
ABSTRACT
Primary tumors often give rise to disseminated tumor cells (DTC’s), which acquire full malignancy after invading distant site(s). Thus, DTC’s may be a productive target for preventing prostate cancer metastasis progression. Our prior research showed that PHSCN peptide (Ac-PHSCN-NH2) targets activated α5β1 integrin to prevent invasion and metastasis in preclinical adenocarcinoma models, and disease progression in Phase I clinical trial. Here, we report that d-stereoisomer replacement of histidine and cysteine in PHSCN produces a highly potent derivative, Ac-PhScN-NH2 (PhScN). PhScN was 27,000- to 150,000-fold more potent as an inhibitor of basement membrane invasion by DU 145 and PC-3 prostate cancer cells. A large increase in invasion–inhibitory potency occurred after covalent modification of the sulfhydryl group in PHSCN to prevent disulfide bond formation; while the potency of covalently modified PhScN was not significantly increased. Thus PhScN and PHSCN invasion inhibition occurs by a noncovalent mechanism. These peptides also displayed similar cell surface binding dissociation constants (Kd), and competed for the same site. Consistent with its increased invasion–inhibitory potency, PhScN was also a highly potent inhibitor of lung extravasation and colonization in athymic nude mice: it was several hundred- or several thousand-fold more potent than PHSCN at blocking extravasation by PC-3 or DU 145 cells, and 111,000- or 379,000-fold more potent at inhibiting lung colonization, respectively. Furthermore, systemic 5 mg/kg PhScN monotherapy was sufficient to cause complete regression of established, intramuscular DU 145 tumors. PhScN thus represents a potent new family of therapeutic agents targeting metastasis by DTC’s to prevent parallel progression in prostate cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung metastasis is found in 40–49 % of metastatic prostate cancer patients at autopsy, with higher rates when multiple organs contain metastases [1, 2]. Invasion is a key process in metastatic progression because it enables tumor cells to spread into the tissue surrounding the primary tumor, enter the circulatory or lymphatic systems, and extravasate at distant sites [3, 4]. Subsequently, microvascular endothelial cell invasion of metastatic nodules supports their growth by promoting angiogenesis [5]. The α5β1 integrin fibronectin receptor has been shown to play a key role in both metastatic invasion during extravasation, and angiogenic invasion [3–5]. Hence, inhibiting the function of α5β1 integrin may inhibit extravasation to prevent lung colony formation, as well as reducing subsequent metastatic progression. The availability of a potent, nontoxic inhibitor of α5β1 integrin–mediated invasion could thus greatly improve outcome in patients with metastatic prostate cancer.
We previously devised the acetylated, amidated PHSCN peptide (Ac-PHSCN-NH2), as a potent inhibitor of α5β1 integrin–mediated invasion by suspended prostate adenocarcinoma, and microvascular endothelial cells [3, 5]. It was licensed as ATN-161, and its efficacy confirmed by others for a variety of cancers [6–10]. In Phase I clinical trial, ATN-161 was well tolerated; and about one-third of patients receiving thrice–weekly, systemic ATN-161 monotherapy, at dosages of 0.5–16 mg/kg, exhibited prolonged stable disease [11].
Here, we present data showing that substitution of d-isomers of histidine and cysteine in PHSCN, forming Ac-PhScN-NH2 (PhScN), increases its in vitro basement membrane invasion–inhibitory potency, relative to PHSCN, by many orders of magnitude: 27,000- and 150,000-fold, for DU 145 and PC-3 cells, respectively. We also found that PHSCN derivatives incapable of disulfide bond formation [S-acetylated PHSC(S-OAc)N, S-methylated PHSC(S-Me)N, and S-acetamidomethylated PHSC(S-acm)N] have similarly increased potencies. Moreover, while these covalently modified PHSCN derivatives have greatly increased invasion–inhibitory potencies, relative to PHSCN, the covalently modified PhSc(S-acm) N peptide had an insignificant increase in potency, relative to PhScN. Hence, the productive mechanism is noncovalent and disulfide bond formation between PHSCN and its integrin target is a side reaction that significantly reduces its potency.
In order to predict therapy response two fundamental models of metastasis have been proposed, the linear progression and the parallel progression models [12]. In the linear progression model, tumor ontogeny proceeds to full malignancy within the microenvironment of the primary tumor, after which metastatic dissemination occurs. In the parallel progression model, the primary tumor continuously gives rise to DTC’s, which acquire full malignancy by somatic progression and metastatic growth at distant site(s). Thus, DTC’s may be a productive target for preventing metastasis progression.
Since established tumors may constantly give rise to DTC’s which intravasate, later extravasating to colonize the lung, we confirmed the necessity of α5β1 integrin function for lung extravasation by DU 145 and PC-3 cells. Next, we investigated the efficacy of a single exposure to PhScN on suspended DU 145 and PC-3 cells prior to tail vein injection as an inhibitor of lung extravasation. Because many DTC’s may never give rise to actual lung colonies, the extravasated cells were also allowed to grow into discernable lung colonies without further treatment, to measure the effect of PhScN exposure on successful lung colonization by DTC’s. We found that, consistent with its increased potency as an inhibitor of α5β1–mediated invasion, PhScN also exhibits greatly increased efficacy as a lung extravasation and colonization inhibitor. A brief prebinding of PhScN to suspended DU 145 or PC-3 cells, just before tail vein injection substantially decreases extravasation, and results in a 379,000-fold reduction in DU 145 and an 111,000-fold reduction in PC-3 lung colonies after 6 weeks of growth without further treatment. Furthermore, to show that 5 mg/kg PhScN monotherapy is well tolerated, and to compare its antitumorigenic potency with that of 5 mg/kg PHSCN, DU 145 cells were injected intramuscularly and tumors allowed to grow for 6 weeks prior to initiation of 5 weeks of monotherapy. PhScN monotherapy was very well tolerated, and was sufficient to cause complete tumor regression, at both macroscopic and microscopic levels. These results indicate that the greatly increased efficacy of PhScN as lung extravasation and colonization inhibitor derives from a noncovalent interaction with its target, and suggest that determination of the PhScN target site on α5β1 integrin could result in a potent new family of therapeutic agents for prostate cancer.
Materials and methods
Cell lines and cell culture
DU 145 [13] and PC-3 [14] metastatic human prostate cancer cells were obtained from American Type Culture Collection (Manassas, VA). They were cultured as recommended, aliquoted, and frozen in liquid N2. Single aliquots were subsequently resuscitated as needed, and cultured as recommended. No aliquot of cells was cultured for more than 4 months, and the morphologies of all cultures were routinely checked by phase contrast microscopy. For all assays in serum–free (SF) medium, cells were first serum–starved overnight.
Peptide synthesis
N-terminal–acetylated, C-terminal–amidated PhScN, PHSCN, hSPNc, and HSPNC peptides, and the PhScNGGK–MAP polylysine dendrimer, and associated cysteine or biotinylated (–Bio) peptide derivatives were synthesized and purified to 95 % by either Peptide 2.0 (Chantilly, VA), Peptisyntha, Inc. (Torrence, CA) or by the University of Michigan Peptide Synthesis Core (Ann Arbor, MI). Stock concentrations of peptides were prepared fresh prior to use by gravimetric measurement from lyophilized material.
In vitro invasion assays
Naturally SF, selectively permeable basement membranes from sea urchin embryos (SU-ECM) were utilized as in vitro invasion substrates, as described [3–5, 15–19]. All cells were serum–starved prior to addition of 10 % FBS to stimulate invasion. For assays evaluating the effects of blocking anti α5β1 MAb on invasion, serum–starved, suspended cells were incubated for 30 min on ice in serum–containing medium with 10 or 50 μg/ml anti α5β1 JBS5 [20] function–blocking monoclonal antibody (MAb) (Serotec, Oxford, England), prior to placement on SU-ECM basement membranes. Peptides were briefly prebound to suspended cells prior to placement on basement membranes. Data, mean invasion percentages, were analyzed using Prism software (GraphPad Software, San Diego, CA) as a function of log (inhibitor) versus normalized data, variable slope.
Determination of dissociation constants, Kd, and inhibition constants, Ki, for cell surface binding by the PhScN and PHSCN peptides
Dissociation constants of the PHSCN and PhScN peptides, and their relevant S-acm cysteine derivatives, to cell surface receptors of PC-3 and DU 145 were performed by centrifugation [21] using lengthened, biotinylated (–Bio) versions of the N-acetylated, C-amidated peptides (Ac-PHSCNGGK–Bio and Ac-PhScNGGK–Bio) as previously described [8]. Because PHSCN and PhScN bind to activated α5β1 integrin, maximal α5β1 activation was promoted by addition of manganese to the binding buffer [22]. Unlike DU 145 cells, PC-3 cells were found to have variances in binding affinity, suggesting disparities in activation states between cell preparations. Therefore all binding experiments were performed as pairs to ensure that the activation states were consistent.
In brief, 125,000–150,000 cells, suspended in binding buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.1 % bovine calf serum, and 2 mM MnCl2), were incubated with varying concentrations of biotinylated peptides for 2 h at 4 °C. Cells were pelleted, and the pellets were washed twice with ice–cold binding buffer, prior to incubation at 4 °C for 30 min with Streptavidin–peroxidase (Sigma). After three additional washes, the cell pellets were incubated with o-phenylenediamine substrate, stabilized by the addition of HCl and the absorbance values recorded at 490 nM.
Competition assays were similarly performed by adding a constant concentration of the biotinylated, labeled compound with varying amounts of unlabeled competitor, and the equilibrium time was extended to 3 h [23]. Binding data were analyzed using Prism software (GraphPad software, San Diego, CA), and binding curves were fit using non-linear regression approaches [23].
Fluorescent DiI labeling of cells
Confluent DU 145 or PC-3 cells were washed with Hanks buffered salt solution (HBSS), harvested with 0.25 trypsin/1 % EDTA rewashed in HBSS, and orange–fluorescently labeled in 6 ml SF medium with 25 μl of the lipophilic carbocyanine vital dye DiI, 1,1′-dilinoleyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate (Invitrogen), for 20 min in dark at 37 °C, as described [24, 25]. DiI has been utilized as a vital dye in numerous studies, for example, to define neural crest invasive/migratory pathways in developing embryos [26]. After labeling, cells were pelleted at 1,000 rpm for 2 min, prior to resuspension in 6 ml of medium, and allowed to recover for 72 h prior to use.
Mice
Female Foxn1nu athymic nude mice (Harlan) were housed in pathogen-free conditions according Association for the Assessment and Accreditation for Laboratory Animal Care guidelines. Studies were performed with approved institutional animal care and use protocols, and adhere to ARRIVE guidelines. No body weight loss or morbidity was associated with the study protocols.
Extravasation of DU 145 or PC-3 cells in the lungs of athymic mice
To compare the effects of blocking anti α5β1 MAb on the accumulation of labeled cells in lung tissue after intravenous injection, DiI–labeled cells were incubated for 30 min on ice with 10 or 50 μg/ml anti α5β1 JBS5 [20] anti α5β1 function–blocking MAb (Abcam, Cambridge MA). A total of 10,000 pretreated cells in 0.1 ml HBSS were injected into the tail vein of each 8-week, nude athymic mouse (Harlan Laboratories). Each treatment group consisted of 10 mice. All mice were euthanized 24 h later, and their lungs were removed. The frozen–tissue samples were then prepared and analyzed at 400-fold magnification with a Zeiss Scanning Laser Confocal microscope (LSM510). A total of 25 10-μm sections were analyzed for one lung in each mouse, for treatment groups consisting of 10 mice each. The sections were taken at intervals of 100 μm; hence, the total thickness of tissue analyzed for each mouse was 2.5 mm. The invaded DiI–labeled DU 145 and PC-3 cells in the lungs were scored using fluorescence microscopy, and the data are presented as mean ± SEM as described [15, 16].
Similar methods were utilized to compare the potencies of a single, systemic PhScN or PHSCN pretreatment on extravasation into lung tissue. Suspended, DiI–labeled cells were prebound with 1, 10 or 100 ng/ml Ac-PhScN-NH2; 10, 100, or 1,000 ng/ml Ac-PHSCN-NH2; 1,000 ng/ml Ac-HSPNC-NH2, for 10 min at 37 °C, or left untreated. Groups of ten, eight week–old, nude mice received one systemic pretreatment with the appropriate peptide concentration by tail vein injection. Immediately after pretreatment, 10,000 cells, prebound with the appropriate peptide concentration or with HBSS only (untreated), were injected into the tail vein of each mouse in 0.1 ml HBSS. To enable the effects of single exposures on extravasation to be quantitated, mice received no other systemic treatments. All mice were euthanized 24 h later, and their lungs were removed. The frozen–tissue samples were then prepared and analyzed as above and as previously described [15, 16]. A total of 25 10-μm sections were analyzed for one lung in each mouse, for treatment groups consisting of 10 mice each. The sections were taken at intervals of 100 μm; hence, the total thickness of tissue analyzed for each mouse was 2.5 mm. Slides were sealed by Mounting Medium for fluorescence with DAPI conjugated with green fluorescence labeled actin (Vector Laboratories, Burlingame, CA). All red DiI–labeled DU 145 and PC-3 cells in one lung of each mouse were scored with a Zeiss Scanning Laser Confocal microscope (LSM510) using a 40× objective, and the data are presented as mean ± SEM as previously described [15, 16].
Lung colonization assays
To compare the potencies of Ac-PhScN-NH2 and Ac-PHSCN-NH2 as inhibitors of productive extravasation, resulting in lung colony formation, suspended, DiI–labeled, peptide–treated cells were prepared and injected as described above for assessment of extravasation. Mice received a single systemic pretreatment with the appropriate peptide concentration by tail vein injection. Immediately after pretreatment, 10,000 cells, prebound with the appropriate peptide concentration or with HBSS only (untreated), were injected into the tail vein of each of 10 mice per treatment group. To enable the effects of single exposures on extravasation leading to colony formation to be quantitated, no additional systemic peptide treatment occurred during lung colony growth. Six weeks after injection, mice were euthanized, and lungs were removed, fixed, prepared, and stored. Twenty sections of 10-μm thickness, each taken at 200 μm intervals, were evaluated for each treatment group. Hence, a total tissue thickness of 4 mm was analyzed in one lung of each mouse. Slides were sealed by Mounting medium with green fluorescence labeled actin (Vector Laboratories, Burlingame, CA). The total numbers of fluorescent lung colonies, each composed of more than 50 DiI–labeled DU 145 or PC-3 cells (red), were scored using a Zeiss Scanning Laser Confocal microscope (LSM510) with a 40× field, and the data are presented with mean ± SEM as previously described [15, 16].
Tumorigenesis assays
Suspended DU 145 cells (1.5 × 106 per mouse) were injected intramuscularly into the right hind legs of a total of 60 athymic nude mice. Tumors were allowed to grow for 6 weeks, prior to the initiation of systemic therapy. All tumors were palpable, but not measurable, when systemic therapies were initiated. Dosages of 5 mg/kg Ac-PhScN-NH2, Ac-PHSCN-NH2 or Ac-HSPNC-NH2, each in a total volume of 0.05 ml per injection, were administered thrice–weekly via tail vein injection as described [3]. Untreated mice received 0.05 ml normal saline on the same injection schedule as the PhScN–, PHSCN–, and HSPNC–treated mice. After 5 weeks of treatment, all DU 145 tumors or injection sites were harvested, fixed, sectioned, and stained with hematoxylin and eosin. A total of 20 sections of 5–micron thickness, each separated by 200 microns, were assessed for tumor cells throughout each tumor or injection site. Hence, a total tissue thickness of 4 mm was analyzed in each injection or tumor site in each mouse. A total of 15 mice were evaluated for each treatment group, including the untreated mice.
Data analysis
Dose response data for extravasation and lung colony formation were analyzed by the Combination index (CI) method of Chou-Talalay [27], as described [28]. The analysis was based on the multiple drug effect equation derived from the median effect principle of the mass-action law. The median–effect equation, y = log(fa/fu), with respect to x = log(dose), defines the dose and effect relationship in the absence of reaction rate constants, where fa is the fraction of cells affected (invasion–inhibited), and fu is the fraction of cells unaffected (invaded). The x intercept represents the IC50 value. CI and DRI values were determined for Ac-PhScN-NH2, relative to Ac-PHSCN-NH2.
Results
Increased invasion inhibitory potency of the PhScN peptide, Ac-PhScN-NH2, and PhScN dendrimer, Ac-PhScNGGK-MAP
Naturally SF, selectively permeable SU-ECM basement membranes have previously been used to identify plasma fibronectin (pFn) as the invasion–inducing protein in serum. Moreover, the PHSRN sequence of the pFn cell binding domain has been shown to be sufficient for α5β1 integrin–mediated invasion induction in human prostate and breast cancer cell lines [3, 18], in normal prostate and mammary epithelial cells [3, 18], in normal microvascular endothelial cells [5], and in normal human keratinocytes and fibroblasts [17]. Thus, quantitating invasion with naturally SF basement membranes (SU-ECM) has been an effective assay for evaluating the potencies of invasion inhibitors.
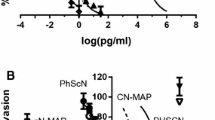
The mean percentages of cells invaded, after placement on SU-ECM in the presence of serum and varying concentrations of PhScN or PHSCN, or scrambled sequence controls are shown in Fig. 1a, b for DU 145 and PC-3, respectively. The PhScN peptide was 4–5 orders of magnitude more potent than PHSCN as an in vitro basement membrane invasion inhibitor. High concentrations of the scrambled sequence controls, HSPNC or hSPNc, had no significant inhibitory effect on invasion. The IC50 values derived from the Hill–Slope plots (Fig. 1a, b) are listed in Table 1. As shown by the dose reduction index (DRI) values, the PhScN peptide was 104–105-fold more potent at blocking FBS–induced invasion. Quantitatively similar results were obtained for SF invasion induction by stimulation with a peptide consisting of the PHSRN sequence of the pFn cell binding domain, Ac-PHSRN-NH2 (not shown).

Hill–slope plots of increased invasion inhibitory potency of the d-His, d-Cys–containing Ac-PhScN-NH2 peptide and Ac-PhScNGGK-MAP dendrimer for DU 145 and PC-3 cells induced by serum (FBS). Panels a DU 145 cells and Panels b PC-3 cells. Specific agents are listed on the right. X axes, log peptide concentration in pg per ml; Y axes, mean relative percentages of invaded cells (±SD). IC50 and DRI values are summarized in Tables 1 and 2
Because our prior studies demonstrated that a multivalent presentation of the PHSCN sequence on the polylysine multiantigenic peptide (MAP) dendrimer resulted in a 100- to 6,700-fold increase in in vitro invasion–inhibitory potency (on a molar basis) as well as similarly increased potencies for inhibition of lung extravasation and colonization by both prostate and breast cancer cell lines [15, 16], we determined the invasion–inhibitory potencies of the Ac-PhScNGGK-MAP dendrimer. Similarly, we found that a multivalent presentation of the PhScN sequence produced an additional 106-fold increase in potency, with IC50’s of 0.16 and 0.08 ag/ml for DU 145 and PC-3 cells respectively (Fig. 1a, b; Table 1). Since our previous work for PHSCNGGK-MAP (7,500 Da) for prostate [15], and breast [16] adenocarcinoma cell lines demonstrated a significantly smaller increase in potency, a 10- to 100-fold reduction in IC50, relative to the PHSCN monomer (598 Da), the greatly increased potency (106) for PhScNGGK-MAP, relative to PhScN, indicates PhScN is an even more attractive candidate than PHSCN for a multivalent presentation (Table 1).
Increased invasion inhibitory potency of PHSCN and PhScN peptides, modified to prevent disulfide bond formation with target
Induction and inhibition of α5β1–mediated invasion are rapid responses in vitro, complete in less than 4 h [4, 18]. The inhibition mechanism of PHSCN or PhScN with its disulfide–rich target, α5β1 integrin, could be either noncovalent, or covalent–by forming a stable mixed disulfide, or by inducing disulfide rearrangement, which may or may not include the loss of the intermediate mixed disulfide. It has been proposed that PHSCN inhibits invasion via disulfide bonding with α5β1 integrin, forming a mixed disulfide [6]. To determine the importance of disulfide bonding, the effects on invasion–inhibitory potencies of substituting commercially available derivatives of l-cysteine in the PHSCN peptide, S-acetylated (S-OAc) or S-methylated (S-Me), were tested. Commercially available derivatives of d-cysteine were limited to S-acetamidomethyl (S-acm). For invasion inhibition comparison and additional Kd determination studies, the biotin–labeled forms of both PHSCN and PhScN and their S-acm derivatives were acquired.
As shown in Fig. 2a, b for DU 145 and PC-3 cells respectively, and summarized in Table 1, the invasion–inhibitory potencies of PHSCN derivatives incapable of disulfide bond formation, PHSC(S-OAc)N or PHSC(S-Me)N, were increased on average 104–106- or 103–104-fold, respectively. The increased potency of the blocked l-cysteine derivatives is similar to that determined for d-cysteine containing PhScN (104–105-fold), and establishes that the productive mechanism of inhibition is noncovalent.

Hill–Slope plots of increased invasion inhibitory potency of the biotinylated and/or cysteine modified derivatives of PHSCN and PhScN for DU 145 and PC-3 cells induced by serum (FBS). Panels a, c DU 145 cells and Panels b, d PC-3 cells. Specific agents are listed on the right. X axes, log peptide concentration in pg per ml; Y axes, mean relative percentages of invaded cells (±SD). IC50 and DRI values are summarized in Tables 1 and 2
In addition, a single substitution of d-cysteine, without d-histidine (Ac-PHScN-NH2), was only a 100-fold more potent than PHSCN (data not shown); whereas, d-amino acid substitutions of both the histidine and cysteine residues in PhScN, as shown above, resulted in a 104–105-fold increase in potency, indicating that the orientation of the histidine and cysteine side chains on the same side of the peptide must also play an important role in the invasion–inhibitory mechanism.
Comparison of the IC50 values in Tables 1 and 2 show that the biotin–labeled Ac-PHSCNGGK-Bio and Ac-PhScNGGK-Bio peptides (1,100 Da) are similar to the unlabeled parent peptides (598 Da), demonstrating that the additional mass on the C-terminus does not interfere with invasion inhibition. This allows for direct comparison of the effects of S-acm modification to both l- and d-cysteine on invasion inhibition and on the dissociation constant (Kd) determinations discussed below.
The S-acm invasion inhibition results, for DU 145 and PC-3 cells respectively (Fig. 2c, d; Table 2), showing a 104-fold increase in potency for the l-cysteine containing, biotin–labeled peptide, were similar to those obtained for S-Me and S-OAc derivatives, 104–106-fold; again demonstrating that the blocked l–cysteine is a substantially more potent inhibitor, and that the productive inhibitory mechanism of PHSCN is actually noncovalent. In contrast, the S-acm modification of the d–cysteine residue in the PhScN peptide resulted in only a minor 10- to 20-fold increase in potency–which is well within the range of error–indicating that the status of the d-cysteine residue (blocked versus unblocked) is irrelevant to the potency of the PhScN peptide, further demonstrating the noncovalent nature of the invasion–inhibitory mechanism for PhScN.
Moreover, attempts to trap a covalent mixed disulfide with PHSCNGGK–bio on the surface of live cells for visualization on a western blot required a significantly longer incubation time to occur compared to the 4 h required for the SU-ECM in vitro invasion inhibition assays to be complete [3, 4] (data not shown). Although inconclusive, this suggests formation of a mixed disulfide is a slow, nonproductive side reaction. Thus, combined with the invasion inhibition results for the cysteine–blocked derivatives, the mechanism suggested by the covalent, mixed disulfide, observed between purified, disulfide–rich α5β1 integrin and the PHSCN peptide [6], is incomplete.
Comparison of dissociation constants, Kd, for cell surface binding by biotinylated PhScN or PHSCN peptides
Results of in vitro invasion assays, showing the greatly increased inhibitory potency of the PhScN peptide, suggested that it might also exhibit a reduced dissociation constant (Kd). Hence, the Kd of biotinylated PhScN, PHSCN and their S-acm derivatives were determined by the methods previously utilized for PHSCN during its development for clinical trial [6].
As shown in Table 2, the Kd values for the PhScN peptide (Ac-PhScNGGK–Bio) were 0.26 and 0.10 μM, and for the PHSCN peptide (Ac-PHSCNGGK–Bio) were 0.35 and 0.09 μM for DU 145 and PC-3 cells, respectively. While the Kd of PhScN appears to be similar or to have improved only slightly in comparison to PHSCN, the lack of any significant change to the Kd suggests that the improved invasion inhibitory potency (Table 1) is due to elimination or reduction of the unproductive, covalent side reaction; and does not appear to result from tighter binding, due to orienting the d–cysteine to the opposite side of the peptide ligand.
Both the l and d cysteine S-acm derivatives showed a significant increase in their Kd values, indicating the binding was not as tight as with the unmodified l- and d- cysteines. The binding data suggest that, although the blocked cysteine derivatives of PHSCN and PhScN are more potent invasion inhibitors (Tables 1 and 2), there are steric hindrances to binding caused by covalent modification of the cysteine. These data illustrate that the endoproteinase–resistant, d–cysteine substituted peptide, PhScN, better fulfills the goal of increased potency than covalent modification of the l–cysteine of the PHSCN peptide.
Competition binding assays of PHSCN and PhScN
The composition of the peptide compounds PHSCN and PhScN and the similarities of their Kd’s suggest that they may also interact with the same binding site. Results of competition assays, in which cells were incubated with a constant concentration of labeled Ac-PHSCNGGK–Bio and varying amounts of unlabeled PhScN or PHSCN peptides, are shown for DU 145 and PC-3 cells in Fig. 3a, b respectively.

Competition binding assays for PHSCN and PhScN peptides with suspended DU 145 and PC-3 cells. Panels a and b 0.4 μM Ac-PHSCNGGK-bio (labeled) was incubated with varying amounts of unlabeled Ac-PhScN-NH2 or Ac-PHSCN-NH2; Panel a, DU 145 cells, Kd (labeled) = 0.4 μM; Panel b PC-3 cells, Kd (labeled) = 0.1 μM; K d (labeled) = IC50/([labeled]/K d (labeled)) + 1. Circles, unlabeled Ac-PhScN-NH2; squares, unlabeled Ac-PHSCN-NH2. Panels c and d varying amounts of labeled Ac-PHSCNGGK-bio were incubated in the absence (Kd, circles) or presence (Kdapp, triangles) of a constant concentration of unlabeled Ac-PhScN-NH2. Panel c, DU 145 cells 0.6 μM Ac-PhScN-NH2. Panel d, PC-3 cells, 1.8 μM Ac-PhScN-NH2; K i = [competitor]/(K dapp /K d −1). X axes, μM Ac-PhScN-NH2, μM Ac-PHSCN-NH2 or Ac-PHSCNGGK-bio; Y axes, % relative A490, ± SD. Plots A and B were fit using a competitive inhibition equation. Plots C and D were fit using a total binding equation to account for nonspecific binding
The dissociation of the labeled material occurred over approximately two logs in competitor concentration, demonstrating that PHSCN and PhScN are competing for the same binding site [23]. The inhibition constants (Ki) were calculated from the relationship of IC50 to Ki defined by Cheng and Prusoff [29] using Eq. 1. The Ki values for the unlabeled PHSCN and PhScN competitors were found to be 0.74 and 0.18 μM for DU 145; and 0.17 and 0.26 μM, for PC-3 cells, respectively. The Ki values of the unlabeled competitors displayed the same relationship observed for the Kd values of the biotin–labeled PHSCN and PhScN peptides (Table 2). Both Ki and Kd values suggest that PhScN binds slightly better than PHSCN to DU 145 cells; and that they have similar binding properties to PC-3 cells.
To confirm competition for the same binding site, the reverse experiment was performed where cells were incubated with varying concentrations of the labeled PHSCNGGK-Bio peptide in the absence and in the presence of a constant concentration of unlabeled PhScN (Fig. 3c, d). This experiment was performed as described above for Kd determination, and generates Kd and Kdapp, representing the Kd values of the PHSCNGGK–Bio determined in the absence and presence of unlabeled competitor, respectively. Subsequently, using Eq. 2 [30] enables the calculation of Ki for unlabeled PhScN.
The Ki values determined for unlabeled PhScN (Fig. 3c, d) were 0.17 and 0.36 μM for DU 145 and PC-3 cells, respectively; and these agree with the values 0.18 and 0.26 μM determined from the reverse experiment above (Fig. 3a, b). Both types of competition binding experiments demonstrate that PHSCN and PhScN compete for the same binding site on both DU 145 and PC-3 cells.
The α5β1 integrin fibronectin receptors of DU 145 and PC-3 function in lung extravasation in vivo
Since PHSCN has been shown to inhibit α5β1-mediated invasion specifically, in our lab [3–5, 15, 16] and by others [6–9], we confirmed the role of α5β1 integrin for extravasation into lung tissue by prebinding the blocking JBS5 anti α5β1 MAb [20] to suspended cells, just prior to tail vein injection into nude mice. As shown in Fig. 4, prebinding 10 μg/ml JBS5 reduced extravasation by about 75 %. Moreover, prebinding 50 μg/ml JBS5 had a greater inhibitory effect of about 90 %; while prebinding 50 μg/ml of the IgG1 isotype control did not significantly inhibit extravasation. These results confirm the key role of α5β1 integrin during lung extravasation, and suggest that since PhScN is a potent inhibitor of α5β1-mediated basement membrane invasion in vitro, it might also be a highly potent inhibitor of lung extravasation and colonization.

Inhibition of DU 145 and PC-3 lung extravasation by prebinding JBS5 blocking anti α5β1 MAb. Relative extravasation of cells per lung section after prebinding with JBS5 anti α5β1 MAb or IgG1 antibody concentrations (μg/ml). JBS5 denotes the CD49e MAb clone JBS5 anti α5β1 MAb. X axis, concentration JBS5 or IgG1; Y axis, mean percentage of extravasated cells, ±SEM
Increased inhibitory potency of PhScN for lung extravasation and colonization by DU 145 and PC-3 cells in athymic nude mice
Since PHSCN has been shown to inhibit α5β1 integrin–mediated invasion specifically [3, 6–9, 15, 16], to reduce or prevent metastasis [3, 4, 6–9, 15, 16], and to enhance the radio response of breast cancer cells [10], and since it demonstrated efficacy in Phase 1 clinical trial [11], we compared the efficacies of the PhScN and PHSCN peptides as inhibitors of lung extravasation and colonization in athymic, nude mice. To assess their relative potencies, treatment groups consisting of 10 athymic nude mice, each received a single systemic pretreatment with the appropriate peptide concentration by tail vein injection. Then, DiI–labeled DU 145 or PC-3 cells, briefly prebound to the appropriate concentrations of the PhScN or PHSCN peptides, were injected into the tail veins of athymic, nude mice; and the lungs were extracted after 24 h. Since many extravasated cancer cells may not be able to colonize the new site effectively [31], the effects of PhScN or PHSCN pretreatment on lung colonization were also evaluated after the cells were allowed to grow for 6 weeks. The mice received no further systemic treatment during the 6 weeks.
Figure 5 compares qualitatively the efficacies of single PHSCN or PhScN pretreatments on lung extravasation, and on subsequent colonization as an indication of successful extravasation. Panel 5a, shows the dose response for mean numbers of extravasated cells per lung tissue section per mouse. Means of 1,802 (±170 SEM) for DU 145, and 2,097 (±100 SEM) for PC-3, were observed in the untreated controls. Means of 1,359 (±67 SEM) for DU 145 and 1,963 (±94 SEM) for PC-3 were observed for the scrambled sequence control, 100 ng/ml Ac-HSPNC-NH2, demonstrating its lack of inhibitory effect. Panel 5b, shows that the mean number of colonies per lung section per mouse after 6 weeks growth for each untreated control was 22.3 ± 3.3 SEM and 31.3 ± 6.2 SEM, for DU 145 and PC-3, respectively. Similar values were obtained with the scrambled sequence control, 100 ng/ml Ac-HSPNC-NH2, of 19.8 ± 4.5 and 22.9 ± 5.2, SEM. Sections were randomized prior to scoring to minimize bias. The SEM ranged from 25 to 30 % of the mean numbers for both extravasated cells and lung colonies in all treatment groups for both cell lines.

Effects on lung extravasation and colony formation by DU 145 and PC-3 cells after exposure to varying concentrations of PHSCN or PhScN. a Mean number of cells extravasated per section of lung tissue after 24 h (±SEM). b Mean number of colonies formed per section of lung tissue after 6 weeks of growth (±SEM). X axes, PHSCN or PhScN concentrations. Y axis, Fig. 6 a, mean number of DU 145 or PC-3 cells extravasated per section (± SEM). Y axis, Fig. 6 b, mean number of DU 145 or PC-3 colonies per section (± SEM). Con., untreated control; HSPNC, scrambled sequence control, 100 ng/ml; PHSCN, 10, 100, or 1,000 ng/ml; PhScN, 1, 10, 100 ng/ml
At 24 h post injection in the untreated controls, ~2,000 cells per section, in each of 25 sections per lobe, appeared to have extravasated for both cell lines (Fig. 5a). At concentrations of either 1,000 ng/ml PHSCN or 1 ng/ml of PhScN, the numbers of extravasated cells observed are similar and reflect about 90 % inhibition, suggesting that PhScN is at least 1,000-fold more potent than PHSCN at preventing extravasation into lung for both cell lines.
Because many extravasated cells could fail to give rise to colonies and hence be irrelevant for metastatic progression, colonies were allowed to grow without further treatment for 6 weeks in duplicate mice. In untreated mice about 25 colonies formed per section, as shown in Fig. 5b. Thus, about 1 % of extravasated DU 145 or PC-3 cells formed colonies in untreated mice. At the highest concentration of PHSCN, 1,000 ng/ml, ~6 colonies per section were observed, for a colonization efficiency of 0.3 %, relative to untreated cells extravasated, and an inhibition rate of about 76 % relative to untreated colony formation. At the lowest concentration of PhScN, 10 ng/ml, ~3 colonies per section were observed, for a colonization efficiency of 0.15 %, and an inhibition rate of about 88 %. These results suggest that PhScN is significantly greater than 100-fold more potent than PHSCN at preventing lung colonization for both cell lines. Analyses of the dose response curves and extrapolation to similar efficacies were performed to allow for a more precise determination of the increased potency.
Quantitation of the inhibitory potency of PhScN for lung extravasation and colonization by DU 145 and PC-3 cells in athymic nude mice
A single PHSCN treatment at a dose range of 10–1,000 ng/ml affected (inhibited) 50–90 % of the cells for both cell lines; whereas a single PhScN treatment at a dose range of 1–100 ng/ml inhibited extravasation by 90–98 %. Conversion of the numbers of cells extravasated to ratios of affected cells to unaffected cells (fa/fu) enables construction of a dose response curve using the median affect and subsequent extrapolation of 50 % inhibition (IC50) and the DRI, which are shown in Fig. 6a, b for DU 145 and PC-3, respectively. The IC50 values were determined from the extrapolated x–intercepts, and are shown for each curve. The DRI represents a ratio of the IC50’s. Typical examples of the confocal images analyzed are presented in Fig. 6c. Each panel depicts sectioned lung tissue, containing variable numbers of extravasated cells after pretreatment with 10 ng/ml PhScN or PHSCN peptides. As shown in Fig. 6a, b PhScN appears to be 15,668- or 272-fold more potent than PHSCN as an inhibitor of DU 145 or PC-3 lung extravasation, respectively.

Increased extravasation inhibition by PhScN prebinding, relative to PHSCN peptide. a Median–affect plot for DU 145 cell extravasation into lung after prebinding with PHSCN or PhScN, with IC50’s. b Median affect plot for PC-3 extravasation into lung after prebinding with PHSCN or PhScN, with IC50’s. X axes, log peptide concentration in ng/ml; Y axes, mean log fraction affected/fraction unaffected (fa/fu) ± SEM. c Typical examples of the confocal images analyzed to provide these data are shown. All extravasated cells in sectioned lung tissue after pretreatment with 10 ng/ml PhScN or 10 ng/ml PHSCN peptides, or with serum–containing medium without peptide are indicated Asterisk in these panels. Control denotes cells pretreated with serum–containing medium without peptide. DiI-labeled DU 145 or PC3 cells are shown in orange/red. The DAPI in the Mounting Medium labels the nuclei blue; and the tissues appear green
Similarly, a single treatment with 100 or 1,000 ng/ml PHSCN inhibited colony formation by 60–80 %; whereas a single treatment with PhScN at 10 or 100 ng/ml reduced lung colony formation by 90–95 %. Conversions to a ratio of affected (inhibited) colonies to unaffected colonies (fa/fu) allows for quantitation of the IC50 and DRI), and are shown in Fig. 7a, b for DU 145 and PC-3 cells, respectively. Typical examples of the confocal images analyzed to provide these data are presented in Fig. 7c. The data indicate that PhScN is substantially more potent than PHSCN as an inhibitor of lung colonization. The increased potency (DRI) of PhScN relative to PHSCN for DU 145 lung colonization is 379,000-fold; and is similarly elevated, 111,000-fold, for PC-3 cells.

Increased inhibition of lung colonization by PhScN prebinding, relative to PHSCN peptide. a Median–affect plot for DU 145 lung colony formation, with IC50’s. b Median–affect plot for PC-3 lung colony formation, with IC50’s. X axes, log peptide concentration in ng/ml; Y axes, log (fa/fu) ± SEM. c Examples of DiI–labeled DU 145 and PC-3 colonies (orange/red) in sectioned lung tissues (green) after 6 weeks of growth
The increased potency (DRI) values of PhScN, relative to PHSCN, for both DU 145 and PC-3 lung colonization (105) are quite similar to that obtained for in vitro invasion assays (104–105), as shown in Table 1. The DRI of PhScN, relative to PHSCN for DU 145 (104) and PC-3 (102) cell extravasation, appears to be somewhat less than that observed in in vitro invasion assays (104–105), especially for PC-3 cells. This may reflect a higher prevalence of PC-3 cells that cannot develop into colonies, but are retained in the lung tissue a day after intravenous injection. Increased PC-3 cell retention, relative to DU 145, could indicate a reduced activation state of the α5β1 integrin for PhScN peptide binding on the batches of DiI–labeled PC-3 cells analyzed. It may also reflect differences in rate of extravasation by the untreated or HSPNC–treated controls. None of these factors would be expected to influence relative differences in lung colonization because 6 weeks are required for lung colonies to grow.
These results demonstrate the greatly increased potency of the PhScN peptide as an inhibitor of lung colonization. Since mice received only a single systemic treatment just prior to injection, these results suggest that PhScN is a highly potent inhibitor of lung extravasation and colonization by DTC’s. These values also suggest that among the cells scored as extravasated many are incapable of colony formation, especially for PC-3 cells.
Complete regression of DU 145 primary tumors in PhScN–treated mice
To determine whether PhScN monotherapy is well–tolerated, and to compare its antitumorigenic potency with that of PHSCN, intramuscular DU 145 tumors were grown in the right hind legs of 60 nude mice for 6 weeks without treatment. Hence, all tumors were allowed to grow until palpable, prior to the initiation of systemic, thrice–weekly, intravenous 5 mg/kg PhScN, PHSCN, or HSPNC therapies. Then, therapies were performed in treatment groups of 15 mice each; while the fourth group of 15 mice received normal saline only (untreated). All treatments continued for 5 weeks. PhScN monotherapy appeared to be very well tolerated, as all PhScN–treated mice exhibited no ill effects from their therapy. As shown in Fig. 8a, 5 weeks of thrice–weekly, systemic 5 mg/kg PhScN monotherapy either prevented growth or caused total regression of DU 145 tumors; while 5 mg/kg PHSCN monotherapy resulted in a 40 % reduction in mean tumor volume (mm3). Moreover, DU 145 tumorigenesis was virtually identical in the HSPNC–treated and in the untreated groups. The typical appearances of the DU 145 injection or tumor sites in the PhScN, PHSCN, and untreated treatment groups are shown in Fig. 8b. Consistent with the total regression of all tumors, no tumor cells in any section were observed in the PhScN treatment group; instead, normal musculature and vascularization were seen. In contrast, the tumor sites in both the PHSCN and untreated treatment groups contained abundant tumor cells. Sections from the HSPNC treatment group exhibited identical appearance to the untreated and PHSCN–treated groups (not shown). These results show that 5 mg/kg PhScN monotherapy is very well tolerated. Furthermore, consistent with its potency as an inhibitor of α5β1–mediated invasion, which drives angiogenesis [5], these results show that 5 mg/kg systemic PhScN monotherapy has potent antitumorigenic effects.

Increased potency of PhScN as a systemic antitumorigenic agent for DU 145 flank tumors in nude mice. a Mean changes in tumor volume during 5 weeks of systemic PhScN, PHSCN, HSPNC, or control monotherapies. X axis, treatment dates. Y axis, mean increases in tumor volume (mm3) ± SEM. Mean increases in tumor volume are shown because all tumors were palpable, but not measurable, when systemic therapies were initiated. b Typical sections stained with Hematoxylin and Eosin (H & E) are shown to compare the appearances of DU 145 tumor sites after 5 weeks of systemic therapy with 5 mg/kg PhScN or 5 mg/kg PHSCN, as compared to untreated mice
Discussion
There is an urgent need for a potent, well–tolerated, systemic therapy to prevent or limit successful metastatic seeding of the lung in prostate cancer. Because α5β1 integrin–mediated invasion by prostate and breast adenocarcinoma cells to cause systemic dissemination, and by microvascular endothelial cells to promote angiogenesis, are key features of metastatic disease progression, we have pursued α5β1 integrin as a therapeutic target in our previous studies [3–5, 15–19].
Using metastatic human prostate cancer cell lines, we showed here that the PhScN peptide is 4–5 orders of magnitude more potent than PHSCN as an inhibitor of α5β1 integrin–mediated invasion of naturally occurring basement membranes in vitro. Since Ac-PHSCN-NH2 (ATN-161) has been reported to form covalent, disulfide bond(s) with the purified α5β1 integrin target [6], we utilized PHSCN derivatives incapable of disulfide bond formation to confirm the role of disulfide bond formation in the inhibition of serum–induced invasion in vitro. We found, instead, that the S-OAc PHSCN peptide, which is incapable of disulfide bond formation, was up to a million–fold more potent than PHSCN as an invasion inhibitor. Substantial increases in potency were also observed for S-Me PHSCN peptide. Also, the S-acm PHSCN peptide was similarly increased In potency; whereas the S-acm PhScN peptide showed at most a 10–20-fold increase in potency over PhScN. These results suggest that the primary invasion–inhibitory mechanism is actually noncovalent, for both PHSCN and PhScN.
Results were also obtained showing that the biotinylated PhScN derivative (PhScNGGK–Bio) has a similar to slightly reduced dissociation constant, relative to PHSCNGGK-Bio, for suspended DU 145 and PC-3 cells. Furthermore, competition–binding assays showed that the PHSCN and PhScN peptides compete for the same binding site on intact cells. Side chain S–modification to either the l- or d-cysteine significantly increased their dissociation constants, suggesting steric hindrances. Thus, substitution of histidine and cysteine with their d-isomers appears to be the most effective way to eliminate or substantially reduce nonproductive covalent side interactions.
Although multiple systemic PhScN treatments, alone or in combination with other agents and/or radiation, would be utilized in an actual therapy to prevent metastasis progression, we modeled PhScN as an inhibitor of DTC invasion by assessing the efficacy of a single exposure on lung extravasation and colonization by suspended DU 145 and PC-3 cells. The efficacy of a single PhScN pretreatment over a range of concentrations was observed to be 2–4 orders of magnitude greater than PHSCN pretreatment at inhibiting extravasation into lung tissue, and over 100,000- to 300,000-fold more potent at inhibiting subsequent lung colonization. Thus, our results show that PhScN is an efficacious agent for preventing or greatly reducing successful metastatic lung extravasation, as shown by the greatly reduced rates of subsequent lung colony formation. Furthermore, multiple administrations of systemic PhScN monotherapy over a 5-week period was observed to be well tolerated, and caused complete macroscopic and microscopic primary tumor regression in all 15 mice, treated thrice–weekly with 5 mg/kg PhScN. Since we have shown that α5β1–mediated, angiogenic invasion is required for tumor growth [5], these results confirm the greatly increased potency of PhScN as an α5β1 inhibitor in vivo.
Because a single PhScN pretreatment is a potent inhibitor of successful lung extravasation, and multiple systemic PhScN treatments result in complete regression of all established DU 145 tumors, our results suggest that PhScN may be an effective therapy for reducing or preventing parallel progression of DTC’s in prostate cancer. Since radiation, which is commonly used to treat a variety of carcinomas, has been shown to induce surface α5β1 integrin and interstitial collagenase matrix metalloproteinase-1 (MMP-1) upregulation, as well as α5β1–mediated invasion by pancreatic adenocarcinoma cells [19], PhScN may also be a useful adjunct to radiation therapy.
While over 90 % of prostate cancer cases are localized at the time of diagnosis, biochemical failure happens in 20–30 % of patients, often in the first two years after prostatectomy [32, 33]. Furthermore, biochemical failure may occur 10–15 years after primary treatment; and there is a mean time of 8 years between biochemical failure and the appearance of clinical metastasis [32]. These results suggest that dispersed, DTC’s can persist in an occult state for a lengthy period prior to metastatic prostate cancer progression. Thus, detection of circulating tumor cells in the peripheral blood has been accepted as a reliable prognostic tool for prostate cancer, especially in prostate cancer patients harboring bone and/or soft–tissue metastatic disease [34]. Moreover, it has also been reported that 70 % of men undergoing radical prostatectomy had DTC’s detected in their bone marrow prior to surgery, suggesting that these cells escape early in disease progression. Moreover, patients exhibiting persistence of DTCs in bone marrow aspirates after surgery had a nearly 7-fold higher incidence of biochemical failure than patients who were DTC negative [35]. These results suggest that the ability to prevent successful extravasation into lung tissue, as indicated by lung colony formation, could prevent subsequent metastasis progression.
Peptide pharmaceuticals like PhScN have a number of advantages. They can have high potency and specificity, and are simple in structure. Their small size allows them to penetrate tissues that may not be reached by larger molecules. However, a major problem with these agents is their rapid degradation by proteolytic enzymes. Aminopeptidases and carboxypeptidases mediate degradation starting at the peptide’s N- and C-termini, respectively; while endopeptidases cleave within the peptide. Thus, N-acetylation and C-amidation render the PhScN peptide resistant to exoproteolytic degradation.
Since all amino acids in mammalian proteins are l-stereoisomers, endo–peptidases and endoproteinases have evolved to degrade proteins formed from l-amino acids only. Few enzymes that hydrolyze peptide bonds involving d-amino acids have, in fact, been discovered or characterized in any multicellular organism [36]. Thus, another potential advantage of the PhScN peptide is that d-His, d-Cys substitution at alternate residues, thereby removing endopeptidase/endoproteinase–sensitive bonds joining l-amino acids, render it more resistant to degradation by endoproteinases. Endoproteinases are present at high levels in tumors and increase with malignancy [37–39]. The strategy of incorporating d-amino acid residues to increase potency by reducing or preventing proteolytic breakdown has been used previously by others to engineer potent inhibitors of serine or cysteine proteinases [40]. Inclusion of d-amino acids in anticoagulant heparin or thrombin peptide inhibitors also greatly increases their inhibitory potencies [41, 42]. d-amino acid–containing inhibitors of HIV viral entry can have up to 40,000-fold greater antiviral potency [43]. Also, a peptide consisting of 10 d–amino acids, the HYD1 peptide, has been shown to be an α6β1 and α3β1 integrin–targeted inhibitor of PC-3 and DU 145 tumor cell migration and invasion on laminin-5 in vitro, at 75 μg/ml [44].
Our results demonstrate that suppression of covalent, disulfide bond formation with the α5β1 fibronectin receptor target is likely the key factor in the greatly increased efficacy of PhScN as an inhibitor of human metastatic prostate cancer cell extravasation and lung colonization in nude mice. Furthermore, the endoproteinase–resistance conferred by the inclusion of alternate d-amino acids (d-His and d-Cys) in the PhScN peptide, could greatly increase its systemic stability, especially in the presence of an invasive tumor. We have shown that a 5-amino acid peptide, consisting of the invasion–inducing sequence of plasma fibronectin (Ac-PHSRN-NH2), is sufficient for serum–free invasion induction in prostate cancer [3], breast cancer [18], and pancreatic cancer [19], as well as in microvascular endothelial cells [5], fibroblasts [17], and keratinocytes [17]. The PHSRN sequence has been shown to interact exclusively with a specific site in the N-terminal region of the α5 integrin subunit, the similarities in sequence suggest this region may also contain the PHSCN/PhScN binding site. Experiments to determine the specific target site are underway.
Abbreviations
- SF:
-
Serum–free
- FBS:
-
Fetal bovine serum
- Bio:
-
Biotin
- CI:
-
Combination index
- IC50:
-
Concentration for 50 % inhibition
- DRI:
-
Dose reduction index
- HBSS:
-
Hanks buffered salt solution
- DTC:
-
Disseminated tumor cells
- ELISA:
-
Enzyme–linked immunoabsorbant assay
- DiI:
-
1,1′-dilinoleyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate
- MAP:
-
Multiantigenic peptide
- MAb:
-
Monoclonal antibody
- SEM:
-
Standard error of mean
- O.C.T.:
-
Optimal cutting temperature
- FITC:
-
Fluorescein isothiocyanate
- Me:
-
Methyl
- OAc:
-
Acetyl
- acm:
-
Acetamidomethyl
- μg:
-
Microgram
- ng:
-
Nanogram
- pg:
-
Picogram
- fg:
-
Femtogram
- ag:
-
Attogram
References
Bubendorf L et al (2000) Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol 31(5):578–583
Saitoh H et al (1984) Metastatic patterns of prostatic cancer. Correlation between sites and number of organs involved. Cancer 54(12):3078–3084
Livant DL et al (2000) Anti-invasive, antitumorigenic, and antimetastatic activities of the PHSCN sequence in prostate carcinoma. Cancer Res 60(2):309–320
Zeng Z-Z et al (2006) Role of focal adhesion kinase and phosphatidylinositol 3′-kinase in integrin fibronectin receptor-mediated, matrix metalloproteinase-1 dependent invasion by metastatic prostate cancer cells. Cancer Res 66(16):8091–8099
Zeng Z–Z et al (2009) (alpha)5(beta)1 integrin ligand PHSRN induces invasion and (alpha)5 mRNA in endothelial cells to stimulate angiogenesis. Transl Oncol 2:8–20
Donate F et al (2008) Pharmacology of the novel antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2): observation of a U-shaped dose-response curve in several preclinical models of angiogenesis and tumor growth. Clin Cancer Res 14:2137–2144
van Golen KL et al (2002) Suppression of tumor recurrence and metastasis by a combination of the PHSCN sequence and the antiangiogenic compound tetrathiomolybdate in prostate carcinoma. Neoplasia 4(5):373–379
Khalili P et al (2006) A non-RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol Can Ther 5:2271–2280
Stoeltzing O et al (2003) Inhibition of integrin alpha5beta1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int J Cancer 104:496–503
Nam JM et al (2010) Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res 70(13):5238–5248
Cianfrocca ME et al (2006) Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a beta integrin antagonist, in patients with solid tumours. Br J Cancer 94(11):1621–1626
Klein CA (2009) Parallel progression of primary tumours and metastases. Nat Rev Cancer 9(4):302–312
Stone KR et al (1978) Isolation of a human prostate carcinoma cell line (DU 145). Int J Cancer 21(3):274–281
Kaighn ME et al (1979) Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol 17(1):16–23
Yao H et al (2010) Increased potency of the PHSCN dendrimer as an inhibitor of human prostate cancer cell invasion, extravasation, and lung colony formation. Clin Exp Metastasis 27(3):173–184
Yao H et al (2011) The PHSCN dendrimer as a more potent inhibitor of human breast cancer cell invasion, extravasation, and lung colony formation. Breast Cancer Res Treat 125:363–375
Livant DL et al (2000) The PHSRN sequence induces extracellular matrix invasion and accelerates wound healing in obese diabetic mice. J Clin Invest 105(11):1537–1545
Jia Y et al (2004) Integrin fibronectin receptors in matrix metalloproteinase-1-dependent invasion by breast cancer and mammary epithelial cells. Cancer Res 64(23):8674–8681
Yao H et al (2011) Role of alpha5beta1 integrin upregulation in radiation-induced invasion by human pancreatic cancer cells. Transl Oncol 4(5):282–292
Peled A et al (2000) The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 95(11):3289–3296
Hulme EC (1992) Centrifugation binding assays. In: Hulme EC (ed) Receptor-Ligand Interactions: A Practical Approach. Oxford University Press, Oxford, pp 235–246
Mould AP et al (1998) Regulation of integrin function: evidence that bivalent-cation-induced conformational changes lead to the unmasking of ligand-binding sites within integrin alpha5 beta1. Biochem J 331(Pt 3):821–828
Motulsky HJ, Neubig RR (2010) Analyzing binding data. Curr Protoc Neurosci Chapter 7: Unit 7.5
Godement P et al (1987) A study in developing visual systems with a new method of staining neurones and their processes in fixed tissue. Development 101(4):697–713
Molnar Z, Blakey D, Bystron I (2006) Tract-tracing in developing systems and in postmortem human material using carbocyanine dyes. In: Záborszky L, Lanciego JL, Wouterlood FG (eds) Neuroanatomical tract-tracing 3: molecules, neurons, and systems, 3rd edn. Springer Science + Business Media Inc, Boston
Collazo A, Bronner-Fraser M, Fraser SE (1993) Vital dye labelling of Xenopus laevis trunk neural crest reveals multipotency and novel pathways of migration. Development 118(2):363–376
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55
Ren H et al (2009) Differential effect of imatinib and synergism of combination treatment with chemotherapeutic agents in malignant glioma cells. Basic Clin Pharmacol Toxicol 104(3):241–252
Cheng Y, Prusoff WH (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22(23):3099–3108
Matthews JC (1993) Fundamentals of receptor. Enzyme and transport kinetics. CRC Press Inc, Boca Raton, pp 64–94
Gupta GP, Massague J (2006) Cancer metastasis: building a framework. Cell 127(4):679–695
Pound CR et al (1999) Natural history of progression after PSA elevation following radical prostatectomy. JAMA 281(17):1591–1597
Amling CL et al (2000) Long-term hazard of progression after radical prostatectomy for clinically localized prostate cancer: continued risk of biochemical failure after 5 years. J Urol 164(1):101–105
Thalgott M et al (2013) Detection of circulating tumor cells in different stages of prostate cancer. J Cancer Res Clin Oncol 139(5):755–763
Morgan TM et al (2009) Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res 15(2):677–683
Yamada R, Kera Y (1998) d-amino acid hydrolysing enzymes. EXS 85:145–155
Clevers H (2004) At the crossroads of inflammation and cancer. Cell 118(6):671–674
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420(6917):860–867
Scherer RL, McIntyre JO, Matrisian LM (2008) Imaging matrix metalloproteinases in cancer. Cancer Metastasis Rev 27(4):679–690
Annedi SC et al (2006) Engineering d-amino acid containing novel protease inhibitors using catalytic site architecture. Bioorg Med Chem 14(1):214–236
Friedrich R et al (2008) Structure of a novel thrombin inhibitor with an uncharged D-amino acid as P1 residue. Eur J Med Chem 43(6):1330–1335
Wang J, Rabenstein DL (2006) Interaction of heparin with two synthetic peptides that neutralize the anticoagulant activity of heparin. Biochemistry 45(51):15740–15747
Welch BD et al (2007) Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci USA 104(43):16828–16833
Sroka TC, Pennington ME, Cress AE (2006) Synthetic d-amino acid peptide inhibits tumor cell motility on laminin-5. Carcinogenesis 27(9):1748–1757
Acknowledgments
The authors wish to thank Dr. David Ballou and Dr. Eric Carter in the Department of Biological Chemistry, University of Michigan for helpful suggestions in the development and analysis of the binding assays and data. We also wish to thank Dr. Ted Lawrence, Dr. Daniel Hamstra and Dr. Yi Sun in the Department of Radiation Oncology, University of Michigan for providing thoughtful insight to the preparation of this manuscript. This research was supported by the National Institutes of Health, R01 CA119007, “PHSCN Therapies to Prevent Prostate Cancer Progression”, with fiscal assistance from the Office of Technology Transfer, University of Michigan Medical School Office of Research, Ann Arbor, Michigan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Veine, D.M., Yao, H., Stafford, D.R. et al. A d-amino acid containing peptide as a potent, noncovalent inhibitor of α5β1 integrin in human prostate cancer invasion and lung colonization. Clin Exp Metastasis 31, 379–393 (2014). https://doi.org/10.1007/s10585-013-9634-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-013-9634-1