifera (Linnaeus, 1758) (Bivalvia: Unionoidea). J Mollus Stud 67:27–35
Gunderson DR (1980) Using r-K selection theory to predict natural mortality. Can J Fish Aquat Sci 37:2266–2271
Guo Q, Taper M, Schoenberger M, Brandle J (2005) Spatial-temporal population dynamics across species range: from centre to margin. Oikos 108:47–57
Hansen MM, Nielsen EE, Ruzzante DE, Bouza C, Mensberg KLD (2000) Genetic monitoring of supportive breeding in brown trout (Salmo trutta L.), using microsatellite DNA markers. Can J Fish Aquat Sci 57:2130–2139
Harley EH (2001) AGARst. A program for calculating allele frequencies, GST and RST from microsatellite data, version 2.0. University of Cape Town, Cape Town
Hedgecock D (1994) Does variance in reproductive success limit effective population size of marine organisms? In: Beaumont A (ed) Genetics and evolution of aquatic organisms. Chapman and Hall, London, pp 122–134
Hedrick PW (2005) Large variance in reproductive success and the Ne/N ratio. Evolution 59:1596–1599
IUCN (2004) The IUCN red list categories and criteria. http://www.iucn.org/themes/ssc/red_list_2004/GSAexecsumm_EN.htm
Lesica P, Allendorf FW (1995) When are peripheral populations valuable for conservation. Conserv Biol 9:753–760
Luikart G, Sherwin W, Steele BM, Allendorf FW (1998) Usefulness of molecular markers for detecting population bottlenecks via monitoring genetic change. Mol Ecol 7:963–974
Lydeard C, Minton RL, Williams JD (2000) Prodigious polyphyly in imperilled freshwater pearly-mussels (Bivalvia: Unionidae): a phylogenetic test o species and generic designations. In: Harper EM, Taylor JD, Crame JA (eds) Evolutionary biology of the Bivalvia. The Geological Society, Geological Society Special Publication, No. 177, London, pp 145–168
Machordom A, Araujo R, Erpenbeck D, Ramos MA (2003) Phylogeography and conservation genetics of endangered European Margaritiferidae (Bivalvia: Unionoidea). Biol J Linn Soc 78:235–252
Morán P, Pérez J, García Vazquez E (2005). Genetic variation in endangered populations of Atlantic salmon (Salmo salar L.) from North-western Spain. J Fish Biol 67:207–213
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Pedersen A, Loeschcke V (2001) Conservation genetics of peripheral populations of the mygalomorph spider Atypus affinis (Atypidae) in northern Europe. Mol Ecol 10:1133–1142
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK. A computer program for detecting recent reductions in the effective population size using allele frequency data. J Heredity 90:502–503
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004) GeneClass2: a software for genetic assignment and first generation migrants detection. J Heredity 95:536–539
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49:1280–1283
Reis J (2003) The freshwater pearl mussel [Margaritifera margaritifera (L.)] (Bivalvia, Unionoida) rediscovered in Portugal and threats to its survival. Biol Conserv 114:447–452
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Roe KJ, Hoeh WR (2003) Systematics of freshwater mussels (Bivalvia: Unionoida). In: Lydeard C, Lindberg DR (eds) Molecular systematics and phylogeography of Mollusks. Smithsonian series in comparative evolutionary biology. Smithsonian Books, Washington DC, pp 91–122
Roff DA (1984) The evolution of life histories parameters in teleosts. Can J Fish Aquat Sci 41:989–1000
Roff DA (1992) The evolution of life histories: theory and analysis. Chapman and Hall, New York
Roff DA (2001) Life history evolution. Sinauer Associates, Massachusetts
Rohlf FJ (1992) NTSYS-PC. Numerical taxonomy and multivariate analysis system. Exeter Software, New York
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
San Miguel E, Monserrat S, Fernández C, Amaro R, Hermida M, Ondina P, Altaba CR (2004) Growth models and longevity of freshwater pearl mussels (Margaritifera margaritifera) in Spain. Can J Zool 82:1370–1379
Schwaegerle KE, Garbut K, Bazzaz FA (1986) Differentiation among nine populations of Phlox. I. Electrophoretic and quantitative variation. Evolution 40:506–517
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47:264–279
Solbrig OT, Rollins RC (1977) The evolution of autogamy in Leavenworthia. Evolution 31:265–281
van Oosterhout C, Hutchinson WF, Wills DPM et al (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13:921–935
Velasco JC, Araujo R, Bueno R, Laguna A (2002) Discovered the southernmost known European of the freshwater pearl mussel Margaritifera margaritifera, L. (Bivalbia, Unionida) in the Iberian Peninsula (Agueda River, Salamanca). Iberus 20:94–108
Vucetich JA, Waite TA (2003) Spatial patterns of demography and genetic processes across the species range: null hypothesis for landscape conservation genetics. Conserv Genet 4:639–645
Weir BS, Cockerham C (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Winnepennincks B, Backeljau T, De Wachter R (1993) Extraction of high molecular weight DNA from molluscs. Trends Genet 9:407
Young MR, Cosgrove PJ, Hastie LC (2001) The extent of, and causes for, the decline of a highly threatened naiad: Margaritifera margaritifera. In: Bauer G, Wächtler K (eds) Ecology and evolution of the freshwater mussels Unionida. Springer-Verlag, Berlin, pp 337–358
Ziuganov V, Zotin A, Nezlin L, Tretiakov V (1994) The freshwater pearl mussels and their relationships with salmonid fish. VNIRO Publishing House, Moscow
Ziuganov V, Beletsky VV, Neves RJ, Tretiakov VA, Mikhno IV, Kaliuzhin SM (1998) The recreational fishery for Atlantic salmon and the ecology of salmon and pearl mussels in the Varzuga River, northwest Russia. Virginia Polytechnical Institute and State University, Blacksburg
Ziuganov V, San Miguel E, Neves RJ, Longa A, Fernández C, Amaro R, Beletsky V, Popkovitch E, Kaliuzhin S, Johnson T (2000) Life span variation of the freshwater pearlshell: a model species for testing longevity mechanisms in animals. Ambio 29:102–105
Acknowledgements
This study was supported by funds from the Xunta de Galicia (Project No. PGIDIT02RFO26101PR). We would like to thank to J. Geist for kindly supplying the ZI sample and individual genotypes as reference for microsatellite analysis, but also for discussion on freshwater pearl mussel molecular ecology. We are also very grateful to R. Vilas for the interesting comments and criticisms on the manuscript. Valuable technical assistance was provided by S. Sánchez. We are grateful to all the forest guards and the biologist J. Latas (Consellería de Medio Ambiente, Xunta de Galicia) for assisting with locating sampling sites.
Author information
Authors and Affiliations
Corresponding author
Appendix A
Appendix A
Allele frequencies at eight polymorphic microsatellites in NW Iberian freshwater pearl mussel populations. MarMa4315 and MarMa4322 loci were monomorphic across studied populations.
Locus | Allele | EO | MD | TA | AG | MR | TR | Miño | SA |
|---|---|---|---|---|---|---|---|---|---|
MarMa1632 | 40 | 25 | 23 | 21 | 28 | 20 | 24 | 23 | |
164 | 1 | 1 | 1 | 0 | 1 | 0.9 | 0.95 | 1 | |
166 | 0 | 0 | 0 | 1 | 0 | 0.1 | 0.05 | 0 | |
MarMa3050 | 40 | 25 | 25 | 21 | 29 | 20 | 24.5 | 24 | |
85 | 0 | 0 | 0.06 | 0 | 0 | 0 | 0 | 0.042 | |
87 | 0.688 | 1 | 0.92 | 1 | 1 | 0.975 | 0.988 | 0.104 | |
89 | 0.313 | 0 | 0.02 | 0 | 0 | 0.025 | 0.013 | 0.854 | |
MarMa3621 | 40 | 25 | 23 | 20 | 26 | 20 | 23 | 23 | |
187 | 0.013 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
189 | 0.813 | 1 | 0.217 | 1 | 0.788 | 0.75 | 1 | 1 | |
191 | 0.175 | 0 | 0.391 | 0 | 0.192 | 0.2 | 0 | 0 | |
193 | 0 | 0 | 0.391 | 0 | 0 | 0 | 0 | 0 | |
195 | 0 | 0 | 0 | 0 | 0.019 | 0.05 | 0.035 | 0 | |
MarMa4143 | 39 | 22 | 19 | 21 | 18 | 19 | 18.5 | 23 | |
194 | 0 | 0 | 0 | 0 | 0 | 0.053 | 0.027 | 0 | |
198 | 0 | 0 | 0 | 0.024 | 0 | 0 | 0 | 0 | |
204 | 0.846 | 1 | 0.947 | 0.976 | 0.528 | 0.947 | 0.738 | 1 | |
206 | 0 | 0 | 0.053 | 0 | 0.472 | 0 | 0.236 | 0 | |
208 | 0.141 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
216 | 0.013 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
MarMa4277 | 39 | 24 | 21 | 21 | 25 | 20 | 22.5 | 22 | |
172 | 0 | 0 | 0 | 0 | 0.02 | 0 | 0.01 | 0 | |
178 | 0 | 0 | 0.071 | 0 | 0 | 0 | 0 | 0 | |
180 | 0.308 | 0.188 | 0.476 | 1 | 0.82 | 0.525 | 0.673 | 0.636 | |
182 | 0 | 0.042 | 0.381 | 0 | 0 | 0.45 | 0.225 | 0.091 | |
184 | 0.679 | 0 | 0 | 0 | 0 | 0 | 0 | 0.273 | |
186 | 0.013 | 0 | 0 | 0 | 0.04 | 0.025 | 0.033 | 0 | |
188 | 0 | 0.771 | 0.071 | 0 | 0.06 | 0 | 0.03 | 0 | |
190 | 0 | 0 | 0 | 0 | 0.06 | 0 | 0.03 | 0 | |
MarMa4726 | 40 | 25 | 22 | 21 | 29 | 20 | 24.5 | 23 | |
158 | 0 | 0 | 0 | 0 | 0.017 | 0.25 | 0.134 | 0 | |
160 | 1 | 1 | 1 | 1 | 0.983 | 0.75 | 0.867 | 1 | |
MarMa4859 | 38 | 24 | 22 | 20 | 23 | 19 | 21 | 20 | |
164 | 0.013 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
170 | 0.013 | 0 | 0 | 0.025 | 0 | 0 | 0 | 0 | |
172 | 0.079 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
174 | 0.224 | 0 | 0.295 | 0 | 0 | 0.053 | 0.027 | 0 | |
176 | 0.079 | 0.271 | 0.341 | 0 | 0 | 0.132 | 0.066 | 0.05 | |
178 | 0.039 | 0.667 | 0.114 | 0 | 0 | 0 | 0 | 0.075 | |
180 | 0 | 0.063 | 0.159 | 0.975 | 0.457 | 0.237 | 0.347 | 0 | |
182 | 0 | 0 | 0.091 | 0 | 0.043 | 0.026 | 0.035 | 0.1 | |
184 | 0.25 | 0 | 0 | 0 | 0 | 0.211 | 0.106 | 0.775 | |
186 | 0.132 | 0 | 0 | 0 | 0.413 | 0.342 | 0.378 | 0 | |
188 | 0.171 | 0 | 0 | 0 | 0.087 | 0 | 0.044 | 0 | |
MarMa5167 | 40 | 17 | 21 | 21 | 24 | 18 | 21 | 20 | |
140 | 1 | 0.029 | 0.929 | 1 | 0.646 | 0.194 | 0.42 | 0 | |
142 | 0 | 0.971 | 0.048 | 0 | 0 | 0 | 0 | 1.000 | |
144 | 0 | 0 | 0.024 | 0 | 0.354 | 0.806 | 0.58 | 0 |
Rights and permissions
About this article
Cite this article
Bouza, C., Castro, J., Martínez, P. et al. Threatened freshwater pearl mussel Margaritifera margaritifera L. in NW Spain: low and very structured genetic variation in southern peripheral populations assessed using microsatellite markers. Conserv Genet 8, 937–948 (2007). https://doi.org/10.1007/s10592-006-9248-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-006-9248-0