
Abstract
Pontoporia blainvillei (Gervais & d’Orbigny, 1844), the franciscana dolphin, is the most endangered small cetacean in the Western South Atlantic. It is an endemic species with a coastal and estuarine distribution that has been divided into four Franciscana Management Areas (FMAs). We used the mitochondrial DNA control region to conduct a phylogeographic analysis to evaluate the population structure of the franciscana and the influence of paleoceanographic events on its biogeographic history. We found nine populations along the entire distribution (ΦST = 0.41, ΦCT = 0.38, p < 10–5), with estimated migration rates resulting in less than one female per generation. Populations from FMAIII and FMAIV in the south (including the Río de La Plata Estuary) showed higher long-term migration rates and effective population sizes than northern populations. The phylogeographic analysis supports the franciscana origin in the Río de La Plata Estuary, with further dispersal south and northwards. The first lineage split happened around 2.5 Ma, with lineage radiation throughout the Pleistocene until recent fragmentation events shaped current-day populations. We suggest that Pleistocene glaciations influenced the dispersion and population structure of the franciscana. Specifically, that the shift of the Brazil-Malvinas Confluence drove the dispersion northwards. Then, low sea-level periods caused either the isolation in estuarine refugia or local extinctions, followed by re-colonizations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pontoporia blainvillei (Gervais & d’Orbigny, 1844), the franciscana dolphin, is an endemic species with a geographic distribution that extends from the state of Espírito Santo, Brazil (18° 25’ S) to Chubut province, Argentina (41° 10’ S) (Bastida et al. 2007). The species occurs in waters typically shallower than 30 m (Danilewicz et al. 2009) but is also found up to the 50 m isobath (Crespo et al. 2010). Due to their coastal and estuarine habits, franciscanas inhabit areas that are highly impacted by anthropogenic activities, and thus major concerns to their conservation are habitat loss and degradation, contamination, and especially incidental mortality in fishing gillnets (Secchi et al. 2003a, 2021; Crespo et al. 2010; Lailson-Brito et al. 2011; Alonso et al. 2012; Gago-Ferrero et al. 2013). P. blainvillei is currently the most threatened small cetacean in the Western South Atlantic, listed as “vulnerable” in the Red List of the International Union for Conservation of Nature (Zerbini et al. 2017) and considered “Critically Endangered” by the Brazilian Government (Instituto Chico Mendes de Conservação da Biodiversidade 2018). Annual mortality in franciscana populations reaches up to 2–5% (Secchi et al. 2003a, 2021; Crespo et al. 2010; Negri et al. 2012). According to the International Whaling Commission Scientific Committee (Donovan and Bjørge 1995), a 2% mortality rate may not be sustainable for cetacean populations. It impacts the size and connectivity among populations and possibly results in the loss of the species evolutionary potential (Hamilton et al. 2001; Mendez et al. 2008). It is also noteworthy that P. blainvillei belongs to a relic lineage, with its closest living relative being the Amazon river dolphin, Inia geoffrensis (Cassens et al. 2000; Hamilton et al. 2001) that occurs in the Amazon and Orinoco River basins.
Secchi et al. (2003b) compiled all available information at the time, based on the species’ geographic distribution, contaminant and parasite loads, vital rates, external morphology, and mitochondrial DNA (mtDNA) data, and proposed four Franciscana Management Areas (FMA). The four FMA would range from: (1) the coast of Espírito Santo to the north of Rio de Janeiro, Brazil (FMAI); (2) from south of Rio de Janeiro to the north of Santa Catarina, Brazil (FMA II); (3) from the central coast of Santa Catarina in Brazil to Uruguay (FMA III); and (4) from Buenos Aires to Chubut in Argentina (FMA IV) (Fig. 1). In addition, based on the high level of genetic divergence found in mitochondrial DNA data analyses, Cunha et al. (2014) proposed two Evolutionarily Significant Units (ESU) for the species, ESU North (from Espírito Santo to northern Rio de Janeiro; 18-22° S) and ESU South (from south of Rio de Janeiro to Argentina; 22-38° S).

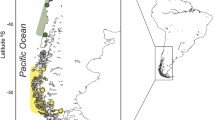
Pontoporia blainvillei sample sizes and localities. Inside the parentheses, the first number represents sample sizes from the literature and the second number represents the new samples. Colours follow FMA subdivisions from the literature. FMAIa: ES (Espírito Santo); FMAIb: RJN (northern Rio de Janeiro); FMAIIa: RJS (southern Rio de Janeiro) and SPN (northern São Paulo); FMAIIb: SPC (central São Paulo), SPS (southern São Paulo), PR (Paraná), SCN (northern Santa Catarina) and BAB (Babitonga bay); FMAIII: RS (Rio Grande do Sul), URAO (Atlantic Ocean of Uruguay) and RP (Río de La Plata); FMAIVa: SCL (San Clemente) and SB (San Bernardo); FMAIVb: was not sampled (Cabo San Antonio/ East Buenos Aires); FMAIVc: NC (Necochea), CL (Claromecó) and BB (Bahía Blanca); FMAIVd: MH (Monte Hermoso); FMAIVe: RN (Río Negro)
Subsequently, studies based on mtDNA and microsatellite analyses refined the Secchi et al. (2003b) subdivisions and suggested the existence of up to ten genetic populations within the FMAs: FMAIa (Espirito Santo), FMAIb (Northern Rio de Janeiro), FMAIIa (Southern Rio de Janeiro to Northern São Paulo), FMAIIb (Central São Paulo to Northern Santa Catarina), FMAIII (Southern Santa Catarina to Uruguay), FMAIVa (San Clemente), FMAIVb (Cabo Santo Antonio to East of Buenos Aires), FMAIVc (Necochea to Claromecó), FMAIVd (Monte Hermoso to Southwest of Buenos Aires) and FMAIVe (Río Negro) (Mendez et al. 2010; Costa-Urrutia et al. 2012; Cunha et al. 2014; Gariboldi et al. 2015, 2016). Nevertheless, these subdivisions are considered provisional, mainly as a conservation precaution, because most studies were conducted in a microscale, focusing on one FMA. Taken together, there are some gaps in geographic sampling and small sample sizes for some localities. Thus, if one single study analyzed all samples together, the population structure could be different than previously suggested.
Although several studies have examined the population structure of the species, both at macro and microscales, an in-depth phylogeographic analysis has not yet been undertaken to understand how and when the colonization of the Atlantic coast by this species occurred and how dispersal and fragmentation have shaped its current populations. The most accepted hypothesis, based on phylogenetic analyses and fossil data, is that the species evolved from an ancestor that lived in a continental sea (the Paranense Sea; Von Ihering 1927) and later migrated to the Atlantic via the Río de La Plata Estuary (Hamilton et al. 2001). Preliminary macroscale population genetic data seemed to support such scenario by showing a gradient of genetic diversity and structure in localities from the Río de La Plata Estuary northwards to Espírito Santo (Cunha et al. 2014).
In this context, we analyzed mtDNA control region sequences from 391 individuals to further evaluate the genetic diversity across the current two ESUs and four FMAs proposed for P. blainvillei. We applied phylogeographic analyses to test the hypothesis proposed by Hamilton et al. (2001) and inferred the influence of paleoceanographic changes on the species biogeographic history. Our study provides additional information about the historical demography of each population and their long-term connectivity, which are useful for the conservation of any threatened species (Hickerson et al. 2009).
Materials and Methods
Sampling
A total of 391 samples were used in this study (Fig. 1). New samples were collected from 83 franciscana carcasses that had washed ashore along the Brazilian coast. An additional 308 samples were obtained from previously published studies (Secchi et al. 2003b; Lazaro et al. 2004; Mendez et al. 2008; Costa-Urrutia et al. 2012; Cunha et al. 2014; Gariboldi et al. 2015, 2016), collected from 19 sampling localities distributed along the coasts of Brazil, Uruguay, and Argentina: Espírito Santo (ES): 14; North of Rio de Janeiro (RJN): 10; South of Rio de Janeiro (RJS): 2; North of São Paulo (SPN): 8; Central region of São Paulo (SPC): 22; South of São Paulo (SPS): 4; Paraná (PR): 1; North of Santa Catarina (SCN): 9; Baía da Babitonga (BAB): 18; Rio Grande do Sul (RS): 14; Atlantic Ocean region of Uruguay (URAO): 2; Río de la Plata (RP): 52; San Clemente (SCL): 4; San Bernardo (SB): 2; Necochea (NC): 31; Claromecó (CL): 81; Monte Hermoso (MH): 15; Bahía Blanca (BB): 4; Río Negro (RN): 15 (Fig. 1, Online Resource 1). Each individual’s locality was attributed according to its stranding location. Therefore, individuals collected inside the Baía da Babitonga were considered to be from BAB, and those collected in the northern coastline of Santa Catarina were considered to be from SCN. All individuals considered to be from RS were collected along the southern coast of Rio Grande do Sul. There is a discontinuity in our sampling from central Santa Catarina to southern Rio Grande do Sul because franciscana sequences from this area are unpublished. However, a study including those sequences did not find genetic structuring in this area; instead, it suggested that FMAIII should comprise the entire coast from central Santa Catarina to Rio Grande do Sul (Santos 2011). The new samples were collected from animals that died on different sites and/or dates. Therefore, sampling is unlikely to be biased towards related individuals. Sampling permits were issued by the Brazilian Environmental Agencies (SISBIO 16586-2, 11980-1, 13303-14, 31226-11, 17418-6 and 64724-6).
DNA Sequencing
DNA of the 83 new samples was isolated through a phenol–chloroform protocol with proteinase K (Sambrook et al. 1989). The mitochondrial DNA control region of the new 83 samples was amplified by Polymerase Chain Reaction (PCR) using primers RCPb-F 5’- CTC CTA AAT TGA AGA GTC TTC G – 3’ and RCPb-R 5’ – CCA TCG AGA TGT CTT ATT TAA GAG G – 3’ following Cunha et al. (2014). Final concentrations used in PCR reaction volumes of 15 µL were: 1 unit of GoTaq polymerase (Promega); Buffer 1X (Promega); 0.20 mM dNTPs; 2.5 mM MgCl2 and 0.5 mM of each primer. PCR cycling was 3 min at 94 °C; 35 cycles of 1 min at 92 °C, 1 min at 48 °C and 1 min at 72 °C; plus 5 min of final extension at 72 °C. PCR products were purified and sequenced in both directions in an ABI3500 automated sequencer. Sequences were edited with program SeqMan 7 (Swindell and Plasterer 1997) and aligned in Geneious (Kearse et al. 2012). Previously published mtDNA control region sequences with more than 455 bp (N = 308) were retrieved from GenBank (Online Resource 1) and included in the alignment. An Inia geoffrensis sequence was included as outgroup (GenBank accession number: AF521123).
Reconstruction of Paleodrainages
Paleodrainages were reconstructed to test if past river mouth configurations influenced the franciscana’s geographic distribution and population structure. Paleodrainage boundaries between contemporary basins were estimated for the Last Glacial Maximum (LGM) with ArcGIS Pro 2.5 (Esri Inc. 2020), using Hydrological and Spatial tools following Thomaz and Knowles (2018). Topographical and bathymetric information from a digital elevation model (DEM) from the General Bathymetric Chart of the Oceans (GEBCO_2014) was used at 30 arc-second resolutions (c. 1 km; http://www.gebco.net/).
With a Contour tool, a base contour line at -125 m was created to estimate the maximum extent of land exposed during the Pleistocene. For each cell, we determined the flow direction by its slope using the Flow Direction tool. Based on this flow direction, we used the Basin tool to identify the ridgelines, and the paleodrainages were delineated by these inferred ridges. We plotted the populations considering the currently proposed FMA subdivision as well as the new populations identified in the present study.
Genetic Diversity and Population Structure
The Analysis of Molecular Variance (AMOVA) was conducted in the Arlequin v3.5 program (Excoffier and Lischer 2010). We tested the population structure hypotheses previously proposed (Secchi et al. 2003b; Costa-Urrutia et al. 2012; Cunha et al. 2014; Gariboldi et al. 2016) and whether the population structure could be explained by paleodrainages (Thomaz et al. 2017). In addition, as this study is the first to include most of the sampling localities across the franciscana total geographic distribution, we performed an exploratory analysis investigating possible groupings of geographically adjacent localities, varying the number of populations (K) from two to eighteen. In the first run all nineteen localities were considered populations in order to lump the localities with sample size below four with sampling localities with the lowest genetic and geographic distance. Thus, we were not able to consider the subdivision of FMAIVb in our analysis, as we only had samples from SB, which were grouped with SCL from FMAIVa. The following localities were lumped: RJS + SPN, SPS + PR, SCL + SB, and BB + RN.
Considering the best population scenario found in the AMOVA analysis, the computation and testing of pairwise FST and ΦST fixation indices, estimation of haplotype and nucleotide diversity, and the neutrality tests Tajima’s D and Fu’s Fs were performed in the Arlequin v3.5 program (Excoffier and Lischer 2010). For ΦST analyses, the Tamura and Nei mutation model was set as indicated by corrected Akaike Information Criterion (AICc) calculations performed with jModelTest 2.1.7 (Posada 2008). Mantel test was also conducted in the Arlequin v3.5 program (Excoffier and Lischer 2010) considering the distances between the populations from the best AMOVA scenario and 1000 permutations. The geographic distances between populations were calculated with ArcGIS Pro 2.5 (Esri Inc. 2020) as the minimum distance by sea between each other.
A median-joining haplotype network was built with PopArt (Leigh and Bryant 2015), in which nine sequences from Mendez et al. (2010) were included as unknown locations in Argentina. These sequences were included in order to provide the most integrated network which would also be comparable to previous studies.
Phylogeny and Divergence Times
We used BEAST v1.10.4 (Drummond et al. 2012) to estimate phylogeny and divergence times under an uncorrelated log-normal relaxed clock with the mutation rate of 1%/Ma, estimated for the control region of cetaceans (Hoelzel et al. 1991), and the GTR + I + G mutation model, as indicated by corrected Akaike Information Criterion (AICc) calculations performed with jModelTest 2.1.7 (Posada 2008). As our analyses contained a mixture of intra and inter-species sequences, although mostly containing franciscana sequences, a coalescent Bayesian skyline prior was used for rates of cladogenesis, as recommended by Ritchie et al. (2016). The number of grouped intervals (m) was set to five, and five independent runs of one hundred million Markov Chain Monte Carlo (MCMC) steps were performed to achieve reliable parameters estimates (Effective Sampling Size > 200). The analysis was performed including one sequence of each haplotype to determine the chronology of populations, FMAs and ESUs divergence events. BEAST analysis was run in the Cipres Science Gateway server (Miller et al. 2010).
Migration Rates
Long-term asymmetrical migration rates between populations from the FMAs and the best AMOVA scenario and their effective population sizes (Ne) were estimated using the program Migrate 4.4 (Beerli 2006). Migrate provides M, the mutation-scaled immigration rate (M = m/μ), and θ, mutation-scaled population size (θ = Neμ), where m is the immigration rate, μ is the mutation rate of the studied gene (μ = 1 × 10−8 was used for the mtDNA control region) and Ne is the effective population size. The estimated θ was used to calculate Ne (Ne = θ/μ) and the number of migrants per generation was calculated multiplying M by θ (Mθ = Neµ*m/μ). We performed the Bayesian inference with three independent runs for each analysis, which consisted of one long chain with 50 million-recorded parameter and genealogy changes after discarding the first 10,000 genealogies as burn-in with a random-subset of 20 individuals per location. To improve the estimation of marginal likelihood we used a static heating scheme with four chains with temperatures 1.00, 1.50, 3.00 and 1,000,000. Prior distributions for M and θ were percent values 10 and 50, respectively. The log marginal likelihood values were used to compare models and to calculate the probability of each model following P(model) = exp(lnmLmodel- a)/∑jexp(lnmLmodelj—a), where lnmL is the log marginal likelihood and a is the largest value among the log marginal likelihoods of all models.
Biogeographic History
We performed a biogeographical inference using “BioGeoBEARS” (Matzke 2013a) implemented in R v.4.1.0 (R Development Core Team 2021). We pruned our time-calibrated Bayesian phylogeny by selecting a single haplotype to represent each population from the best AMOVA scenario and Inia geoffrensis for the outgroup, resulting in a tree with ten terminals that was used for the “BioGeoBEARS” analyses. We used "BioGeoBEARS" to calculate the log-likelihood (lnL) and the corrected Akaike Information Criterion (AICc) to choose the best fitting biogeographical model. For this we considered the six “BioGeoBEARS” models: likelihood-based Dispersal-Extinction Cladogenesis (Ree and Smith 2008; Matzke 2013b), and DEC considering founder-event (Matzke 2013b, 2014); DIVAlike, a likelihood version of the DIVA model (Ronquist and Sanmartín 2011), and DIVAlike considering founder-event (DIVAlike + J - Matzke 2013b, 2014); and BAYAREAlike which is a likelihood version of the BAYAREA (Landis et al. 2013), and BAYAREAlike considering founder-event (BAYAREAlike + J - Matzke 2013b, 2014). The DEC model presumes that lineages that derived after cladogenesis will inherit a single-range area, which can be a subset of the ancestor’s range. The DIVAlike model permits derived lineages to inherit more than one area as their range, but it cannot be a subset of the ancestor’s range (Ronquist and Sanmartín 2011). The BAYAREAlike presumes that at cladogenesis there is no range evolution, i.e. that the derived lineages inherit the same range of the ancestral state (Landis et al. 2013). The parameter “J” adds founder-event to each of the mentioned models (DEC + J, DIVAlike + J, and BAYAREAlike + J - Matzke 2013b, 2014). We set the parameter max_range_size to five and included the null range parameter allowing the ranges to consist of zero areas.
Results
Population Structure and Genetic Diversity
Analyses were conducted using an alignment of 391 sequences with 455 base pairs. Global AMOVA showed a considerably high degree of structuring (ΦST = 0.36; p < 10–5). As we increased the number of samples from FMAI and FMAII the ΦST at ESU North had a significant decrease from ΦCT = 0.72 (Cunha et al. 2014) to ΦCT = 0.46 (Online Resource 2), and the ΦST at ESU South increased from ΦCT = 0.19 (Cunha et al. 2014) to ΦCT = 0.23 (Table 1), while ΦCT between ESU South x ESU North was only slightly lower (ΦCT = 0.42, Table 1) than previously reported (ΦCT = 0.44, Cunha et al. 2014). Most pairwise comparisons were significant and showed ES (FMAIa) with the highest ΦST values in relation to all other populations (ΦST from 0.57 to 0.86, Table 2). RJN (FMAIb), the other population from the ESU North, also presented high ΦST values.
The paleodrainage reconstruction recovered 40 paleodrainages throughout the study area, with franciscanas present in 18 of those (Fig. 2). But the AMOVA result for the paleodrainages scenario (ES / RJN / RJS + SPN / SPC / SPS + PR / SC + BAB / RS / URAO + RP / SCL + SB / NC + CL + MH / BB + RN) was not statistically significant (ΦCT = 0.24, p = 0.10, Online Resource 2).

Reconstruction of the Western South Atlantic coastal area from Espírito Santo in Brazil to Río Negro in Argentina during Pleistocene glaciations. Light grey indicates areas of the continental shelf that were exposed during periods of low sea level (-125 m). Red indicates the area within the 30 m isobaths, and the 1000 m isobath is shown in green. The 40 inferred paleodrainages are delimited with light blue contour lines. Circles indicate the mouth of the larger river within each of the paleodrainages, with the colour code used for the corresponding population (FMA subdivision). Numbers indicate the paleodrainages names from which samples were analyzed
Considering the results of AMOVA for all tested scenarios (Table 1, Online Resource 2) our analyses indicate the existence of at least nine populations (ES, RJN, RJS + SPN, SPC + SPS + PR, SCN, BAB, RS + URAO + RP + SCL + SB + NC + CL, MH and RN + BB, ΦCT = 0.38, p < 10–5, Table 1). This scenario is in accordance with pairwise ΦST analysis (Table 2). Some other scenarios had similarly high significant ΦCT (ΦCT = 0.37 and 0.38, Online Resource 2), but neither showed further divisions supported by pairwise ΦST nor any other previous genetic study (i.e. SPC as a population on its own). They also presented negative ΦSC values, which lead to artificially inflated ΦCT. Those scenarios were thus not considered.
The best supported scenario of nine populations defines the populations considered in all our analyses with the following configuration: ES, RJN, RJS + SPN, SPC + SPS + PR, SCN, BAB, RS + URAO + RP + SCL + SB + NC + CL, MH, and RN + BB. This scenario agrees with micro-scale studies using microsatellites that recognized SCN and BAB as different from each other (Cunha et al. 2020a) and MH and BB + RN as unique populations (Gariboldi et al. 2016). In a fine-scale analysis of franciscanas from the Río de La Plata estuary and adjacent coastal waters, microsatellite data also revealed differentiation between SCL and RS + URAO + RP (Costa-Urrutia et al. 2012), not supported by our mtDNA data. Our data, nevertheless, show genetic differentiation between this RS + URAO + RP + SCL + SB + NC + CL population and two other populations in FMAIV: MH and BB + RN, corroborating Gariboldi et al. (2016).
Haplotype and nucleotide diversities in each population varied between 0.05–0.89 (± 0.0001) and 0.00022–0.01061 (± 0.00021), respectively (Table 3). The ES population presented the lowest nucleotide and haplotype diversity (Table 3). The Mantel test did not support the existence of isolation by distance in the species (R2 = 0.09, p = 0.05; Online Resource 3).
Forty-four substitutions were observed, defining 63 haplotypes, of which four had not been previously reported. These sequences have been submitted to the GenBank database under accession numbers OK188832-35. Haplotypes H3, H9, and H10, more frequently found in the south, are in the center of star-shaped topologies of the median-joining network, suggestive of population expansions (Sherry and Roger 1994). It is important to highlight that the Río de La Plata Estuary is the only area where all those three haplotypes are present. The most frequent haplotype (H3) was found in the majority of populations, except Espírito Santo and the North of Rio de Janeiro (which together form the North ESU). On the other hand, the second most common haplotype (H1) is exclusive to the North ESU (Fig. 3). In past studies (Cunha et al. 2014; Gariboldi et al. 2015) haplotype H4 was exclusive to SCN and BAB, but here it is shared with SPC; H22 was observed only in RP and here it is shared with SCN; and H9 was formerly found in RS, RP, SB and BB and here is also found in SCN.

Median-joining network of P. blainvillei control region haplotypes (N = 400, 455 bp). Circle size is proportional to frequency. The number of mutations is represented by lines crossing the branches. New haplotypes (H61, H62, H63, and H64) are highlighted in bold. FMAIa: ES (Espírito Santo); FMAIb: RJN (northern Rio de Janeiro); FMAIIa: RJS (southern Rio de Janeiro) and SPN (northern São Paulo); FMAIIb: SPC (central São Paulo), SPS (southern São Paulo), PR (Paraná), SCN (northern Santa Catarina) and BAB (Babitonga bay); FMAIII: RS (Rio Grande do Sul), URAO (Atlantic Ocean of Uruguay) and RP (Río de La Plata); FMAIVa: SCL (San Clemente) and SB (San Bernardo); FMAIVb: was not sampled (Cabo San Antonio/ East Buenos Aires); FMAIVc: NC (Necochea), CL (Claromecó) and BB (Bahía Blanca); FMAIVd: MH (Monte Hermoso); FMAIVe: RN (Río Negro)
H14 is a haplotype from the North ESU (ES) that groups with the South ESU. Cunha et al. (2014) chose to leave H14 out of their population structure analyses because it was a singleton and the sequence could no longer be confirmed. In this study, a new haplotype (H63), closely related to H14, was observed in RJN and SPC. However, even with the inclusion of those haplotypes (H14, H63), AMOVA and ΦST analyses showed that ES and RJN are different from the remaining FMA, and the scenario of two groups, encompassing the two ESU, is the one that best explains the total genetic variance (ΦCT = 0.42; p = 0.002). Considering the new haplotypes found, H63 is shared between RJN and SPC, and H62 is shared by SPC, SPN, and RJS, while H61 is exclusive to ES and H64 to SCN.
Migration Rates and Effective Population Size
As demographic analyses were conducted with mtDNA, our results reflect long-term female migration and ancestral Ne estimates. Populations closer to the Río de La Plata Estuary seem to have kept stable and larger effective population sizes and higher migration rates than populations from other FMA. The higher genetic diversity found in FMAIII (Table 3; Cunha et al. 2014) supports the hypothesis that P. blainvillei would have been in the Río de La Plata Estuary region for longer than anywhere, and that its colonization happened from there northwards and southwards, as proposed by Hamilton et al. (2001).
Importantly, immigrants in most populations are below one female per generation, the only two exceptions being RS + URAO + RP + SCL + SB + NC + CL and MH. Estimates also suggest that populations ES, RJN, SCN, and BB + RN provide more emigrants than receive immigrants, and MH receives immigrants but hardly provides emigrants.
Estimations show greater migration rates between geographically closer populations, but populations from FMAIII and FMAIV presented higher migration rates and larger ancestral effective population sizes than northern populations from FMAII and FMAI (Fig. 4 and Table 4). Considering the connection between northern and southern populations, population BAB would be the only one from the northern group to have received migrants from southern populations (RS + URAO + RP + SCL + SB + NC + CL) while RS + URAO + RP + SCL + SB + NC + CL received migrants from RJS + SPN, SPS + SPC + PR, SCN, BAB. Besides, population ES (North ESU) presented the lowest migration rates (below 0.2; Table 4). Additionally, the ES population presents low haplotype and nucleotide diversity (Fig. 1 and Table 3; Cunha et al. 2014; de Oliveira et al. 2020). The overall low migration rates and restricted gene flow between FMAI/FMAII and FMAIII/FMAIV indicate a past divergence between the populations analyzed, as indeed was suggested by phylogeographic analyses.

Long-term migration estimates for Pontoporia blainvillei populations. Arrows indicate the directionality of gene flow (where present). Numbers above or below the arrows represent the migration rate. Effective population size is represented by θ inside each population circle. ES: Espírito Santo; RJN: northern Rio de Janeiro; RJS: southern Rio de Janeiro; SPN: northern São Paulo; SPC: central São Paulo; SPS: southern São Paulo; PR: Paraná; SCN: Santa Catarina; BAB: Babitonga bay; RS: Rio Grande do Sul; URAO: Atlantic Ocean of Uruguay; RP: Río de La Plata; SCL: San Clemente; SB: San Bernardo; NC: Necochea; CL:Claromecó; MH: Monte Hermoso; BB: Bahía Blanca; RN: Río Negro
Diversification and Biogeographic Patterns
The time-calibrated phylogenetic tree of haplotypes recovered four clades consistent with the four main haplogroups defined in the network (A, B, C, and E), and some low-frequency haplotypes found in FMAI, II, and III, related to clade E (Fig. 5). Despite the lack of reciprocal monophyly, those clades roughly correspond to the four original FMA described by Secchi et al. (2003b). The phylogenetic reconstruction indicates that the radiation of Pontoporia present-day lineages would have begun around 2.5 Ma, and the split between ESU North and ESU South would have happened around 1.8 Ma.

Bayesian phylogenetic tree of haplotypes of Pontoporia blainvillei based on the mitochondrial DNA control region. Capital letters refer to haplogroups. Red dashed lines delimit the clades. Posterior probability values above 0.5 are shown next to the nodes. Circles next to the haplotypes refer to Franciscana Management division labelled in b (FMAI: blue; FMAII: green; FMAIII: yellow; FMAIV: red). Median-joining networks next to the clades refer to P. blainvillei sample localities labelled in a. Time scale is in Million years (Ma), the horizontal bar on the axis represents in blue Pleistocene (2.8—0 Ma), in red Pliocene (5—2.8 Ma), and Miocene in green (25—5 Ma)
Of the six biogeographical models evaluated using BioGeoBears, the best-supported model was DIVAlike + J (AICc: 24.0, Table 5) which explicitly models founder events, narrow and widespread vicariance, and narrow sympatry (widespread and subset sympatry are not allowed in this model). The DIVAlike + J model indicates that all lineages shared a common ancestor before around 2.7 Ma, probably living in the area around the Río de La Plata Estuary (Fig. 6a, e). At approximately 2.7 Ma, the ancestral lineage split in two, one that would originate FMAII/FMAI (hereafter termed “northern group”), and the other would give rise to FMAIV/FMAIII (hereafter termed “southern group”). This first split is likely associated with the colonization of the northern area by the FMAII/FMAI lineage, and the spread of lineage FMAIII/IV in the southern area (Fig. 6a, f). The divergence between FMAIV and FMAIII and between FMAII and FMAI followed (Fig. 6a, g). Finally, the fragmentation of the populations within the FMAs would be the most recent event, taking place during Late Pleistocene (1–0.1 Ma, Fig. 6a).

Graphical summary of changes in the distribution of Pontoporia blainvillei major genetic lineages over time based on the best-fit model, DIVALIKE + J, from BioGeoBEARS analysis. Cladogram a at the top left shows the sequential order of splitting events in the nodes, and the cladogram b at the top right shows hypothetical haplotype frequencies in the nodes. Combinations of areas are indicated as 1 (pink): Inia geoffrensis + FMAIV + FMAIII; 2 (orange): FMAIV + FMAIII; 3 (ciano): FMAII + FMAI. Map c represents the current FMA division: blue: FMAI (ES: Espírito Santo and RJN: northern Rio de Janeiro); green: FMAII (RJS: southern Rio de Janeiro, SPN: northern São Paulo, SPC: central São Paulo, SPS: southern São Paulo, PR: Paraná, SCN: northern Santa Catarina and BAB: Babitonga bay); yellow: FMAIII (RS: Rio Grande do Sul, URAO: Atlantic Ocean of Uruguay and RP: Río de La Plata); red: FMAIV (SCL: San Clemente, NC: Necochea, CL: Claromecó, BB: Bahía Blanca, MH: Monte Hermoso and RN: Río Negro). Maps d-f and at the bottom indicate the sequential order of P. blainvillei geographic distribution following cladogram at combined areas 1, 2 and 3 until the current FMA subdivision
Discussion
We evaluated the genetic diversity, population structure and migration rates across the current two ESUs and four FMAs proposed for P. blainvillei. Through in-depth phylogeographic analyses, we estimated populations’ divergence times and inferred the influence of paleoceanographic events on the species biogeographic history. Phylogeographic and historical demographic analyses support the hypothesis that the species origin was in the Río de La Plata Estuary, with further dispersal south and northwards, followed by fragmentation. We reconstructed the species’ evolutionary history from the first lineage splitting, around 2.5–2.7 Ma. The lineage radiation probably occurred throughout the Pleistocene, and current-day populations were recently fragmented. Our analyses detected nine franciscana populations based on the mitochondrial DNA control region: ES (FMAIa), RJN (FMAIb), RJS + SPN (FMAIIa), SPC + SPS + PR (FMAIIb), SCN (FMAIIc), BAB (FMAIId), RS + URAO + RP + SCL + SB + NC + CL (FMAIII/IV), MH (FMAIV) and RN + BB (FMAIV). Results also suggest that the FMAIII and FMAIV populations have higher long-term migration rates between them and with the geographically closer populations from FMAII, and larger effective population sizes than northern populations, but most populations have negligible migration rates (i.e. less than one effective migrant female per generation). Considering that those estimates reflect historical patterns, including a long period of the species microevolutionary history when effective sizes were larger due to the lack of human interference, it is reasonable to suppose that contemporary migration rates are much smaller. In other words, current migration rates are insufficient to compensate for mortality rates in each population, and therefore, they need independent management.
Population Structure
Our population structure analyses agreed on several points with the macroscale study by Cunha et al. (2014), but noteworthy differences stand out. For instance, our re-analyses with the inclusion of new samples revealed new haplotypes from ES and RJN, which reinforced the paraphyly of the ESU North in relation to ESU South, formerly suggested by only one haplotype.
This is the first study based on mtDNA to find evidence of fine-scale genetic structure between FMAIII and IV and within FMAIV, with the finest geographic coverage provided to date. Cunha et al. (2014) detected five genetic populations: ES, RJN, RJS + SPN, SPC + SPS + PR + SCN, and RS + UR + AR, which would correspond to FMAIa, FMAIb, FMAIIa, FMAIIb, and FMAIII/IV. Their study was mainly based on mtDNA, but the authors took into consideration in their FMA proposal previous regional studies that used microsatellites and reported fine-scale structure within FMAIII and IV, as discussed below.
Additionally, the most likely scenario for AMOVA and pairwise ΦST analyses indicates that FMAII includes not only two but four genetically distinct populations. Besides SPN + RJS (FMAIIa) and SPC to PR (FMAIIb), which were previously suggested by Cunha et al. (2014), our macroscale analyses support the distinction between SCN (FMAIIc) and BAB (FMAIId). Therefore, our data corroborate the lack of panmixia in the area from the south of RJ to the north of SC, as previously suggested by Cunha et al. (2014), and recently confirmed with the increase in the number of samples from RJS (Cunha et al. 2020b), but also show a greater level of population fragmentation, in which SCN appears as a different population in relation to SPC + SPS + PR. Also, population BAB comprises an isolated group of franciscanas restricted to an estuarine area, the Babitonga Bay, characterized by calm and shallow waters free from potential predators as sharks and killer whales. This is possibly the most threatened local population, given its small size and the intense human activity, related mainly to harbour development and fishing gillnets, that are major threats to the species (Cremer and Simões-Lopes 2005, 2008). The population differentiation of franciscanas from Babitonga Bay (BAB) in relation to coastal areas in northern Santa Catarina (SCN) was already verified using mitochondrial data and microsatellites (Cunha et al. 2020a). However, the fact that BAB was detected as a unique population in the macro-scale analyses presented here emphasizes the need to preserve this small resident population. Using mtDNA and microsatellites, Costa-Urrutía et al. (2012) found similar evidence between franciscanas from the Río de La Plata Estuary and Samborombon Bay. These scenarios corroborate the hypothesis that environmental discontinuities led to franciscanas’ population fragmentation (Mendez et al. 2010) and highlight the essential role of estuarine habitats in this process.
Concerning the populations from both sides of the Río de La Plata Estuary, our analyses provide evidence of genetic differentiation between FMAIII and FMAIV from AMOVA and ΦST analyses. However, we could not find evidence of a subdivision where the current limit between the two FMA is settled, in the Río de La Plata Estuary. In fact, our analyses cannot reject panmixia in the area from RS to NC, which include FMAIII and FMAIV. The differentiation between FMAIII and FMAIV is supported by external morphology analyses (Barbato et al. 2012), infection levels, diet composition (Secchi et al. 2003b), and haplotype frequencies in microscale studies (Lazaro et al. 2004; Mendez et al. 2008).
In FMAIV our findings corroborate Gariboldi et al. (2016) where they found the same division (MH and BB + RN) not only with mtDNA but also with microsatellites. Further differentiation within FMAIV was reported by Mendez et al. (2008, 2010) based on analyses of both mtDNA and microsatellites. We were not able to use Mendez et al. (2010) sequences in our population structure analyses due to the lack of information regarding samples’ localities. However, the sampling localities from which no identified sequence was available to us were Cabo Santo Antonio (CSA), East of Buenos Aires (BAE), and Southwest of Buenos Aires (BA-SW). Thus, FMAIVb was the only locality previously identified as a unique population that we did not include in our analyses. If we consider the microscale analyses from Mendez et al. (2010), Costa-Urrutia et al. (2012), and Gariboldi et al. (2015, 2016), franciscanas are probably divided into 12 populations (ES, RJN, RJS + SPN, SPC + SPS + PR, SC, BAB, RS + URAO + RP, SCL, CSA + BAE, NC + CL, MH, and RN), of which ES is the genetically most differentiated (Online Resource 2, Table 2). But since our aim here was to investigate the phylogeography of franciscanas, and our resolution was limited to that provided by the mtDNA control region, we adopted the nine population-scenario that we could detect using our data.
Mitochondrial DNA analyses reflect evolutionary events that took place before some more recent fragmentation episodes that are also relevant for species conservation, and that can be detected, for instance, using microsatellites. So finer genetic differentiation assessed in regional studies must not be neglected, because it provides evidence that the detected populations act as independent demographic units. Genetic divisions that are detectable at macro-scale mtDNA analyses must be regarded as a minimum population structure.
It should be noted that our population structure results may have been overestimated because sampling is not spatially continuous and some areas have small sample sizes. However, we should acknowledge that satellite-tagging records suggest that franciscanas movements are limited to 70–90 km (Bordino et al. 2008; Wells et al. 2013), supporting a scenario of fine-scale genetic differentiation.
Finally, our results refer only to female population structure because mtDNA is maternally inherited. But two aspects deserve consideration. First, from the conservation standpoint, the delimitation of populations (or Management Units) should be based on mtDNA when females are philopatric (Avise 1995; Dizon et al. 1997), which seems to be the case in franciscanas (Costa-Urrutia et al. 2012; Cunha et al. 2020a). This reasoning makes sense because if females do not disperse, they would not recolonize a locally extirpated population. Thus, each genetically differentiated female population must be protected. And secondly, although the use of nuclear DNA for all areas along the species’ distribution is recommended, studies at a regional scale that used both mtDNA and microsatellites found coincident results between the two types of markers (Costa-Urrutia et al. 2012; Gariboldi et al. 2016).
Phylogeography: Timing and Geological Setting of the Micro-evolution of Franciscanas
The phylogenetic reconstruction of franciscana lineages based on mtDNA (Fig. 5) reflects somewhat ancient divergences, but those lineages are probably not old enough to have achieved reciprocal monophyly. Thus, in the northern portion of the species distribution, where population effective sizes have been smaller, lineage sorting was probably more efficient and the tree agrees well with the FMA division described by Secchi et al. (2003b). On the other hand, in the south, where effective sizes and migration rates have been larger, lineages have not reached reciprocal monophyly. This is unsurprising in studies dealing with microevolution.
Despite this limitation, the phylogenetic reconstruction provided the opportunity to date some splitting events, offering a timeframe for the interpretation of franciscana’s phylogeography. Dating estimates suggest that the first divergence happened around 2.7–2.5 Ma. It separated one lineage that would survive in all FMA (clades A, B, and C), and another one that is currently not found in FMAI (D and clade E). The first lineage would split around 2.0–1.5 Ma into clades A, B, and C, which roughly correspond to FMAI, FMAII, and FMAIII/IV, respectively. More recent fragmentation events cannot be detected in the phylogenetic tree or haplotype network, but are clearly shown in population structure results, as we discussed above.
The analysis using BioGeoBEARS shed more light on the species’ past. This analysis is suited for intraspecific differentiation and incorporates distribution data to genetic information. Considering the nine-population scenario detected in our population structure analyses, the best model indicated by BioGeoBEARS shows a first split dated at around 2.7 Ma (Fig. 6a). In the oldest inferred microevolutionary event, the ancestral population, which lived in the Río de La Plata Estuary, diverged into two populations. One of them would originate FMAIII and FMAIV populations in the south (“southern group”), and the other would originate FMAII and FMAI in the north (“northern group”). This split was probably related to dispersal followed by long-distance isolation or another phenomenon that led to restrictions to gene flow between the ancestral population and the group that first dispersed northwards. The next split in this lineage (around 0.5 Ma) would have been between a population that was ancestral to both ES and RJN populations (FMAI) and the other population from the “northern group” (FMAII). The most recent divergence was between populations within FMAII and FMAI, at 1.0–0.1 Ma.
The “southern group” also split into two lineages, around 2.0–1.75 Ma, one that today includes franciscanas distributed between the coast of RS and SCL (FMAIII and FMAIV), and the other that gave rise to the present-day populations found at the species’ extreme south reaches (RN + BB and MH, around 1.5–1.0 Ma). This suggests that franciscanas had already arrived at their present-day southern distribution limit at that time.
The inferred timings of the observed divergence events reinforce the notion that the phylogeography of franciscanas was influenced by Pleistocene paleoceanographic events. During the Pleistocene, a minimum of seven glaciations took place and influenced not only the currents but the sea level (lowering up to 100-140 m) and temperature in the South Atlantic Ocean (Rabassa et al. 2005). As a result, Pleistocene glacial cycles resulted in a reduction of habitable area and had a significant impact on coastal marine life (Ludt and Rocha 2015). Pleistocene sea level fluctuations occurred around every 41–100 thousand years (kyr) as a result of changes in climate cycles and were intercalated with higher temperature and sea-level periods lasting around 10 kyr (Elderfield et al. 2012). Thus, it was probably a time of repeated separation (during low sea levels), and mixing (during high sea levels) for marine populations (Davies 1963; Ludt and Rocha 2015).
Habitat preferences are strongly correlated to how species responded to changes during the Pleistocene (Ludt and Rocha 2015). The split date of FMAI/FMAII and FMAIII/FMAIV corresponds to the early Pleistocene when the Quaternary glaciations began. In the Western South Atlantic, the encounter of southward flowing Brazil Current and northward-flowing Malvinas/Falkland Current, known as the Brazil-Falklands/Malvinas Confluence, is usually close to the Río de La Plata region, being responsible for high primary productivity in the area. But data indicate that during the Pleistocene glacial periods the Brazil-Falklands/Malvinas Confluence shifted to the north (Gartner 1988; Rabassa et al. 2005; Gu et al. 2019). Assuming that the high productivity in the Río de La Plata Estuary was important to sustain the largest and oldest franciscana population up to the present, we suppose that franciscanas may have dispersed northwards following the changes in the primary productivity as the Brazil-Falklands/Malvinas Confluence was displaced.
In addition, studies in the Coastal Plain of Rio Grande do Sul found fossil records of Pontoporia in the Barrier-Lagoon System III (Ribeiro et al. 1998) and Holocene barrier IV (Cruz et al. 2017). These findings indicate the presence of the Pontoporia in this region during transgressive events of interglacial periods (Ribeiro et al. 1998; Cruz et al. 2017). Besides Pontoporia, the most common marine fossils registered by Cruz et al. (2017) were from Sciaenidae, Teleostei fishes important to the franciscana diet (Tellechea et al. 2017; Henning et al. 2018). Therefore, it is likely that the productivity in southern Brazil coastal lagoons influenced the species dispersion to this area and thus northwards.
Our results also help to explain the pattern of morphological differentiation detected by Pinedo (1991), who identified two franciscana morphotypes, a large form that ranges from Argentina to the Rio Grande do Sul and a smaller one that occurs from north of Santa Catarina to Rio de Janeiro. The Cape of Santa Marta, located in the south of the Santa Catarina coast, might have been a barrier to gene flow since it is responsible for deflecting Malvinas Current offshore (Peterson and Stramma 1991; Martins et al. 2021). Besides, from the Cape of Santa Marta to the south of RS, estuaries are intercalated with open sea areas that also might be a barrier to gene flow (Martins et al. 2021). According to our results, the differentiation between the larger southern form and the small northern form probably coincides with the split of FMAI/FMAII and FMAIII/FMAIV during the early Pleistocene after franciscanas dispersed northwards occupying the area from north of Santa Catarina to Espírito Santo.
Eventually, during episodes of sea level lowering, some groups may have become isolated in estuarine/coastal habitats related to the paleodrainages. In this context, it is crucial to consider that franciscanas not only have a coastal habit, being rarely seen in depths over 30m, but are also frequently related to estuaries. The reconstruction of the coast and the continental shelf during the Pleistocene glacial periods (with a sea level of -125 m; Fig. 2) suggests that habitat contraction was a critical factor influencing the phylogeography of franciscanas. During glacial periods the available habitat (bordered by the 30 m isobath in Fig. 4) was restricted to a narrow strip from RJ to RS. During these periods franciscanas probably concentrated in this area, creating the opportunity for secondary contact between the two more ancient franciscana lineages, which could explain the existence of shared haplotypes among FMAII, III, and IV.
The coastal region from SCN to RJS seems to be an area with few barriers to gene flow and higher panmixia in many marine taxa. Data indicate that most tropical species that live in this area have occupied it recently, probably because of the gradual warming after the last glacial maxima (Martins et al. 2021). The few exceptions to this pattern are the species that are not exclusively tropical, as P. blainvillei (Martins et al. 2021). As our data indicate, franciscanas were seemingly able to colonize this region earlier, during the Pleistocene. Later, franciscana populations became fragmented possibly due to environmental/ecological differences, which may be reflected, for instance, in differences in diet found across this region (Henning et al. 2018).
On the other hand, populations in both sides of this central area, i.e. from RJN to ES, and southwards from RS to RN, would not have had suitable habitat during glacial periods, since the depth in these areas reached 1000 m very close to shore. Thus, three possibilities exist for each of those populations: they may have been colonized after the last glacial period, they may have been extirpated (and recolonized later), or they may have persisted in small numbers in refugia, such as the mouth of larger rivers. This latter hypothesis is possible because paleodrainage reconstruction shows that most of these areas had at least one large river mouth, as shown in Fig. 4. Marine estuaries have been proposed to have acted as glacial refugia for coastal species during the Pleistocene, which would have resulted in marine-estuarine endemism at a local level (García 2012). We propose that the same phenomenon may have shaped intraspecific differentiation in franciscanas. Furthermore, the population at the northern extreme (ES) had the lowest genetic diversity, long-term migration rates, and ancestral effective population size. Even in periods of higher sea level the ES population probably had limited contact with nearby populations.
It is important to be aware that genealogies based on a single locus are subject to stochasticity in the lineage divergence process. Considering that each locus is an independent replicate of the coalescent process, maximizing the number of loci would increase the accuracy of those estimates (Arbogast et al. 2002).
Final Considerations
In summary, we suggest that P. blainvillei habitat preference for estuaries and shallower waters was probably the principal driver on population dispersal and contraction cycles. The fragmentation that led to current populations would have occurred during Pleistocene paleoceanographic events such as sea level fluctuations.
Even though our population structure analysis has limitations, such as lack or small sampling in some areas and being based in a single mtDNA locus, it is the most geographically comprehensive analysis conducted to date. Thus, the population structure of franciscanas still needs further investigation improving sampling size and geographic coverage and incorporating more molecular markers. However, our findings highlight the importance of considering that the franciscana FMA division originally proposed by Secchi et al. (2003b) probably contemplates early divergence events that took place more than 1.8 Ma. Analyses that assess population divergence in more recent times need to be taken into account in conservation programs aimed at the regional level because those analyses are more closely related to the species’ present-day demography. In this regard, besides the fine-scale divisions already proposed by previous genetic studies (Mendez et al. 2008, 2010; Costa-Urrutia et al. 2012; Cunha et al. 2014; Gariboldi et al. 2015; 2016; Cunha et al. 2014, 2020a, b) we recommend north of Santa Catarina (FMAIIc) and Babitonga bay (FMAIId) to be considered unique populations.
Data Availability
DNA sequences used in the current study are available in the GenBank repository. New haplotype sequences generated during this study are deposited with the accession codes OK188832, OK188833, OK188834 and OK188835. The authors declare that all data supporting the findings of this study are available within the article [and its supplementary information files].
References
Arbogast BS, Edwards SV, Wakeley J, Beerli P, Slowinski JB (2002) Estimating divergence times from molecular data on phylogenetic and population genetic timescales. Annu Rev Ecol Syst 33:707-740. https://doi.org/10.1146/annurev.ecolsys.33.010802.150500
Alonso MB, Eljarrat E, Gorga M, Secchi ER, Bassoi M, Barbosa L, Bertozzi CP, Marigo J, Cremer M, Domit C, Azevedo AF, Dorneles PR, Torres JPM, Lailson-Brito J, Malm O, Barceló D (2012) Natural and anthropogenically-produced brominated compounds in endemic dolphins from Western South Atlantic: Another risk to a vulnerable species. Environ Pollut 170:152–160. https://doi.org/10.1016/j.envpol.2012.06.001
Avise JC (1995) Mitochondrial-DNA polymorphism and a connection between genetics and demography of relevance to conservation. Conserv Biol 9:686-690
Barbato BHA, Secchi ER, Di Beneditto APM, Ramos RMA, Bertozzi C, Marigo J, Bordino P, Kinas PG (2012) Geographical variation in franciscana (Pontoporia blainvillei) external morphology. J Mar Biol Assoc UK 92:1645–1656. https://doi.org/10.1017/S0025315411000725
Bastida R, Rodríguez D, Secchi ER, da Silva VMF (2007) Mamíferos Acuáticos de Sudamérica y Antártida. Vázquez Manzini Editores, Buenos Aires, Argentina
Beerli P (2006) Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22:341-345. https://doi.org/10.1093/bioinformatics/bti803
Bordino P, Wells R, Stamper S, Andrew M (2008) Satellite tracking of franciscana dolphins Pontoporia blainvillei in Argentina: preliminary information on ranging, diving and social patterns. IWC Scientific Committee Meeting. SC60/SM14.
Cassens I, Vicario S, Waddell VG, Balchowsky H, Belle D Van, Ding W, Fan C, Mohan RSL, Simões-Lopes PC, Bastida R, Meyer A, Stanhope MJ, Milinkovitch MC (2000) Independent adaptation to riverine habitats allowed survival of ancient cetacean lineages. Proc Nat Acad Sci USA 97:11343–11347. https://doi.org/10.1073/pnas.97.21.11343
Costa-Urrutia P, Abud C, Secchi ER, Lessa EP (2012) Population genetic structure and social kin associations of franciscana dolphin, Pontoporia blainvillei. J Hered 103:92–102. https://doi.org/10.1093/jhered/esr103
Cremer MJ, Simões-Lopes PC (2005) The occurrence of Pontoporia blainvillei (Gervais & d’Orbigny) (Cetacea, Pontoporiidae) in an estuarine area in southern Brazil. Rev Bras Zool 22:717–723. https://doi.org/10.1590/S0101-81752005000300032
Cremer MJ, Simões-Lopes PC (2008) Distribution, abundance and density estimates of franciscanas, Pontoporia blainvillei (Cetacea: Pontoporiidae), in Babitonga bay, southern Brazil. Rev Bras Zool 25:397–402. https://doi.org/10.1590/S0101-81752008000300003
Crespo EA, Pedraza SN, Grandi MF, Dans SL, Garaffo GV (2010) Abundance and distribution of endangered Franciscana dolphins in Argentine waters and conservation implications. Mar Mamm Sci 26:17–35. https://doi.org/10.1111/j.1748-7692.2009.00313.x
Cruz EA, Dillenburg SR, Buchmann FS (2017) Description and controls on distribution of Pleistocene vertebrate fossils from the central and southern sectors of the Coastal Plain of Rio Grande do Sul, Brazil. Rev Bras Paleontolog 19:425–438. https://doi.org/10.4072/rbp.2016.3.08
Cunha HA, Medeiros BV, Barbosa LA, Cremer MJ, Marigo J, Lailson-Brito J, Azevedo AF, Solé-Cava AM (2014) Population structure of the endangered franciscana dolphin (Pontoporia blainvillei): Reassessing management units. PloS One 9:e85633. https://doi.org/10.1371/journal.pone.0085633
Cunha HA, Dias CP, Alvarenga LC, Wells RS, Cremer MJ (2020a) Microscale population structure and kinship analyses suggest philopatry of both sexes in franciscanas (Pontoporia blainvillei). IWC Scientific Report. SC/68B/SDDNA/04
Cunha HA, Bisi TL, Bertozzi CP, Dias CP, Santos-Neto EB, Manhães BMR, Oliveira-Ferreira N, Montanini GN, Ikeda J, Carvalho RR, Azevedo AF, Lailson-Brito J (2020b) Population differentiation of endangered franciscanas in Southeastern Brazil: new genetic, contaminant and stable isotope data support subdivision of FMAII. IWC Scientific Committee. SC/68B/SDD 1–10
Danilewicz D, Secchi ER, Ott PH, Moreno IB, Bassoi M, Borges-Martins M (2009) Habitat use patterns of franciscana dolphins (Pontoporia blainvillei) off southern Brazil in relation to water depth. J Mar Biol Assoc UK 89:943–949. https://doi.org/10.1017/S002531540900054X
Davies JL (1963) The antitropical factor in cetacean speciation. Evolution 17:107–116
de Oliveira VKM, Faria DM, Cunha HA, dos Santos TEC, Colosio AC, Barbosa LA, Freire MCC, Farro APC (2020) Low genetic diversity of the endangered franciscana (Pontoporia blainvillei) in its northernmost, isolated population (FMAIa, Espírito Santo, Brazil). Front Mar Sci 7:608276. https://doi.org/10.3389/fmars.2020.608276
Dizon AE, Perrin WF, Amos W, Baker CS, Chivers SJ, Costa AS, Curry BE, Gaggiotti O, Hoelzel AR, Hofman R, LeDuc RG, Loughlin TR, Lux CA, O’Corry-Crowe GM, Rosel PE, Rosenberg A, Scribner KT, Taylor BL (1997) Report of the Workshop In: Dizon AE, Chivers SJ, Perrin, WF (eds) Molecular Genetics of Marine Mammals. The Society for Marine Mammalogy, Lawrence, Kansas, pp 3-48
Donovan GP, Bjørge A (1995) Dall’s porpoise, Phocoenoides dalli - Introductory remarks. In: Bjørge A, Donovan GP (eds) International Whaling Commission Special Issue, pp 378–380
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1970. https://doi.org/10.1093/molbev/mss075
Elderfield H, Ferretti P, Greaves M, Crowhurst S, McCave IN, Hodell D, Piotrowski AM (2012) Evolution of Ocean Temperature and Ice Volume Through the Mid-Pleistocene Climate Transition. Science 337:704–709. https://doi.org/10.1126/science.1221294
Environmental Systems Research Institute (2020) ArcGIS Pro. Redlands, California
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Gago-Ferrero P, Alonso MB, Bertozzi CP, Marigo J, Barbosa L, Cremer M, Secchi ER, Azevedo A, Jr. JLB, Torres JPM, Malm O, Eljarrat E, Díaz-Cruz MS, Barceló D (2013) First determination of UV filters in marine mammals. Octocrylene levels in franciscana dolphins. Envir Sci Tec 47:5619–5625. https://doi.org/10.1021/es400675y
García G (2012) Phylogeography from south-western Atlantic Ocean: Challenges for the Southern Hemisphere. In: Anamthawat-Jónsson, K (ed) Current Topics in Phylogenetics and Phylogeography of Terrestrial and Aquatic Systems, InTech Open, London, pp 13–32. https://doi.org/10.5772/36390
Gariboldi MC, Túnez JI, Dejean CB, Failla M, Vitullo AD, Negri MF, Cappozzo HL (2015) Population genetics of franciscana dolphins (Pontoporia blainvillei): Introducing a new population from the southern edge of their distribution. PLoS One 10:e0132854. https://doi.org/10.1371/journal.pone.0132854
Gariboldi MC, Túnez JI, Failla M, Hevia M, Panebianco MV, Paso Viola MN, Vitullo AD, Cappozzo HL (2016) Patterns of population structure at microsatellite and mitochondrial DNA markers in the franciscana dolphin (Pontoporia blainvillei). Ecol Evol 6:8764–8776. https://doi.org/10.1002/ece3.2596
Gartner S (1988) Paleoceanography of the mid-Pleistocene. Mar Micropaleontol 13:23–46
Gu F, Chiessi CM, Zonneveld KAF, Behling H (2019) Shifts of the Brazil-Falklands/Malvinas Confluence in the western South Atlantic during the latest Pleistocene–Holocene inferred from dinoflagellate cysts. Palynology 43:483–493. https://doi.org/10.1080/01916122.2018.1470116
Hamilton H, Caballero S, Collins AG, Brownell RL (2001) Evolution of river dolphins. Proc Roy Soc B - Biol Sci 268:549–556. https://doi.org/10.1098/rspb.2000.1385
Henning B, Carvalho BS, Pires MM, Bassoi M, Marigo J, Bertozzi C, Araújo MS (2018) Geographical and intrapopulation variation in the diet of a threatened marine predator, Pontoporia blainvillei (Cetacea). Biotropica 50:157–168. https://doi.org/10.1111/btp.12503
Hickerson M, Carstens BC, Crandall KA (2009) Phylogeography’s past, present, and future: 10 years after Avise, 2000. Mol phylogenet evol 54:291-301. https://doi.org/10.1016/j.ympev.2009.09.016
Hoelzel ARR, Hancock JMM, Dover GAA (1991) Evolution of the cetacean mitochondrial D-Loop region. Mol Biol Evol 8:475–493.
Instituto Chico Mendes de Conservação da Biodiversidade (2018) Livro Vermelho da Fauna Brasileira Ameaçada de Extinção: Volume II - Mamíferos. Livro Vermelho da Fauna Brasileira Ameaçada de Extinção, Brasília, Brazil, pp. 143–148
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A, Valencia A (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Lailson-Brito J, Dorneles PR, Azevedo-Silva CE, Azevedo A de F, Vidal LG, Marigo J, Bertozzi C, Zanelatto RC, Bisi TL, Malm O, Torres JPM (2011) Organochlorine concentrations in franciscana dolphins, Pontoporia blainvillei, from Brazilian waters. Chemosphere 84:882–887. https://doi.org/10.1016/j.chemosphere.2011.06.018
Landis MJ, Matzke NJ, Moore BR, Huelsenbeck JP (2013) Bayesian analysis of biogeography when the number of areas is large. Syst Biol 62:789–804. https://doi.org/10.1093/sysbio/syt040
Lazaro M, Lessa EP, Hamilton H (2004) Geographic genetic structure in the franciscana dolphin (Pontoporia blainvillei). Mar Mamm Sci 20:201–214. https://doi.org/10.1111/j.1748-7692.2004.tb01151.x
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110-1116. https://doi.org/10.1111/2041-210X.12410
Ludt WB, Rocha LA (2015) Shifting seas: The impacts of Pleistocene sea-level fluctuations on the evolution of tropical marine taxa. J Biogeogr 42:25–38. https://doi.org/10.1111/jbi.12416
Martins N, Macagnan L, Cassano V, Gurgel C (2021) Barriers to gene flow along the Brazilian coast: a synthesis and data analysis. Authorea Preprints. https://doi.org/10.22541/au.161358135.51187023/v1
Matzke NJ (2013a) BioGeoBEARS: Biogeography with Bayesian (and likelihood) evolutionary analysis in R scripts. http://CRAN.R-project.org/package=BioGeoBEARS
Matzke NJ (2013b) Probabilistic historical biogeography: new models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Front Biogeogr 5:242–248. https://doi.org/10.21425/F5FBG19694
Matzke NJ (2014) Model selection in historical biogeography reveals that founder-event speciation is a crucial process in island clades. Syst Biol 63:951–970. https://doi.org/10.1093/sysbio/syu056
Mendez M, Rosenbaum HC, Bordino P (2008) Conservation genetics of the franciscana dolphin in Northern Argentina: Population structure, by-catch impacts, and management implications. Conserv Genet 9:419–435. https://doi.org/10.1007/s10592-007-9354-7
Mendez M, Rosenbaum HC, Subramaniam A, Yackulic C, Bordino P (2010) Isolation by environmental distance in mobile marine species: Molecular ecology of franciscana dolphins at their southern range. Mol Ecol 19:2212–2228. https://doi.org/10.1111/j.1365-294X.2010.04647.x
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 2010 Gateway Computing Environments Workshop. pp 1-8.
Negri MF, Denuncio P, Panebianco MV, Cappozzo HL (2012) Bycatch of franciscana dolphins Pontoporia blainvillei and the dynamic of artisanal fisheries in the species’ southernmost area of distribution. Braz J Oceanogr 60:149–158
Peterson RG, Stramma L (1991) Upper-level circulation in the South Atlantic Ocean. Progr Oceanogr 26: 1–73
Pinedo MC (1991) Development and variation of the franciscana Pontoporia blainvillei. Unpublished thesis, University of California, Santa Cruz, USA
Posada D (2008) jModelTest: Phylogenetic model averaging. Mol Biol Evol 25:1253–1256. https://doi.org/10.1093/molbev/msn083
R Development Core Team R (2021) R: A Language and Environment for Statistical Computing (RDC Team, Ed.). R Foundation for Statistical Computing 1:409. http://www.R-project.org/
Rabassa J, Coronato AM, Salemme M (2005) Chronology of the Late Cenozoic Patagonian glaciations and their correlation with biostratigraphic units of the Pampean region (Argentina). J S Am Earth Sci 20:81–103. https://doi.org/10.1016/j.jsames.2005.07.004
Ree RH, Smith SA (2008) Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst Biol 57:4–14. https://doi.org/10.1080/10635150701883881
Ribeiro AM, Drehmer C, Buchmann FS (1998) Pleistocene skull remains of Pontoporia blainvillei (Cetacea, Pontoporiidae) from the coastal plain of Rio Grande do Sul State, Brazil, and the relationships of pontoporids. Rev UnG Geocienc 3:71–77
Ritchie AM, Lo N, Ho SYW (2016) The impact of the tree prior on molecular dating of data sets containing a mixture of inter- and intraspecies sampling. Syst Biol 66:413–425. https://doi.org/10.1093/sysbio/syw095
Ronquist F, Sanmartín I (2011) Phylogenetic methods in biogeography. Annu Rev Ecol Syst 42:441-464. https://doi.org/10.1146/annurev-ecolsys-102209-144710.
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: a Laboratory Manual. Cold Spring Harbor Laboratory Press, New York
Santos EV (2011) Estrutura populacional e história filogeográfica da toninha (Pontoporia blainvillei). Dissertation, Pontifícia Universidade Católica do Rio Grande do Sul
Secchi ER, Ott PH, Danilewicz D (2003a) Effects of fishing bycatch and the conservation status of the franciscana dolphin, Pontoporia blainvillei In: Gales N Hindell MKR (eds) Marine Mammals: Fisheries, Tourism, and Management Issues. CSIRO Publishing Collingwood, Australia, pp 174-191
Secchi ER, Danilewicz D, Ott PH (2003b) Applying the phylogeographic concept to identify franciscana dolphin stocks: implications to meet management objectives. J Cetacean Res Manag 5:61-68
Secchi ER, Secchi ER, Cremer MJ, Danilewicz D, Lailson-Brito J (2021) A synthesis on the ecology, human-related threats and conservation perspectives for the endangered franciscana dolphin. Front Mar Sci 8:617956. https://doi.org/10.3389/fmars.2021.617956
Sherry ST, Roger SAR (1994) Mismatch distributions of mtDNA reveal recent human population expansions. Hum Biol 5:761-775
Swindell SR, Plasterer TN (1997) SEQMAN. Sequence data analysis guidebook:75–89
Tellechea JS, Perez W, Olsson D, Lima M, Norbis W (2017) Feeding habits of franciscana dolphins (Pontoporia blainvillei): Echolocation or passive listening? Aquat Mamm 43:430–438. https://doi.org/10.1578/AM.43.4.2017.430
Thomaz AT, Knowles LL (2018) Flowing into the unknown: Inferred paleodrainages for studying the ichthyofauna of brazilian coastal rivers. Neotrop Ichthyol 16:1–13. https://doi.org/10.1590/1982-0224-20180019
Thomaz AT, Malabarba LR, Knowles LL (2017) Genomic signatures of paleodrainages in a freshwater fish along the southeastern coast of Brazil: Genetic structure reflects past riverine properties. Heredity 119:287–294. https://doi.org/10.1038/hdy.2017.46
Von Ihering H (1927) Die Geschichte des Atlantischen Ozeans. Gustav Fisher, Jena, Germany
Wells RS, Bordino P, Douglas DC (2013) Patterns of social association in the franciscana, Pontoporia blainvillei. Mar Mammal Sci 29:E520–E528. https://doi.org/10.1111/mms.12010
Zerbini AN, Secchi E, Crespo E, Danilewicz D, Reeves R (2017) Pontoporia blainvillei (errata version published in 2018). The IUCN Red List of Threatened Species eT17978A123792204. https://doi.org/10.2305/IUCN.UK.2017-3.RLTS.T17978A50371075.en
Acknowledgements
This article is part of the Ph.D. requirements at Programa de Pós Graduação em Biodiversidade e Biologia Evolutiva of the Universidade Federal do Rio de Janeiro. A previous version of this manuscript has been reviewed by C.G. Schrago (Universidade Federal do Rio de Janeiro), F. Henning (Universidade Federal do Rio de Janeiro), and S.M.Q. Lima (Universidade Federal do Rio Grande do Norte), to whom the authors are grateful for their suggestions. The authors also thank P. Beerli (Florida State University) for all his inputs with Migrate analysis, N. Matzke for advice about BioGeoBEARS, and A. Thomaz (University of Michigan) for her contribution regarding the paleodrainages reconstructions. This study was made possible by the support of the Organization for the Conservation of South American Aquatic Mammals—YAQU PACHA e.V., Nuremberg Zoo (Germany). This study was part of project Conservação da Toninha, which is an environmental offset measure established through a Consent Decree/Conduct Adjustment Agreement between PetroRio and the Brazilian Ministry for the Environment.
Funding
Conselho Nacional de Desenvolvimento Científico e Tecnológico, PQ 313577/2020–0, Marta J Cremer, 310597/2018–8, Eduardo R Secchi; Fundo Brasileiro para a Biodiversidade, Edital 01/2016 Projeto 081/2016; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, 88882.425727/2019–01, Luana Nara
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below are links to the electronic supplementary material.
10914_2022_9607_MOESM1_ESM.pdf
Supplementary file1 (PDF 164 KB) Online resource 1: Frequency of occurrence and sampling sites of the mtDNA control region haplotypes used in this study
10914_2022_9607_MOESM2_ESM.pdf
Supplementary file2 (PDF 198 KB) Online resource 2: AMOVA results of all population structure scenarios tested for Pontoporia blainvillei, considering all sampling localities, compared to scenarios proposed previously.
10914_2022_9607_MOESM3_ESM.pdf
Supplementary file3 (PDF 71 KB) Online resource 3: Mantel test based on Pontoporia blainvillei control region sequences
Rights and permissions
About this article

Cite this article
Nara, L., Cremer, M.J., Farro, A.P.C. et al. Phylogeography of the Endangered Franciscana Dolphin: Timing and Geological Setting of the Evolution of Populations. J Mammal Evol 29, 609–625 (2022). https://doi.org/10.1007/s10914-022-09607-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10914-022-09607-7