
Abstract
Foskett Spring is a small isolated desert spring in the Warner Basin of Oregon containing an undescribed subspecies of speckled dace protected under the US Endangered Species Act. Uncertainty regarding the taxonomic status of Foskett dace has raised questions about their evolutionary relationship to other more abundant populations in the Warner Basin. To address these questions, we sequenced 1,982 base pairs (bp) of the ND2 and cyt-b genes of mitochondrial DNA (mtDNA) for 85 fish from Foskett Spring, the surrounding Warner Basin, and the adjacent Goose Lake Basin. We observed 58 unique mtDNA haplotypes defined by 96 bp substitutions in both genes. MtDNA sequences were highly divergent and reciprocally monophyletic between dace from the Warner and Goose Lake Basins, with sequence divergences (5 and 3% at ND2 and cyt-b, respectively) in the range usually observed between fish species. In contrast, mean sequence divergence between Foskett dace and other Warner Basin populations was less than 1%, with no evidence of reciprocal monophyly among mtDNA lineages. These results indicate that Foskett dace and other populations in the Warner Basin are approximately equally diverged from one another evolutionarily, suggesting similar times of divergence since the late Pleistocene. We believe further studies are needed to determine if rapid evolution of novel traits have occurred in dace inhabiting the unique ecological setting of Foskett Spring during the past 10,000 years.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Speckled dace (Rhinichthys osculus spp.) are the most broadly distributed native fish in the western United States (Lee et al. 1980). They exhibit extensive morphological diversity and occupy the widest variety of habitats of any fish within their range (Moyle 2002). The evolutionary history of this species has been heavily influenced by climatic and geologic events. These events have resulted in complex patterns of isolation and connection among populations over the past 5 million years (Minckley et al. 1986, Smith et al. 2002).
The variable nature of the species combined with the allopatric distribution of unique morphological types has made speckled dace taxonomy very difficult. Traditional taxonomy, based on analysis of phenotypic variation, has not revealed informative characters needed to assess evolutionary relationships within the species. A lack of informative taxonomic characters has resulted in grouping all forms into a single highly variable species (Hubbs et al. 1974). This taxonomic approach has likely resulted in a polyphyletic grouping of multiple taxa which cannot be resolved because of complications associated with parallel evolution of phenotypic characters (Bernatchez 1995, Moyle 2002).
Two recent molecular systematic studies, based on mtDNA, have provided new insights into the evolutionary history of speckled dace (Oakey et al. 2004; Pfrender et al. 2004). Both studies found speckled dace are characterized by deep genetic divergences among major river basins and high levels of genetic diversity within basins. Highly divergent specked dace lineages are likely the result of geologic processes that have isolated populations within major river basins for the past 5 million years (Smith et al. 2002; Pfrender et al. 2004; Oakey et al. 2004). Historically large effective population size, associated with phenotypic plasticity and high reproductive potential, have allowed large amounts of genetic variation to be maintained within these lineages. These two studies suggest examination of geographic and phylogenetic relationships among mtDNA haplotypes can help resolve evolutionary relationships among various forms of speckled dace.
An undescribed subspecies of speckled dace inhabiting Foskett Spring in southeast Oregon is protected under the ESA (USFWS 1985). Foskett Spring forms a 33 m2 pool that is the source for a 70 m long brook which feeds a series of shallow marshes that disappear into the normally dry Coleman Lake. The spring is located along the western margin of Coleman Lake, a hydrologically separated sub-basin approximately 10 km south of the Warner Basin (Williams et al. 1990). Coleman Lake became isolated hydrologically from the Warner Basin at the end of the Pleistocene when reduced precipitation isolated many lakes and basins in southeast Oregon.
Substantial progress has been made toward the recovery of the threatened Foskett speckled dace (R. osculus ssp.). In 1987, the Bureau of Land Management acquired the spring and the surrounding 65 ha of land, thereby allowing for 28 ha around the spring to be fenced to exclude cattle (Williams et al. 1990). An adult census conducted in 2005 documented 3,147 Foskett speckled dace with half of the population residing in the spring pool (Scheerer and Jacobs 2006). Despite these successful conservation efforts and large population size, Foskett speckled dace remain at risk because their restricted distribution makes them highly vulnerable to catastrophic losses caused by threats such as introduction of nonnative species or climate change (USFWS 1998; Williams et al. 2005).
There are unresolved questions regarding the evolutionary significance of Foskett speckled dace. Speckled dace are the only fish native to the Coleman sub-basin (i.e., Foskett Spring) and biologists have pondered whether their presence may have resulted from anthropogenic transplants, either during Euro-American settlement or previously by Native Americans. From an evolutionarily perspective, biologists have wondered about the significance of Foskett speckled dace relative to the more abundant populations in the Warner Basin.
Our current understanding of the evolutionary relationship of Foskett speckled dace with other Warner Basin populations is based on phenotypic characters. Foskett speckled dace were found to differ slightly from those found in the rest of the Warner Basin, with fish exhibiting a shorter lateral line, larger eye, and a dorsal fin position well behind the pelvic fin but before the beginning of the anal fin (Dr. Carl Bond, Oregon State University, pers. comm., 1990; cited in USFWS 1998). These characters have not been described formally and the degree to which these defining morphological characters are genetically or environmentally influenced is unknown. Foskett speckled dace is thus considered to be an undescribed subspecies until these uncertainties regarding their taxonomic status are resolved.
The overarching goal of this study was to use molecular systematics to gain insights into the origin and evolutionary significance of the Foskett speckled dace. To accomplish this, we compared patterns of molecular variation at two mtDNA genes observed in Foskett specked dace to those of speckled dace inhabiting the Warner and nearby Goose Lake Basins. Finally, to better understand the broader biogeographic structuring and evolutionary relationships of the groups examined in this study, we combined our results with a previous study that examined dace in five other major Oregon drainage systems (Pfrender et al. 2004).
Methods
Population samples
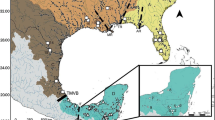
Speckled dace were sampled from Foskett Spring and three streams in the Warner Basin and two streams in the Goose Lake Basin (Fig. 1; Appendix Tables 3, 4). We assumed that each stream and Foskett Spring represented an independent population of dace. Adult fish were captured via electrofishing, minnow traps, and trap nets by personnel of the USFWS and Bureau of Land Management during the summers of 2003 and 2004 at slightly different locations, which are identified in Fig. 1. A small, non-lethal caudal fin clip was collected from each fish and stored in 100% non-denatured ethanol for future DNA analysis.

aMap of study area showing Goose Lake Basin, Warner Basin and the 13 populations examined in Pfrender et al. (2004), the two letter population codes are defined in Table 1. b Location of the six populations from the Goose Lake and Warner Basins examined in this study. Sites indicated by the star were sampled in 2003 and those identified by a circle were sampled in 2004
Data collection
Genomic DNA was extracted from fin tissue of each fish using the Qiagen DNeasy tissue kit following standard procedures (Qiagen, Valencia, CA). The polymerase chain reaction (PCR) was used to amplify the NADH subunit 2 (ND2) and cytochrome b (cyt b) mitochondrial genes for sequence analysis. Primers HA and LA (Dowling et al. 2002) were used to amplify the cyt b gene and primers ILE and ASN (Dr. Thomas Dowling, Arizona State University, pers. comm.) were used to amplify the ND2 gene. To ensure quality DNA sequence data, we designed primers to PCR amplify an internal section of each gene to reduce the size of the sequencing fragments to 650 bp or less; HA-int and LA-int were used for cyt b and ILE-int and ASN-int were used for ND2 (Fig. 2). All PCR reactions were performed in 25 μL reactions. Cyt b reactions contained 3.5 μL extracted DNA, 1× PCR buffer, 1.5 mM MgCl2, 0.2 mM each of dNTP, 0.34 μM forward and reverse primers, and 0.85 units Taq DNA polymerase (Promega, Madison, WI). ND2 reactions contained 3.5 μL extracted DNA, 1× PCR buffer, 2.0 mM MgCl2, 0.2 mM each of dNTP, 0.34 μM each primer, and 0.63 units Taq DNA polymerase. We used a standard PCR profile for both genes of: 94°C for 3 min. followed by 38 cycles of 94°C for 45 s, 50°C for 30 s, 72°C for 45 s., and finishing with a hold of 72 for 8 min. The exceptions were 54°C annealing temperatures for the ASN-ILE primer set and a 56°C annealing temperature for both internal primer sets.

Primers used for amplification and sequencing of the speckled dace cytochrome b and NADH subunit two genes. Primers used for sequencing and primer pairs used for amplification are listed in the text
The PCR products were sent to Nevada Genomics Center (Reno, NV) for purification and sequencing using an ABI 3730 genetic analyzer and Dye Terminator Cycle Sequencing. Complete sequence of the cyt b sequence was obtained using HA, LA and LA-int as sequencing primers, while the ILE, ASN, and ASN-int primers were used to obtain complete sequence data for the ND2 gene. Figure 2 provides a schematic of the two genes and location of primers used for PCR amplification and DNA sequencing.
Data analysis
Sequence statistics
Sequence contigs for cyt b and ND2 genes were generated, collated, aligned, and edited with Sequencher v 3.0 (Gene Codes Corp.). We used DNASP v 4.10.9 (Rozas et al. 2003) and the methods of Nei (1987) to estimate nucleotide diversity (π), nucleotide diversity between samples (d XY), genetic distance between samples (d A), and haplotype diversity (H d). Estimates of d A were calculated as the mean percent sequence differences after correcting for within sample nucleotide diversities. To test for departures from neutral molecular evolution we used the methods of Tajima (1989) to estimate his D statistic for each population. All estimates of Tajima’s D and associated significance values were calculated using Arlequin v 3.11 (Excoffier et al. 2005).
Variation within and among dace populations in the Warner and Goose Lake Basins
To test for genetic subdivision of sequence variation at the ND2 gene among populations in the Warner and Goose Lake Basins we used Arlequin v 3.11 (Excoffier et al. 2005) to conduct an analysis of molecular variance (AMOVA; Excoffier et al. 1992), with populations grouped by basin. The AMOVA allowed us to evaluate variance estimates between basins, among populations within a basin, and within populations. The input matrix for the AMOVA was obtained by calculating the number of differences between haplotypes in pairwise comparisons. Significance of the variance estimates was obtained using a randomization procedure (20,000 permutations).
We also used Arlequin v 3.11 to estimate Φ statistics for sequence variation at the ND2 gene. The Φ statistics are a haplotype correlation measure that are analogous to F statistics (Excoffier et al. 1992). The three Φ statistics are defined as follows: ΦCT is the correlation of random haplotypes within a basin relative to that of random haplotypes drawn from all populations; ΦSC is the correlation of random haplotypes within populations relative to that of random haplotypes drawn from the basin to which that population belongs; and ΦST is the correlation of random haplotypes within populations relative to that of random haplotypes drawn from the whole dataset. The Φ statistics are close to 0 with little genetic subdivision among the groups and close to 1 when there is a high degree of genetic subdivision among the groups. Input matrix for estimation of Φ statistics and randomization procedures used to determine statistical significance were identical to those for estimating variance components for the AMOVA.
Statistical parsimony networks among haplotypes for both genes were computed with the program TCS 1.13 (Clement et al. 2000). These intraspecific gene genealogies were generated using the methods of Templeton et al. (1992). We plotted the parsimony networks accounting for the number of times a haplotype was observed and the locality from which it was sampled.
We examined the distribution of pairwise differences between pairs of sequences (i.e., mismatch distribution) using the methods of Schneider and Excoffier (1999) to better understand the historical demography of populations in the Warner Basin. Rogers and Harpending (1992) showed demographic expansions leave distinctive mismatch distributions in populations. Populations that have undergone a recent demographic expansion are expected to have a unimodal Poisson distribution while populations that have undergone recent bottlenecks or been at equilibrium are expected to have multimodal patterns (Johnson et al. 2007). Harpending (1994) developed the raggedness index (r) to quantify the degree to which a population displayed a unimodal (smooth) or multimodal (ragged) mismatch distribution. Growing populations are expected to have small r values and populations that have been at equilibrium of have experienced a recent bottleneck are expected to have higher r-values. We calculated r-values for observed mismatch distributions at ND2 in the four Warner Basin populations using the program ARLEQUIN v 3.11. We also used this program to generate the expected mismatch distribution for a population that has undergone a demographic expansion. The probability that each population had undergone a sudden demographic expansion was estimated by generating 1,000 bootstrap replicates of the observed mismatch distribution for each population assuming a model of sudden demographic expansion. The proportion of bootstrap replicates that produced a larger r-value than the observed r-value was used to calculate the P-value for the null hypothesis that the population had undergone a sudden demographic expansion.
Fu’s (1997) F s and Ramos-Onsins and Rozas’s (2002) R 2 were used as two additional independent assessments of historical changes in population size. An excess of rare haplotypes and reduction of common haplotypes is a signature of demographic expansion and gives rise to a large negative F s values (Fu 1997). The R 2 statistic is based on comparing the number of singleton mutations with the average number of nucleotide differences observed (Ramos-Onsins and Rozas 2002). Low values of R 2 indicate recent population growth. Evidence for demographic expansion was determined in ARLEQUIN v3.11 for F S and DNASP v 4.10.9 for R 2 by comparing observed values for both parameters with the distribution of F s and R 2 values generated from 1,000 random permutations for each population assuming demographic equilibrium. The P-value of R 2 and F S statistics were obtained as the proportion of permutated statistics less or equal to observed values for each statistic.
Phylogenetic analysis of speckled dace in Oregon
We compared cyt b sequences from Foskett Spring, Warner Basin, and Goose Lake Basin with the five Oregon river basins examined in Pfrender et al. (2004) (Fig. 1). Relationships among unique cyt b haplotypes were inferred by constructing phylogenetic trees using maximum-parsimony (Swofford et al. 1996) and the program Mega v 2.1 (Kumar et al. 2001). The maximum parsimony analysis consisted of a heristic search on unweighted, parsimony-informative characters using the Close-Neighbor-Interchange (CNI) algorithm with a search level of 3. The initial trees for the CNI search were generated through construction of random addition trees with ten replicates (Nei and Kumar 2000). We tested the reliability of the consensus tree using the methods of Felsenstein (1985) with 1,000 bootstrap replicates, this method provides a relative support for the internal branching patterns with values above 50% considered informative and values above 95% generally considered “correct.”
Phylogenetic relationships among unique cyt b haplotypes were also examined using Bayesian methods implemented in the program MrBayes (Huelsenbeck and Ronquist 2001). We selected the best-fit model of evolution for the Bayesian analysis using the hierachial log-likelihood ratio test in the program Modeltest version 3.06 (Posada and Crandell 1998). Runs in MrBayes consisted of four Monte Carlo Markov chains that were 1,000,000 generations long with sampling every 100 generations after a burn in of 80,000 generations. To estimate Bayesian posterior probabilities or Bayesian support values for a given clade we constructed a consensus tree from the last 5,000 trees generated by MrBayes using the strict Majority rule options of PAUP (Swofford 1999). If 95% or more of the sampled trees contained a given clade, we considered it to be significantly supported by our data.
Divergence time between populations and basins
Estimates of divergence times between basins based on cyt b sequence data were gained using the methods of Hedrick et al. (2006) as implemented by Phyltest v 2.0 (Kumar 1996). We used sequence data from blacknose dace (Rhinichthys atratus) as an outgroup for the relative rate analysis when estimating divergence times. These divergence times are most likely over estimates of the true divergence times because they are based on the time to the most common ancestor and the isolation of the populations likely occurred after the origin of the haplotype clades (Pfrender et al. 2004).
Results
Gene diversity
We observed 18 cyt b haplotypes from 43 speckled dace representing six populations from the Warner and Goose Lake Basins. A total of 40 unique ND2 haplotypes were observed in 85 dace from the same six populations. The number of individuals sampled per population ranged from 5 to 8 and 8 to 16 for cyt b and ND2, respectively. Geographical distribution of haplotypes is shown in Appendix Table 3 for ND2 and Appendix Table 4 for cyt b. All unique haplotypes were deposited in GenBank (EU158191–EU158248). The trimmed portions for each gene were 670 bases for cyt b and 1,312 bases for ND2. A total of 107 variable sites were observed for ND2 in the Warner and Goose Lake Basins and 140 variable sites were observed for cyt b when the 18 unique haplotypes observed in the Warner and Goose Lake Basins were combined with the 44 unique haplotypes from Pfrender et al. (2004). A deletion was observed at a non-protein coding portion of ND2 in a single individual from Honey Creek (haplolotype EU158212). The cyt b haplotypes contained no insertions or deletions. Our analysis of Pfrender et al. (2004) results and associated sequences deposited in GenBank revealed a haplotype from the Klamath River Basin that was included in their analysis but was not deposited in GenBank. Pfrender shared the sequence of this haplotype (JK0001) with us and we have included it in our analysis (Dr. Michael Pfrender, Utah State University, pers. comm.).
Variation within populations
Multiple ND2 and cyt b haplotypes were observed in all six populations (Appendix Tables 3, 4) from the Warner and Goose Lake Basins. Mean π at ND2 ranged from 0.42% in Foskett Spring to 0.09% in Twelvemile Creek (Table 2). Thomas Creek had the highest H d estimate of 0.88 while Twelvemile Creek had the lowest H d estimate of 0.59 (Table 2). Our results from cyt b revealed the Warner Basin had the lowest nucleotide diversity (0.23%) which was an order of magnitude lower than the estimate of 2.33% observed in the Klamath River Basin (Table 1). The Klamath River Basin also had the highest H d of 0.95.
Tajima’s D was not significant for any of the population samples (P > 0.05) indicating data for both genes do not depart from expectations for neutral variation or an infinite alleles model of evolution. Observed mismatch distributions at ND2 were unimodal in Deep Creek, Honey Creek, and Twelvemile Creek suggesting these populations have recently undergone sudden demographic expansions. These three populations all had negative F S values indicating evidence for demographic expansion and estimates for R 2 confirmed a genetic signal for sudden demographic expansions in Deep Creek (P < 0.05; R 2 = 0.097) and Honey Creek (P < 0.05; R 2 = 0.098) (Fig. 6). In contrast, the Foskett Spring population exhibited a multimodal mismatch distribution with a positive F S value, a nonsignificant R 2 estimate, and a r value that was significantly different from the mismatch distribution expected for a population that had recently undergone a demographic expansion (P < 0.05; r = 0.261; Fig. 6). These results indicate Foskett dace have been in demographic equilibrium or have experienced a recent population bottleneck.
Geographic variation
Average sequence divergences for ND2 haplotypes observed between populations from the Warner and Goose Lake Basins ranged from 4.9 to 5.0 %; while d XY estimates ranged from 0.24 to 0.54 % between populations within each basin (Table 2). Estimates of d XY for cyt b revealed a similar pattern with divergences between basins ranging from 3.0 to 3.6% while divergences between populations within each basin ranged from 0.18–0.41%. The Foskett Spring population was the most divergent population within the Warner Basin with d A estimates ranging from 0.08 to 0.23% based on the ND2 data (Table 2). All populations within the Warner Basin appeared to be equally diverged based on the d XY and d A estimates of cyt b.
The AMOVA revealed strong and significant genetic differentiation between the two basins. Differences between the Warner and Goose Lake Basins accounted for 93.4% of the total variation (P < 0.0001) which corresponds to a ΦCT estimate of 0.93 (95% CI 0.90–0.96). A small but significant portion of the total variance was accounted for within populations (4.3%; P < 0.001) with a ΦST estimate of 0.96 (95% CI 0.94–0.97). Differences among populations within basins accounted for just 2.3% of the total variation (P < 0.001) with a ΦSC of 0.35 (95% CI 0.25–0.41).
Two divergent haplogroups were observed at both ND2 and cyt b genes, with fixed differences at 47 of 1,312 sites and 18 of 670 sites, respectively (Fig. 3). We designated these haplogroups ND2-W, ND2-G, cyt b-W, and cyt b-G. The ND2-W and cty b-W haplotypes were found exclusively in the Warner Basin, while the ND2-G and cyt b-G haplotypes were only observed in the Goose Lake Basin. In contrast to the reciprocally monophyletic relationships observed between speckled dace from the Warner and Goose Lake Basins, no evidence of reciprocal monophyly was observed among populations within a basin (Fig. 3).

Statistical parsimony networks for 85 ND2 and 18 cyt b haplotypes observed in Warner and Goose Lake Basins of Oregon. Each connection is a single mutational step with black circles representing inferred haplotypes. Color codes represent populations where the haplotypes were sampled and the size of the circles are proportional to the number of individuals with that haplotype. Numbers inside the circles indicate the number of individuals observed with a particular haplotype with blank circles indicating a single individual was observed. No haplotypes were shared among populations in the ND2 network. Note the strong geographic structure between haplotypes in the Warner and Goose Lake Basins with 62 and 18 inferred haplotypes separating the haplogroups observed within each basin for ND2 and cyt b, respectively
Phylogenetic analysis of speckled dace in Oregon
The MP consensus tree (length = 298; CI = 0.644; RI = 0.893) revealed a strong phylogenetic signal in the speckled dace cyt b data. We observed a highly structured phylogeny with monophyletic clades of haplotypes recovered for all major river basins except within the Klamath River and Columbia River Basins. No haplotype was found to occur in more than one major river drainage system. The Warner and Goose Lake Basin haplogroups formed sister clades with the Klamath (A) haplogroup of Pfrender et al. (2004), and these haplogroups were nested within a clade that included the Lower Columbia and Snake River haplogroups (Fig. 4).

Unrooted and unweighted maximum parsimony tree of 62 unique cytochrome b haplotypes observed in speckled dace sampled from seven major basins in Oregon. Localities and number of occurrences for each haplotype are shown in Appendix Table 4. This single most parsimonious tree consists of 298 steps, a consistency index of 0.644, and a retention index of 0.893. Support for the genetic structure was assessed with 1,000 pseudoreplicates of the data and the resulting bootstrap proportions are shown above the branches where the values exceeded 50%. Haplotypes are identified GenBank Accession numbers. Major river basins are shown to the right of the tree
Based on the results of the log-likelihood ratio test we selected the Tamura–Nei equal transversion frequencies model with a gamma distribution describing the rate of variation among sites (TrNef + G). This TrNef + G model assumes a six-step rate matrix ([A–C] = 1.00; [A–G] = 23.72; [A–T] = 1.0; [C–G] = 1.0; [C–T] = 9.24; [G–T] = 1.0), no invariant sites and a gamma distribution shape parameter = 0.2079. Log-likelihood values for sampled trees in MrBayes stabilized after approximately 80,000 generations. Levels of support for internal branches of the Bayesian consensus tree (Fig. 5) were much higher than observed under maximum parsimony analysis (Fig. 4).

Consensus tree from Bayesian analysis of 62 unique cytochrome b haplotypes observed in speckled dace sampled from seven major basins in Oregon. Localities and number of occurrences for each haplotype are shown in Appendix Table 4. Values above the branches represent percentage Bayesian posterior probabilities or Bayesian support values where the values exceeded 50%. Haplotypes are identified GenBank Accession numbers. Major river basins are shown to the right of the tree
The topology of the phylogeny obtained with the Bayesian analysis was largely congruent with the maximum parsimony analysis (Figs. 4, 5). Both analyses identified the Middle Columbia River Basin as the basal clade. Deeper relationships among major river basins were best defined in the Bayesian consensus tree. Deeper clades supported by the Bayesian analysis include a Goose Lake–Klamath (A)–Warner clade. This Goose Lake–Klamath (A)–Warner clade was nested in a larger clade that included the Lower Columbia and Snake River haplogroups (Fig. 5). The position of the Klamath (B) and Coastal haplogroups were not well supported while the position of the Middle Columbia River as the basal clade was well supported (Fig. 5).
Divergence time between basins and populations
Using a cyt b sequence from blacknose dace as a outgroup, rate consistency was not rejected using the relative rate test as implemented in PYLYTEST. Therefore assuming a mutation rate of 5 × 10−9 (Stepine et al. 1997), the estimated divergence time between Warner and Goose Lake Basin dace was 3.55 million years with a 95% confidence interval of 2.83–4.27 million years.
Discussion
Molecular systematic results presented here and those of Pfrender et al. (2004) and Oakey et al. (2004) suggest the evolutionary history of speckled dace is well ordered geographically with high levels of genetic subdivision among major river basins. These findings suggest geographical distribution of speckled dace mtDNA haplotypes is a powerful method for understanding the evolutionary history of this species. We use the results of our genetic analysis to provide insights into the evolutionary significance of the Foskett Spring speckled dace population.
Genetic patterns among major river basins
Our results suggest speckled dace from the Warner Basin and Goose Lake Basin have experienced a long period of evolutionary isolation. Sequence divergence at cyt b dated the divergence time of dace from these two basins at 2.83–4.27 million years ago. A divergence time in this range is consistent with the uplift of the Warner Basin Mountains which likely occurred during this same time frame (Minckley et al. 1986). This deep biogeographical split was also evident in the AMOVA results which indicated differences between basins explained 93% the total variation among ND2 haplotypes. This magnitude of genetic distance between speckled dace from the Warner and Goose Lake Basins is consistent with patterns usually observed between fish species (Smith et al. 2002).
Haplogroups observed for both genes provide additional evidence that speckled dace from the Warner and Goose Lake Basins have been evolving independently for a long period of time. All sequences from Warner Basin dace share a more recent common ancestor with each other than with sequences from Goose Lake dace and all sequences from Goose Lake dace share a more common ancestor with each other than with Warner Basin dace, a pattern termed reciprocal monophyly. The rate at which two groups become monophyletic at mitochondrial loci depends on the number of generations they have been reproductively isolated and the inbreeding size of the female population (N ef); with 2 N ef generations needed on average to reach monophyly (Avise 2000). Although the N ef for speckled dace in each basin is unknown we know it is likely less than 700,000 (assuming a 2-year generation interval and 2.8 million years of reproductive isolation) because dace from the two basins exhibit reciprocal monophyly.
We observed high haplotype diversity and low nucleotide diversity at the cyt b gene in the Warner and Goose Lake Basins (Table 1). This pattern of many haplotypes that are closely related to one another is characteristic of populations that have been isolated for long periods of time (McCusker et al. 2000). Both Warner and Goose Lake are endorheic basins and our data suggest speckled dace populations have been isolated within these basins over the past 2–4 million years. In contrast, the high haplotype and nucleotide diversity observed within the Klamath Basin indicates this basin is a zone of contact for dispersing haplotypes from different refugia (Fry and Zink 1998).
When the cyt b haplotypes observed in the Warner and Goose Lake populations were added to the phylogeny constructed by Pfrender et al. (2004) we observed reciprocal monophyly among major tributaries and small coastal streams but not at larger geographic scales such as the Klamath and Columbia River Basins (Figs. 3, 4, 5). This pattern suggests simple vicariance operating frequently with more complex patterns of dispersal causing short internodes for the basal branches of the phylogeny (Figs. 4, 5). Pfrender et al. (2004) speculated that the inability to resolve relationships at the interbasin level was caused by a lack of information in the DNA sequence data or by a rapid radiation of the species during the Miocene to early Pliocene.
Two distinct lineages of speckled dace observed in the Columbia River, Lower and Middle Columbia (Figs. 4, 5), is consistent with other species of fish from these locations. The Lower and Middle Columbia are divided by the Cascade Mountains and studies with bull trout and rainbow trout have documented a similar phylogeographic pattern which has been attributed to postglacial colonization of the western cascades by a coastal refugium and the eastern Cascades by an interior refugium approximately 10,000 years ago (Taylor et al. 1999; McCusker et al. 2000). Nevertheless, based on the Bayesian consensus tree it was more likely for dace to have recolonized the Willamette River by headwater capture from an adjacent interior basin than via a coastal refugium (Fig. 5).
The Warner and Goose Lake populations formed a moderately supported clade with Klamath A (Figs. 4, 5). The Warner–Goose–Klamath A clade was more closely related to the Snake and Lower Columbia than to the Middle Columbia or coastal drainages (Fig. 5). These findings are consistent with the Oakey et al. (2004) hypothesis that the Pliocence western passage of the Snake River from Idaho to the Pacific Ocean created a connection between Malheur Lakes, Klamath River Basin and the Pit River basin of California. The Warner and Goose Lake Basins are geographically situated in between the Malheur Lakes and Klamath/Pit Basins and our data suggests these two basins were likely associated with this westward-draining Pliocene Snake River as well.
Genetic variation within the Warner Basin
Speckled dace populations in the Warner Basin are closely related but show signs of recent isolation from each other. This pattern of genetic variation is consistent with climatic events that impacted this basin during the Pleistocene. Dace in the Warner Basin were likely more abundant and had greater opportunities for gene flow among populations when this basin contained large glacial lakes during the Pleistocene (Reheis et al. 2002). The natural drying of these glacial lakes during the last 10,000 years has led to isolated populations of dace within Foskett Spring and among disconnected stream segments in the Warner Basin (Minckley et al. 1986).
Levels of genetic divergence observed between dace from Foskett Spring compared to other dace from the Warner Basin were in the range typically observed between conspecific populations (Table 2; Pfrender et al. 2004; Smith et al. 2002). Populations within Warner Basin were much more similar to one another than they were to Goose Lake Basin populations. In fact, the AMOVA results suggest differences among dace within the Warner and Goose Lake Basins only accounted for 6.6% of the ND2 haplotype variation observed in this study. Polyphyletic structuring of the ND2 haplotypes among the Warner Basin populations also suggests these populations have recently become isolated from each other (Neigel and Avise 1986).
Populations of dace in the Warner Basin appear to be recently diverged on an evolutionary timeframe. However, significant genetic subdivision among populations is apparent in these data with a ΦSC of 0.35 (95% CI 0.25–0.41). Each population also had a unique set of ND2 haplotypes (Appendix Table 3). We believe this pattern of haplotype diversity at ND2 is a signature of genetic drift in which speckled dace experienced a historical bottleneck followed by population expansion in these geographically isolated populations over the past 10,000 years. The high level of haplotype diversity in all of the sampled populations and the polyphyletic relationship of haplotypes among populations suggests incomplete lineage sorting among populations of dace in the Warner Basin. High haplotype diversity and incomplete lineage sorting suggests the N ef of dace within the Warner Basin has historically been and continues to be large. However, if Foskett dace continue to remain physically isolated we’d expect reciprocal monophyly to develop in 2 N ef generations (Avise 2000).
Origin of the Foskett Spring dace population
The pattern of diversity observed at ND2 among the four Warner Basin populations is consistent with all populations being natural origin. We base this conclusion on the Foskett Spring sample exhibiting a similar pattern of haplotype diversity to the other Warner Basin populations (Fig. 3). If a small number of dace had been introduced by humans into Foskett Spring we would expect to see low H d and π values which are characteristic genetic signals of a recently founded population. However, the Foskett Spring population had the highest observed π and H d was similar to the other natural populations (Table 2).
Mismatch distribution results also suggest dace are a native to Foskett Spring. If this population was recently founded it would have undergone a recent demographic expansion to reach the current adult census of over 3,000 individuals. The mismatch distribution results for Foskett Spring indicated this population has been at equilibrium or experienced a bottleneck in the recent past (Fig. 6). We believe it is more likely that this population has been at equilibrium because of the relatively high levels of diversity observed at the ND2 gene (Table 2).

Mismatch distribution of ND2 sequences in four speckled dace populations (histogram) and the simulated distribution (line) for a population that has experienced a sudden expansion with the same mean. All populations are from the Warner Basin in Oregon; a Deep Creek, b Foskett Spring, c Honey Creek, and d Twelvemile Creek. Statistical tests for sudden demographic expansion are also reported: raggedness index values (r), Fu’s F S and Ramos-Onsins and Rozas’s R 2. An * denotes a significant result (P < 0.05) for population expansion for F S and R 2. Conversely, for r and * denotes a significant result (P < 0.05) indicating the population has not undergone a demographic expansion
Levels and patterns of genetic diversity observed in dace from Foskett Spring are consistent with the biographic history of the Coleman Lake Subbasin which became isolated hydrologically from the Warner Basin at the end of the Pleistocene. Because Coleman Lake is usually dry, dace are restricted to Foskett Spring which provides a constant water source and a stable habitat for dace. However, the limited size of Foskett Spring places limits on the demographic expansion of the population. Foskett dace were the most genetically distinct of the Warner Basin populations based on d A estimates at ND2 (Table 2), although the variance of these estimates were high (Table 2). The phylogenetic pattern observed at ND2 showed evidence for reproductive isolation and incomplete linage sorting among Warner Basin populations (Fig. 3). These phylogenetic patterns and genetic distance estimates support the hypothesis that the Foskett dace have been isolated from the other populations in the Warner Basin since the end of the Pleistocene.
Subspecies status and conservation of speckled dace
In the broadest sense a subspecies is defined as a population or group of populations that are genetically distinct from but reproductively compatible with other such groups. Two recent studies have provided criteria for defining subspecies. Haig et al. (2006) suggested that at minimum a unit being considered as a subspecies be discrete in relation to the remainder of the species and that it must also be biologically significant to the species. Zink (2004) suggested using mtDNA phyolgenetic data to test the value of named subspecies. This approach uses the mtDNA phylogeny as a test for discreteness and also uses the criterion of reciprocal monophyly to infer biological significance. Reciprocally monophyletic groups often take thousands of years to evolve which led Zink (2004) to conclude they represent, “….significant bouts of independent history and adaptive phenotypic variation.” Both of these studies provide a framework for identifying evolutionary units that are diverging toward full speciation.
Current taxonomic groupings of speckled dace are based primarily on the biological species concept and information on morphology and geography (Hubbs et al. 1974). Low information content associated with these taxonomic characters has resulted in grouping of all forms of speckled dace into a single highly variable species (Oakey et al. 2004). Ichtyologist recognized this grouping was incorrect but could do little until more useful morphological or genetic based taxonomic approaches are developed (Moyle 2002). While waiting for new methods to sort out the species level groupings ichthyologist have used the Linnaean rank of subspecies to document unique local endemics of speckled dace. These subspecies have been designated in relation to the geographic area they are found and information used for their classification has been focused on the local area not the entire speckled dace range (e.g., Hubbs and Kuhne 1937).
Four subspecies of speckled dace are currently protected as endangered under the ESA; the Ash Meadows speckled dace (R. o. nevadensis), Clover Valley speckled dace (R. o. oligoporus), Independence Valley speckled dace (R. o. lethoporus) and Kendall Warm Springs speckled dace (R. o. thermalis). We conducted a literature search on these subspecies and only found enough information on the Kendall Warm Springs dace to evaluate its subspecies status using the criteria of Haig et al. (2006) and Zink (2004).
Kendall Warms Springs dace provide a valuable case study for selecting characters used to identify speckled dace subspecies. This subspecies is the only fish found in Kendall Warm Springs and its outflow ends in a 10 m waterfall at the confluence with the Green River in Northwestern Wyoming. Water temperatures for the spring and its outflow remain at a nearly constant 29°C while temperatures in the Green River fluctuate from 0 to 18°C. Hubbs and Kuhne (1937) described Kendall Warms Springs dace as a subspecies based on its unique ecological setting and morphological differences from the Green River speckled dace (R.o. yarrowi). Subsequent studies revealed that assumed differences in thermal tolerances between the subspecies did not exist (Kaya et al. 1992). There were also concerns that morphological characters used to differenate these subspecies were thermally labile (Kaya et al. 1992).
Uncertainty concerning Kendall Warm Springs subspecies status prompted Kaya et al. 1992 to examine additional characters. Mitochondrial DNA analysis revealed a reciprocal monophyletic relationship between Kendall Warms Springs dace and Green River dace. However, the genetic distances between the subspecies were small and the level of genetic variation in the Kendall Warms Springs dace was low. These findings are consistent with Hubbs and Kuhne (1937) hypothesis that dace colonized Kendall Warm Springs after it was isolated from the Green River by a travertine shelf approximately 10,000 years ago. In addition, Kaya et al. (1992) found pharyngeal tooth counts differ between the subspecies and are not thermally labile. These differences in pharyngeal tooth counts are likely an example of adaptive radiation which has biologically significance to the species because this trait could have a utility requirement associated with feeding in the unique Kendall Warm Spring environment. Our analysis confirmed the taxomonic classification of Kendall Warm Springs dace as a subspecies. This conclusion was well supported by biological (Gould and Kaya 1991), genetic (Kaya et al. 1992), and physical data (Hubbs and Kuhne 1937).
In this study we found no evidence that specked dace in Foskett Spring are reciprocally monophyletic in respect to the other Warner Basin populations. These results suggest it is difficult to justify subspecies status for Foskett Spring speckled dace based on nucleotide variation at two mtDNA genes. Our results do not preclude the possibly that other independent genetically based traits that are associated with morphological or life history differences could have occurred within the past 10,000 years as the result of rapid evolution at these traits in different selective environments. For example, the Foskett Springs dace population has no fish predators while the other Warner Basin dace populations do co-exist with fish predators. Phenotypic divergence of Utah chub life histories strategies has been influenced by the presence or absence of trout predators among geographically isolated populations in the Bonneville Basin (Johnson and Belk 1999). Johnson (2002) suggests selective environments associated with late Pleistocence fragmentation and presence or absence of predators have been critical factors in shaping evolutionary divergence of Utah chub. It is the presence of such traits as defined by Carl Bond that were presumably the basis for designating the Foskett Spring population as an undescribed subspecies.
Additional information is needed to follow-up on the observations of Carl Bond to document traits he described in his 1990 personal communication to the US Fish and Wildlife Service. We suggest conducting similar common garden experiment to those of Kaya et al. (1992) to determine the genetic vs. environmental influence on the phenotypic traits. Foskett dace are a geographically isolated population in a unique ecological setting. However, the degree to which this population is biologically significant to the species is uncertain. Our genetic results and those of Pfrender et al. (2004) and Oakey et al. (2004) suggest biologically significant differences are likely to be present among major watersheds (i.e., Warner and Goose Lake). We believe a comprehensive range wide systematic study of speckled dace is needed to accurately identify the major evolutionary units of this broadly distributed and morphologically diverse species.
References
Avise JC (2000) Phylogeography. Harvard University Press, Boston
Bernatchez L (1995) A role for molecular systematics in defining evolutionary significant units in fishes. Am Fish Soc Symp 17:114–132
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Dowling TE, Tibbetts AC, Minckley WL, Smith GR (2002) Evolutionary relationships of the Plagopterins (Teleostei: Cyprinidae) from Cytochrome b sequences. Copeia 2002:665–678
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) ARLEQUIN, version 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1(4):7–50
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evol Int J org Evol 39:783–791
Fry AJ, Zink RM (1998) Geographic analysis of nucleotide diversity and song sparrow (Aves: Emberizidae) population history. Mol Ecol 7:1303–1313
Fu Y-X (1997) Statistical tests of neutrality against population growth, hitchhiking, and background selection. Genetics 147:915–925
Gould WR, Kaya CM (1991) Pharynegeal teeth in the endangered Kendall Warm Springs dace (Rhinichthys osculus thermalis). Copeia 1991:227–229
Haig SM, Beever EA, Chambers SM, Draheim HM, Dugger BD, Dunham S, Elliott-Smith E, Fontaine JB, Kesler DC, Knaus BJ, Lopes IF, Loschl P, Mullins TD, Sheffield LM (2006) Taxonomic considerations in listing subspecies under the US Endangered Species Act. Conserv Biol 20(6):1584–1594
Harpending RC (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol 66:591–600
Hedrick PW, Lee RN, Hurt CR (2006) The endangered Sonoran topminnow: examination of the species and ESUs using three mtDNA genes. Conserv Genet 7:483–492
Hubbs CL, Kuhne ER (1937) A new fish of the genus Apocope from a Wyoming warm spring. Occas Pap Mus Zool Univ Mich 343:1–21
Hubbs CL, Miller RR, Hubbs LC (1974) Hydrographic history and relict fishes of the north-central Great Basin. Calif Acad Sci Mem 7:1–259
Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inferences of phylogeny. Bioinformatics 17:754–755
Johnson JB (2002) Evolution after the flood: phylogeography of the desert fish Utah chub. Evol Int J org Evol 56(5):948–960
Johnson JB, Belk MC (1999) Effects of predation on life-history evolution in Utah chub (Gila atraria). Copeia 1999:948–957
Johnson JA, Dunn PO, Bouzat JL (2007) Effects of recent population bottlenecks on reconstructing the demographic history of prairie-chickens. Mol Ecol 16(11):2203–2222
Kaya CM, Brussard PF, Cameron DG, Vyse ER (1992) Biochemical genetics and thermal tolerances of Kendall Warm Springs dace (Rhinichthys osculus thermalis) and the Green River speckled dace (R. o. yarrowi). Copeia 1992:528–535
Kumar S (1996) PHYLTEST: Phylogenetic hypothesis testing software, version 2.0. Pennsylvania State University, University Park
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: Molecular evolutionary genetics analysis software. Arizona State University, Tempe
Lee DS, Gilbert CR, Hocutt CH, Jenkins RE, McAllister DE, Stauffer JR (1980) Atlas of North American freshwater fishes. North Carolina Museum of Natural History, Releigh
McCusker MR, Parkinson E, Taylor EB (2000) Mitochondrial DNA variation in rainbow trout (Oncorhynchis mykiss) across its native range: testing biographical hypotheses and their relevance to conservation. Mol Ecol 9:2089–2108
Minckley WL, Hendrickson DA, Bond CE (1986) Geography of western North American freshwater fishes: description and relationships to intracontinential tectonism. In: Hocutt CH, Wiley EO (eds) The zoogeography of North American freshwater fishes. Wiley, New York, pp 519–614
Moyle PB (2002) Inland fishes of California. University of California Press, Berkley
Nei M (1987) Molecular evolutionary genetics. Middle Columbia University Press, New York
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Neigel JE, Avise JC (1986) Phylogenetic relationships of mitrochondrial DNA under various demographic models of speciation. In: Nevo E, Karlin S (eds) Evolutionary processes and theory. Academic Press, New York, pp 515–534
Oakey DD, Douglas ME, Douglas MR (2004) Small fish in a large landscape: Diversification of Rhinichthys osculus (Cyprinidae) in western North America. Copeia 2:207–221
Pfrender ME, Hicks J, Lynch M (2004) Biogeographic patterns and current distribution of molecular-genetic variation among populations of speckled dace, Rhinichthys osculus (Girard). Mol Phylogen Evol 30(3):490–502
Posada D, Crandell KA (1998) Modeltest: testing the model DNA substitution. Bioinformatics 14:817–818
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100
Reheis M, Sarna-Wojcicki AM, Reynolds RL, Repenning CA, Mifflin MD (2002) Pliocene to middle Pleistocene lakes in the western Great Basin: ages and connections (pp 53–109). In: Hershler R, Madsen DB, Currey DR (eds) Great Basin Aquatic Systems History Smithsonian Contributions to Earth Sciences, vol 33, 405 pp
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DNA SP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Scheerer PD, Jacobs SE (2006) Hutton Spring Tui Chub & Foskett Spring Speckled Dace Investigations. Oregon Department of Fish and Wildlife. Fish Research Project E-2-37, Annual Progress Report, Corvallis
Schneider S, Excoffier L (1999) Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Smith GR, Dowling T, Gobalet KW, Lugaski T, Shiazawa D, Evans RP (2002) Biogeography and timing of evolutionary events among Great Basin fishes (pp 175–234). In: Hershler R, Madsen DB, Currey DR (eds) Great Basin aquatic systems history, Smithsonian Contributions to Earth Sciences, vol 33, 405 pp
Stepine CA, Dillion AK, Brooks MJ, Chase KL, Hubers AN (1997) The evolution of blennioif fishes based on the analysis of mitochondrial 12S rDNA. In: Kocher TD, Stepien CA (eds) Molecular systematics of fishes. Academic, San Diego, pp 245–270
Swofford DL (1999) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer Associates, Sunderland
Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996) Phylogenetic inference. In: Hillis DM, Mortiz C, Mable BK (eds) Molecular systematics, 2nd edn. Sinauer Associates, New York, pp 407–514
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Taylor EB, Pollard S, Louie D (1999) Mitochondrial DNA variation in bull trout (Salvelinus confluentus) from northwestern North America: implications for zoogeography and conservation. Mol Ecol 8:1155–1170
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132:619–633
US Fish Wildlife Service (1985) Determination of threatened status for Hutton tui chub and Foskett speckled dace. Fed Regist 50:12302–12306
US Fish and Wildlife Service (1998) Recovery plan for the native fishes of the Warner Basin and Alkali Subbasin. Portland, 86 pp
Williams JE, Stern MA, Munhall AV, Anderson GA (1990) Conservation status of threatened fishes in the Warner Basin, Oregon. Great Basin Naturalist 50(3):243–248
Williams JE, Macdonald CA, Williams CD, Weeks H, Lampman G, Sada DW (2005) Prospects for recovering endemic fishes pursuant to the US Endangered Species Act. Fisheries 30(6):24–29
Zink RM (2004) The role of subspecies in obscuring avian biological diversity and misleading conservation policy. Proc R Soc Lond B Biol Sci 271(1539):561–564
Acknowledgments
Funding for this project was provided by the US Fish and Wildlife Service. D. Campton provided valuable input on development of this project. We would like to thank C. W. Kilpatrick, D. Markle, T. Dowling, M. Pfrender, and C. Hurt for assistance with laboratory methods and data analysis. J. Baumsteiger, C. Allen, R. White, A. Mauer, A. Munhall, and S. Reid collected samples used in this study. D. Campton, P. Crandell, D. Markle, and two anonymous reviewers provided critical review and comments that greatly improved this paper. The findings and conclusions in the article are those of the authors and do not necessarily represent the views of the US Fish and Wildlife Service.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ardren, W.R., Baumsteiger, J. & Allen, C.S. Genetic analysis and uncertain taxonomic status of threatened Foskett Spring speckled dace. Conserv Genet 11, 1299–1315 (2010). https://doi.org/10.1007/s10592-009-9959-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-009-9959-0