
Abstract
The α5β1 integrin fibronectin receptor is an attractive therapeutic target in breast cancer because it plays key roles in invasion and metastasis. While its inactive form is widely expressed, activated α5β1 occurs only on tumor cells and their associated vasculature. The PHSCN peptide has been shown to bind activated α5β1 preferentially, thereby blocking invasion in vitro, and inhibiting growth, metastasis and tumor recurrence in preclinical models. Moreover in a recent Phase I clinical trial, systemic PHSCN monotherapy was well tolerated, and metastatic disease failed to progress for 4–14 months in 38% of patients receiving it. A significantly more potent PHSCN derivative, the PHSCN–polylysine dendrimer (Ac-PHSCNGGK-MAP) has recently been developed. We report that it is 1280- to 6700-fold more potent than the PHSCN peptide at blocking α5β1 mediated SUM-149 PT and MDA-MB-231 human breast cancer cell invasion of naturally occurring basement membranes in vitro. Chou–Talalay analysis of these data suggested that invasion inhibition by the PHSCN dendrimer was highly synergistic. We also report that, consistent with its enhanced invasion-inhibitory potency, the PHSCN dendrimer is 700- to 1100-fold more effective than the PHSCN peptide at preventing SUM-149 PT and MDA-MB-231 extravasation in the lungs of athymic, nude mice. Our results also show that many extravasated SUM-149 PT and MDA-MB-231 cells go on to develop into metastatic colonies, and that pretreatment with the PHSCN dendrimer is more than 100-fold more effective at reducing lung colony formation. Since many patients newly diagnosed with breast cancer already have locally advanced or metastatic disease, the availability of a well-tolerated, nontoxic systemic therapy that can prevent metastatic progression by blocking invasion could be very beneficial.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metastatic breast cancer is currently incurable [1]. Despite therapeutic advances, patients with metastatic disease have poor prognoses, and their median survival time is 2–3 years [2]. Combination therapies can result in objective responses, but tumor resistance usually develops, and disease control cannot be maintained [3]. Breast cancer patients with metastasis to visceral organs, such as lung, have shorter survival times than those with bone metastasis alone [4]. Like patients with hormone receptor-negative tumors or hormone-refractory disease, patients with symptomatic visceral disease are treated with cytotoxic chemotherapetic regimens [5].
Invasion is a key feature of breast cancer metastasis because it promotes tumorigenesis by supporting endothelial cell invasion and neovascularization [6]. Breast cancer cell invasion also enables tumor cells to migrate through the connective tissue surrounding a tumor, enter the circulatory system, and extravasate at distant sites to form metastatic colonies [7–9]. Thus, unrestricted invasion is a very important, debilitating aspect of the metastatic phenotype [10].
Extravasation is a key step in hematogenous cancer metastasis [11]. It occurs after tumor cells arrest in microvasculature or sinusoids. Tumor cell arrest may occur by trapping, without stable adhesion to the microvasculature [12], or may involve preferential adhesion to the endothelial cells of specific sites [13]. In either case, after arrest, invasion of the vascular basement membrane and underlying interstitial connective tissue is required for extravasation [11].
We have devised a peptide of five amino acids that is a potent inhibitor of α5β1 integrin-mediated invasion by breast cancer cells. The acetylated, amidated PHSCN peptide, whose primary structure is Ac-PHSCN-NH2 [6, 8, 9], emerged as an invasion inhibitor during the study of the structure activity relationship of the invasion-inducing PHSRN peptide [14]. The PHSRN sequence is a known α5β1 ligand that interacts with a specific region of the α5 subunit of the α5β1 integrin, and is found in the cell binding domain of fibronectin [15, 16]. In addition to preventing prostate cancer cell invasion and metastasis [8], the PHSCN peptide is also a potent inhibitor of microvascular endothelial cell invasion and angiogenesis [6]. Systemic PHSCN therapy prevented disease progression for prolonged periods in a variety of preclinical models, and in phase 1 clinical trial [8, 17–20].
Thus, Ac-PHSCN-NH2 is a promising lead compound for targeted therapy of invasion and metastasis. To improve its potency, we attached eight Ac-PHSCNGGK peptide moieties to the N-termini of the multiantigenic peptide (MAP) polylysine dendrimer. We report that this creates a significantly more potent inhibitor of α5β1 integrin-mediated SUM-149 PT and MDA-MB-231 human breast cancer cell invasion and matrix metalloproteinase 1 (MMP-1) induction in vitro, and of extravasation and lung colony formation in vivo in athymic nude mice.
Materials and methods
Cell lines and cell culture
MDA-MB-231 metastatic [21] and SUM-149 PT [22] inflammatory human breast cancer cell lines were obtained from American Type Culture Collection (Manassas, VA), or from the laboratory of Dr. Stephen Ethier, respectively. Both cell lines were cultured as recommended, and frozen in liquid N2 in aliquots within 2 months of receipt. Single aliquots were subsequently resuscitated as needed, and cultured as recommended. No aliquot of cells was cultured for more than 4 months, and the morphologies of all cultures were routinely checked by phase contrast microscopy. Growth curves of all cultures were always recorded, and checked for consistency. For all assays in serum-free (SF) medium, SUM-149 PT and MDA-MB-231 cells were first serum-starved overnight.
Peptide and dendrimer synthesis
N-terminal acetylated, C-terminal amidated PHSRN, PHSCN, and HSPNC peptides (Ac-PHSRN-NH2, Ac-PHSCN-NH2, and Ac-HSPNC-NH2) were synthesized, their structures confirmed, and their purities assessed as described [6–9, 14]. Their purities were as follows: Ac-PHSRN-NH2 97%, Ac-PHSCN-NH2 98%, Ac-HSPNC-NH2 91%.
N-terminal acetylated PHSCN and HSPNC MAPs were synthesized by covalently attaching peptide C-termini to the N-termini of a polylysine dendrimer, 8 core MAP (Sigma-Aldrich, Saint Louis MO). MAPs were synthesized by Fmoc solid phase synthesis in a manual procedure with reaction monitoring by Ninhydrin test [23], to allow for complete coupling of each amino acid. Quality control of the MAPs was performed by amino acid analysis [24], followed by Edman sequencing and preview analysis to reveal any deletions in the sequences [25]. Dendrimer purities were estimated to be as follows: Ac-PHSCNGGK-MAP 94%; Ac-HSPNCGGK-MAP 97% (not shown). The MAPs were also evaluated by MALDI for the expected mass of the fully populated dendrimer. The spectra showed the expected mass for the complete MAP and very little evidence of incomplete synthesis.
PHSCN and HSPNC peptides to be attached to polylysine dendrimers or ovalbumin were synthesized with two glycines and a lysine (GGK) on the C-terminal end (PHSCNGGK or HSPNCGGK) to provide a spacer and an attachment site for labeling. Ac-PHSCNGGK-NH2 and Ac-HSPNCGGK-NH2 had functional characteristics identical to Ac-PHSCN-NH2 and Ac-HSPNC-NH2, respectively (not shown). PHSCN-coupled ovalbumin (Ac-PHSCNGGK-Ova) was synthesized by coupling the PHSCNGGK C-terminus to ovalbumin with an attached EDC (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) crosslinker (ThermoFisher Scientific, Waltham MA), according to established procedures [26]. Ac-PHSCNGGK-Ova purity was 90%.
In vitro invasion assays
Naturally serum-free, selectively permeable basement membranes from sea urchin embryos were utilized as in vitro invasion substrates, as described [6–9, 14]. All cells were serum starved prior to addition of 10% FBS or 0.1 μg/ml Ac-PHSRN-NH2 to stimulate invasion. Peptides or dendrimers were briefly prebound to cells in half of the final invasion assay volume, prior to placement on basement membranes in the remaining volume of medium. Invasion assays were incubated for 16 h at 37°C prior to scoring at 400-fold magnification under phase contrast optics, as described [6–9, 14]. Data, mean invasion percentages, were analyzed using Graphpad Prism 5 software (San Diego, CA) as a function of log (inhibitor) versus normalized data, variable slope.
Data analysis
Data were analyzed by the Combination index (CI) method of Chou–Talalay [27], as described in [28]. The analysis was based on the multiple drug effect equation derived from the median effect principle of the mass-action law. The median-effect equation, y = log(f a/f u), with respect to x = log(dose) defines the dose and effect relationship in the absence of reaction rate constants, where f a is the fraction of cells affected (invasion-inhibited), and f u is the fraction of cells unaffected (invaded). The x-intercept represents the IC50 value. CI and DRI values were determined assuming that the monomer represents the single dose, and the dendrimer (with 8 PHSCNGGK moieties) represents the combinatorial dose.
MMP-1 activity assays
Effects of Ac-PHSCNGGK-MAP, Ac-HSPNCGGK-MAP, or Ac-PHSCN-NH2 on MMP-1 activities secreted by adherent cells were analyzed as described [6, 7, 9]. Adherent cells were serum-starved overnight, then pretreated with Ac-PHSCN-NH2, Ac-PHSCNGGK-MAP, or Ac-HSPNCGGK-MAP at 250 μg per 1,000,000 cells for 1 h. After pretreatment, the treatment groups were stimulated with 5 ml 10% FBS or SF medium containing 250 μg per 1,000,000 cells Ac-PHSRN-NH2 for 24 h prior to assay. Treatment groups were run in triplicate. Supernatants from treated cells were collected for analysis. Human MMP-1 ELISA Kit (RayBiotech, Inc., Norcross GA) was used to quantitate MMP-1 activity, according to manufacturer’s instructions. Data are presented as means ± SD, and results were analyzed using Student’s t-test.
Fluorescent DiI and DiO labeling of cells
Confluent SUM-149 PT or MDA-MB-231 cells were washed with Hanks-buffered salt solution (HBSS; Life Technologies, Grand Island NY), harvested with 0.25% trypsin/1% EDTA (Life Technologies), rewashed in HBSS, and fluorescently labeled in 6 ml SF medium with 25 μl of the red lipophilic carbocyanine vital dye DiI, 1,1′-dilinoleyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate (Invitrogen) or 25 μl of the green lipophilic vital dye DiO, 3,3′-Dioctadecyloxacarbocyanine iodide, for 20 min in dark at 37 °C, as described [29, 30]. Cells were pelleted at 1,000 rpm for 2 min, prior to resuspension in 6 ml of medium, and recovery for 72 h. DiI has been utilized as a vital dye in numerous studies, e.g., to define neural crest invasive/migratory pathways in developing embryos [31]. DiI labeling has been reported to persist in living, cultured neurons for a year [32].
Extravasation in the lungs athymic mice
To evaluate the importance of α5β1 integrin in breast cancer cell extravasation, DiI labeled SUM-149 PT or MDA-MB-231 cells were incubated for 30 min on ice with 10 μg/ml or 50 μg/ml anti α5β1 MCA1187 function blocking monoclonal antibody (MAb) (Serotec, Oxford, England), as described [33]. A total of 10,000 pretreated cells in 0.1 ml HBSS were injected into the tail vein of each 8-week, nude athymic mouse (Jackson Laboratories, Bar Harbor ME). Each treatment group consisted of 10 mice. All mice were euthanized 24 h later, and their lungs removed and thoroughly rinsed with phosphate-buffered saline (PBS) prior to fixation with 4% paraformaldehyde in PBS overnight at 4°C. Following fixation, lungs were rinsed at room temperature in PBS, and placed in 20% sucrose overnight at 4°C. The samples were cryoprotected by submerging in optimal cutting temperature (O.C.T.): 20% sucrose mixture (VWR, Batavia IL) overnight at 4°C, prior to freezing and storing at −80°C. Frozen lungs were sectioned with a thickness of 10 μm with a Zeiss Cryostat (M550). All slides were sealed with VECTASHIELD mounting medium with DAPI (VECTOR Laboratories, Burlingame CA). Twenty sections were cut from each lung in all mice of each treatment group of 10 mice, at 200 μm intervals. Thus, a total thickness of 4 mm was analyzed from each lung. All sections were examined at 400-fold magnification with a Zeiss Scanning Laser Confocal microscope (LSM510), as previously described [34].
To compare Ac-PHSCNGGK-MAP and Ac-HSPNCGGK-MAP as extravasation inhibitors, DiI-labeled SUM-149 PT or MDA-MB-231 cells were prebound with appropriate concentrations of dendrimer or peptide in HBSS and incubated at 37°C for 10 min. The concentrations used for prebinding were as follows: Ac-PHSCNGGK-MAP: 10, 1, 0.1, or 0.01 ng/ml; Ac-PHSCN-NH2: 100, 10, 1, or 0.1 ng/ml; Ac-HSPNCGG-MAP: 100 ng/ml. Nude mice (Jackson Laboratories) received an equivalent intravenous dose of dendrimer, peptide (0.0005 to 5.0 μg/kg), or HBSS in 0.1 ml at the appropriate concentrations via tail vein, immediately prior to intravenous injection of DiI labeled, dendrimer- or peptide-prebound SUM-149 PT or MDA-MB-231 cells. Labeled, pretreated cells were intravenously injected in 0.1 ml HBSS, and mice euthanized 24 h later, as described above. Lungs were removed, rinsed, fixed, treated, and frozen as described above. Twenty sections were cut from each lung in all mice of each treatment group of 10 mice, at 200 μm intervals. Thus, a total thickness of 4 mm was analyzed from each lung. All sections were examined at 400-fold magnification with a Zeiss Scanning Laser Confocal microscope (LSM510), as previously described [34].
To determine what fractions of SUM-149 PT and MDA-MB-231 cells were intravascular or extravascular, sections made as described above were stained with rat anti-mouse PECAM-1 (platelet endothelial cell adhesion molecule-1) monoclonal antibody (Millipore, Temecula CA) [35], and FITC-conjugated secondary antibody (Jackson Laboratories, Bar Harbor, Maine). Sections were scored as described above, with SUM-149 PT and MDA-MB-231 cells surrounded by at least 75% of their circumference with anti PECAM staining judged to be intravascular. Cells surrounded by less anti PECAM staining were judged to be extravascular, and hence to have completed extravasation.
Lung colony formation assays
To verify the enhanced potency of Ac-PHSCNGGK-MAP as an inhibitor of lung colony formation, suspended, DiI labeled SUM-149 PT or MDA-MB-231 cells were briefly prebound with 10 ng/ml Ac-PHSCNGGK-MAP, 10 ng/ml and 100 ng/ml Ac-PHSCN-NH2, or 100 ng/ml Ac-HSPNCGGK-MAP. Eight-week-old, nude mice (Jackson Laboratories) received one systemic pretreatment with the appropriate peptide or dendrimer in 0.1 ml HBSS by tail vein injection. Immediately after pretreatment of the mice, SUM-149 PT or MDA-MB-231 cells, prebound with dendrimer or peptide, or with HBSS only, were injected into tail veins. Six weeks after injection, mice were euthanized, and lungs were removed, fixed, prepared, and stored as above.
Sections of 10 μm thickness were cut with a Zeiss cryostat as previously described [36]: 20 sections were cut from each lung in all mice of each treatment group, at 200 μm intervals. Thus, a total thickness of 4 mm was analyzed from each lung. Sections were stained with monoclonal, FITC-conjugated anti β-actin antibody (Chemicon, Temecula CA) and scored at 400-fold magnification, based on the retention of DiI red fluorescence by the prostate cancer cells, as described for other cancer types [36–38]. SUM-149 PT and MDA-MB-231 colonies were scored only if they contained more than 50 cells, according to established criteria [37–39]. Data are presented as mean numbers of colonies per lung section ± SEM of 10 mice per group.
Clonogenic assays
Clonogenic survival assays were performed as described [40].
Results
Inhibition of extravasation by blocking anti α5β1 monoclonal antibody
We evaluated the potential role of α5β1 integrin in extravasation directly by prebinding suspended SUM-149 PT and MDA-MB-231 cells to blocking anti α5β1 MCA1187 MAb [33] prior to intravenous injection into mouse tail veins. A mean of 1741 (±25 SEM) SUM-149 PT or 2119 (±31 SEM) MDA-MB-231 cells per 10 μm section was observed in the untreated groups. As shown in Fig. 1, MCA1187 MAb pretreatment reduced the numbers of SUM-149 PT (Fig. 1a) and MDA-MB-231 (Fig. 1b) cells in lung tissue after 24 h by eight- to ten-fold, suggesting that α5β1 plays a key role in extravasation.

Inhibition of SUM-149 PT and MDA-MB-231 cell extravasation in the lungs of nude mice after 24 h by pretreatment with MCA1187 blocking anti α5β1 MAb. x-axes treatments; y-axes mean percentages of extravasated cells, relative to control (±SEM). a Inhibition of SUM-149 PT extravasation. b Inhibition of MDA-MB-231 extravasation
Invasion inhibition by the PHSCN polylysine dendrimer, Ac-PHSCNGGK-MAP
We utilized naturally serum-free, selectively permeable basement membranes from sea urchin embryos [6–9, 14] as in vitro invasion substrates to compare the invasion inhibitory potencies of the PHSCN polylysine dendrimer (Ac-PHSCNGGK-MAP) and the PHSCN peptide (Ac-PHSCN-NH2). To compare their effects on serum-induced invasion, SUM-149 PT and MDA-MB-231 cells were serum-starved overnight, prior to suspension in medium containing 10% FBS, and prebinding to various concentrations of the PHSCN dendrimer or PHSCN peptide or to maximal concentrations of the specificity controls: the HSPNC dendrimer (Ac-HSPNCGGK-MAP) or HSPNC peptide (Ac-HSPNC-NH2).
Our previous research showed that the PHSRN sequence of the fibronectin cell binding domain [7–9] is specifically responsible for serum- or plasma fibronectin-induced invasion by human breast and prostate cancer cell lines. In addition, we found that the PHSRN peptide is a potent inducer of invasion by normal microvascular endothelial cells, epithelial cells, and fibroblasts [6, 14]. Thus, we also compared the invasion-inhibitory potencies of PHSCN dendrimer and PHSCN peptide on PHSRN-induced invasion by serum-free (SF) SUM-149 PT and MDA-MB-231 cells. Since the PHSCN dendrimer displays the PHSCN sequence on a significantly larger molecule (7575 vs. 598 Da), we also evaluated the effect on invasion inhibitory potency of a modified protein, resulting from coupling a single Ac-PHSCNGGK sequence to ovalbumin (45.0 kDa), to make Ac-PHSCNGGK-Ova (45.9 kDa).
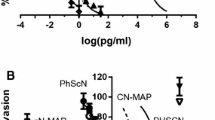
Hill plots depicting the results of these in vitro invasion assays are presented in Fig. 2. The (log) nM concentrations of the PHSCN dendrimer and PHSCN peptide, and the HSPNC dendrimer and PHSCN–ovalbumin negative controls were plotted versus the mean percentages of DU 145 and PC-3 cells invaded, after stimulation with 10% FBS, or with 100 ng/ml Ac-PHSRN-NH2, under serum-free conditions. Irrespective of whether α5β1 mediated invasion was stimulated by the presence of serum or by the PHSRN peptide under serum-free conditions, the PHSCN dendrimer was three orders of magnitude more potent as an invasion inhibitor than the PHSCN peptide. High concentrations of HPSNC dendrimer failed to block invasion. Also, putting a single Ac-PHSCNGGK moiety on a larger molecule, ovalbumin, failed to increase its potency significantly. These data were also plotted according to the median-effect equation, y = log(f a/f u), with respect to x = log(dose), where f a is the fraction of cells affected (invasion-inhibited), and f u is the fraction of cells unaffected (invaded). This analysis indicated a linear coefficient (r) value >0.97, suggesting conformity to the mass-action law principle (not shown).

Hill plots of the increased invasion-inhibitory potency of Ac-PHSCNGGK-MAP as compared with the Ac-PHSCN-NH2 peptide with naturally serum-free, selectively permeable basement membranes from sea urchin embryos as invasion substrates. a Inhibition of serum-induced SUM-149 PT invasion. b Inhibition of serum-induced MDA-MB-231 invasion. c Inhibition of Ac-PHSRN-NH2–induced, SF SUM-149 PT invasion. d Inhibition of Ac-PHSRN-NH2-induced, SF MDA-MB-231 invasion. x-axes log peptide concentration in nM; y-axes mean percentages of invaded cells, relative to uninhibited, FBS- or PHSRN-stimulated controls (±SD). Black circles Ac-PHSCNGGK-MAP, black squares Ac-PHSCN-NH2 peptide, black diamonds Ac-HSPNCGGK-MAP, white circles Ac-HSPNC-NH2 peptide, white squares Ac-PHSCNGGK-Ova
The IC50 values calculated from the Hill plots (Fig. 2), were similar to those derived from the median-affect equation, and are listed in units of nM in Table 1. As shown by the Dose reduction index (DRI) values for SUM-149 PT and MDA-MB-231 cells PHSCN dendrimer were 1280- to 1691-fold, and 6747- to 2066-fold more potent than PHSCN peptide at blocking FBS-induced or SF, Ac-PHSRN-NH2-induced invasion, respectively.
The combination index (CI) and the DRI values, shown in Table 1, allow determination of whether the 8 PHSCN moieties on the dendrimer result in an additive or a synergistic increase in invasion inhibitory potency. Synergism is demonstrated by CI <1 and DRI >1, and additive effects are indicated by CI ≥1 and DRI ≥1 [27]. Thus, in combination, the CI and DRI values listed in Table 1 for both cell lines show the increased efficacy of the PHSCN dendrimer, and indicate that its 8 PHSCN moieties interact synergistically to block invasion for in vitro assays.
Inhibition of serum- or PHSRN-induced MMP-1 secretion by Ac-PHSCNGGK-MAP
Our prior research has demonstrated the induction of MMP-1 secretion and MMP-1-dependent invasion by the PHSRN/α5β1 interaction in mammary epithelial cells, breast cancer cells, microvascular endothelial cells, as well as in prostate epithelial cells and prostate cancer cells [6–9]. We have also observed the inhibition of α5β1-mediated MMP-1 secretion in human prostate cancer cells by the PHSCN peptide [8, 9]. These results suggest that the PHSCN dendrimer should be a significantly more potent inhibitor of α5β1-mediated MMP-1 induction in breast cancer cells by serum, or by the PHSRN peptide in SF conditions. Thus, adherent SUM-149 PT were serum-starved overnight prior to peptide or dendrimer pretreatment and stimulation with medium containing either 10% FBS or Ac-PHSRN-NH2, at 250 μg/1,000,000 cells. These were the same conditions used to stimulate MMP-1 secretion in prior studies [6, 7, 9]. Inhibitors of serum- or PHSRN peptide-induced MMP-1 secretion included the PHSCN peptide, the PHSCN dendrimer, or the HSPNC dendrimer specificity control, all at a concentration of 250 μg/1,000,000 cells. The results of MMP-1 assays for SUM149PT cells are shown in Fig. 3; while the results of MMP-1 assays for MDA-MB-231 cells are shown as Figure 1 of the supplemental data. As shown in Fig. 3, the PHSCN dendrimer was a more potent inhibitor of α5β1-mediated MMP-1 secretion by SUM-149 PT cells than the PHSCN peptide, whether induced by the presence of 10% FBS or by the PHSRN peptide under SF conditions.

Increased potency of Ac-PHSCNGGK-MAP as an inhibitor of α5β1-mediated MMP-1 secretion in vitro by SUM-149PT cells by ELISA detection. x-axis treatment groups in triplicate of Ac-PHSCN-NH2, Ac-PHSCNGGK-MAP, and Ac-HSPNCGGK-MAP at 250 μg/1,000,000 cells; black bars, stimulation with 10% FBS; white bars, stimulation with Ac-PHSRN-NH2 at 250 μg/1,000,000 cells. y-axis, mean % MMP-1 activity relative to stimulated control cells (±SD)
Inhibition of SUM-149 PT or MDA-MB-231 cell extravasation in nude mice by prebinding Ac-PHSCNGGK-MAP or Ac-PHSCN-NH2
The enhanced invasion inhibitory potency of the PHSCN dendrimer, relative to the PHSCN peptide suggested that the PHSCN dendrimer should also be a more potent inhibitor of SUM-149 PT or MDA-MB-231 extravasation into mouse lungs. To compare efficacies of the PHSCN dendrimer and PHSCN peptide as breast cancer cell extravasation inhibitors, SUM-149 PT or MDA-MB-231 cells were labeled with DiI, then briefly prebound to varying concentrations of PHSCN dendrimer, HSPNC dendrimer or PHSCN peptide. After a brief prebinding, the suspended, DiI-labeled SUM-149 PT or MDA-MB-231 cells were injected into the tail veins of athymic, nude mice. Twenty-four hours later, all mice were killed and their lungs removed for analysis of extravasation by fluorescent confocal microscopy. A mean of 1,623 (±54 SEM) SUM-149 PT or 1268 (±59 SEM) MDA-MB-231 cells per 10 micron section was observed when untreated cells were intravenously injected into the tail veins of nude mice. Pretreatment of either cell line, with 0.01–10 ng/ml Ac-PHSCNGGK-MAP or 0.1–100 ng/ml Ac-PHSCN-NH2, resulted in dose dependent reductions of the numbers of cells in lung tissue, and are shown in Fig. 4a, b as a function of concentration (nM). Pretreatment with the scrambled sequence control dendrimer (100 ng/ml Ac-HSPNCGGK-MAP) resulted in a mean of 1,103 (±65) or 975 (±63) when the same number of suspended SUM-149 PT cells or MDA-MB-231 cells were injected, respectively; indicating little or no effect. Typical examples of the data for MDA-MB-231 extravasation are shown in Fig. 4c. Sections containing extravasated SUM-149 PT cells had a very similar appearance (not shown).

Increased extravasation inhibition in the lungs of nude mice after 24 h by Ac-PHSCNGGK-MAP prebinding, relative to the PHSCN peptide. x-axes log nM PHSCN dendrimer or PHSCN peptide. y-axes, log fa/fu ± SEM. Black circles Ac-PHSCNGGK-MAP, black squares Ac-PHSCN-NH2 peptide. a Median-effect plot for the dose response of DiI labeled SUM-149 PT cells. b Median-effect plot for the dose response of DiI labeled MDA-MB-231 cells. c DiI labeled MDA-MB-231 cell extravasation, typical example of a scored section. Arrows indicate all extravasated cells visible in section. Sections showing DiI labeled SUM149PT extravasation had a very similar appearance (not shown)
The IC50 values were determined from the extrapolated x-intercept (Fig. 4a, b) and the DRI and CI values were calculated, as summarized in Table 2. The increased potency of Ac-PHSCNGGK-MAP over the Ac-PHSCN-NH2 peptide (700–1100-fold) for preventing extravasation was similar to that found for in vitro invasion assays with naturally occurring basement membranes. Furthermore, the DRI and CI values shown in Table 2 indicate that extravasation inhibition was highly synergistic.
The complete process of extravasation requires that blood-borne cells cross the endothelial cell layer and its basement membrane to enter the underlying connective tissue. To determine what fractions of SUM-149 PT and MDA-MB-231 cells in the lungs were fully extravasated, and hence outside of the vasculature, a constant number of fluorescent DiI-labeled, Ac-PHSCNGGK-MAP-pretreated (10 ng/ml) or untreated cells were injected in the tail veins of nude mice. To delineate precisely the vascular walls, lung sections were immunofluorescently stained by reaction with rat anti-mouse PECAM-1 antibody [35], and fluorescent secondary antibody. The total numbers of DiI-labeled SUM-149 PT and MDA-MB-231 cells were counted in 20 lung sections from each lung of each mouse injected. Their positions relative to the anti PECAM-stained endothelial cell layer were noted as described above, and the results of the analysis are shown in Fig. 5a. Typical examples of sectioned anti PECAM-1-stained blood vessels, with DiI-labeled SUM-149 PT or MDA-MB-231 cells, inside or outside of the lung vasculature, are shown in Fig. 5b, c, respectively.

Reduced extravascular breast cancer cells in vivo after PHSCNGGK-MAP pretreatment. a Quantitation of intravascular and extravascular SUM-149 PT and MDA-MB-231 cells in 10 μm sectioned mouse lungs. x-axis cell lines, y-axis mean percentages of cells (±SEM). b, c Examples of SUM-149 PT cells (b) and MDA-MB-231 cells (c) in lung sections from mice injected with untreated (Control) or 10 ng/ml Ac-PHSCNGGK-MAP pretreated cells. Sections were stained with anti PECAM-1. Arrows extravascular cells. Stars intravascular cells
The results of this experiment indicate that Ac-PHSCNGGK-MAP pretreatment caused 73.3% (SUM-149 PT) or 82.0% (MDA-MB-231) of DiI-labeled breast cancer cells to remain in the lung vasculature; while 26.7% or 18.0%, respectively, appeared to have extravasated. In contrast, only 12.3% (SUM-149 PT) or 9.3% (MDA-MB-231) of untreated breast cancer cells appeared to remain inside the vasculature; whereas, 87.7% or 90.7%, respectively, appeared to have extravasated. Thus, a single, brief Ac-PHSCNGGK-MAP pretreatment at 10 ng/ml was able to reduce breast cancer cell extravasation in the lungs of nude mice by 3- to 5-fold.
Inhibition of SUM-149 PT or MDA-MB-231 lung colony formation in nude mice by prebinding Ac-PHSCNGGK-MAP or Ac-PHSCN-NH2
Extravasation is a key step in metastasis formation [11, 39]. Based on its increased anti-invasive potency on naturally occurring basement membranes in vitro, the PHSCN dendrimer should be a more potent inhibitor of lung colony formation than PHSCN peptide in nude mice, after intravenous injection with pretreated SUM-149 PT or MDA-MB-231 cells. We found that pretreatment with 10 ng/ml (1.32 nM) Ac-PHSCNGGK-MAP reduced SUM-149 PT lung colony formation by 4.4-fold, relative to untreated controls; whereas, pretreatment with 10 ng/ml (17 nM) Ac-PHSCN-NH2 had a less inhibitory effect: reducing SUM-149 PT lung colonies by 1.9-fold (Fig. 6a). Pretreatment with a 10-fold higher concentration of Ac-PHSCN-NH2 (100 ng/ml, 170 nM) reduced SUM-149 PT lung colony formation by 3.4-fold, similar to the reduction observed after pretreatment with 10 ng/ml (1.32 nM) Ac-PHSCNGGK-MAP. No significant reduction in lung colony formation was obtained by pretreatment of SUM-149 PT cells with Ac-HSPNCGGK-MAP, at 100 ng/ml (170 nM).

Increased inhibition of lung colony formation by Ac-PHSCNGGK-MAP pretreatment, relative to PHSCN peptide pretreatment. a Dose response of DiI labeled SUM-149 PT colonies. b Dose response of DiI labeled MDA-MB-231 colonies. x-axes: log (nM) pretreatment. y-axes: % Colonies/section as compared to untreated controls (±SEM). Black bars Ac-PHSCNGGK-MAP pretreated cells, white bars Ac-PHSCN-NH2 pretreated cells, dark gray bars Ac-HSPNCGGK-MAP pretreated cells. c, d Examples of sectioned lung tissue, stained with FITC-conjugated anti β-actin, obtained from mouse lungs after 6 weeks of colony growth. Pretreatment conditions are indicated, and are as follows: untreated (Control), PHSCNGGK-MAP pretreated (10 ng/ml), and HSPNCGGK-MAP pretreated (100 ng/ml). DiI labeled SUM-149 PT colonies (c) or MDA-MB-231 colonies (d) are shown
After pretreatment with either 10 ng/ml (17 nM) or 100 ng/ml (170 nM) of Ac-PHSCN-NH2, lung colony formation by MDA-MB-231 cells (Fig. 6b) was reduced by approximately 50%. In contrast, a dose of 10 ng/ml (1.32 nM) of Ac-PHSCNGGK-MAP achieved 80% inhibition, similar to that observed for SUM-149 PT cells. The overall reduction of lung colonization by Ac-PHSCNGGK-MAP pretreatment in both cell lines suggests at least 2 orders of magnitude in increased potency for SUM-149-PT cells, and perhaps even more in MDA-MB-231 cells (Fig. 6a, b). This allows estimation of a DRI of approximately 100–1,000-fold.
Since only growing micrometastases are functionally relevant, the total number of cells in each micrometastasis was determined for each treatment group shown in Fig. 6. Each micrometastasis included in the totals contained from 50 to 200 cells (data not shown). No micrometastases were scored unless they contained at least 50 cells, similar to criteria previously employed by others [36–38].
Because it was possible that inhibition of lung colony formation by the PHSCN dendrimer might have been due to reduction in survival of the extravasated cells, the effects on clonogenic survival of several PHSCN dendrimer concentrations were compared to elevated concentrations of HSPNC dendrimer and PHSCN peptide. The effects of various concentrations of Ac-PHSCNGGK-MAP on the clonogenic survival of MDA-MB-231 and SUM149PT cells were compared to the effects of elevated concentrations (100–200 μg per ml) of Ac-HSPNCGGK-MAP or Ac-PHSCN-NH2. As shown in Fig. 7, the presence of 50–200 μg per ml of the PHSCN dendrimer did reduce MDA-MB-231 clonogenic survival in a dose-dependent manner; while elevated concentrations (100–200 μg per ml) of the HSPNCGGK dendrimer or the PHSCN peptide had no effect. However, the concentrations of PHSCN dendrimer required to reduce clonogenic survival were 500- to 20,000-fold higher than the concentrations used in the pretreatment to prevent extravasation and inhibit lung colony growth (0.01–0.1 μg per ml). Thus, inhibition of MDA-MB-231 clonogenic survival by PHSCN dendrimer pretreatment was very unlikely. Quantitatively, similar results were obtained for SUM149PT cells, as shown in Figure 2 of the supplemental data.

Effects of Ac-PHSCNGGK-MAP on clonogenic survival in vitro by MDA-MB-231 cells. x-axis: μg per ml, y-axis mean number of colonies per well (±SD). Black bars Ac-PHSCNGGK-MAP, dark gray bars Ac-HSPNCGGK-MAP, light gray bars Ac-PHSCN-NH2, white bar untreated
Discussion
Chemotherapy is utilized for the treatment of breast cancer patients with symptomatic visceral metastases, and in patients without symptomatic visceral metastases whose tumors are estrogen receptor and progesterone receptor negative, thus ruling out hormonal therapy [41]. Despite therapeutic advances, a majority of patients with node-positive breast cancer eventually develop recurrence, most frequently to the lungs, liver, or bone. Overall, patients with metastatic breast cancer have a median survival of 24 months, but those with visceral metastasis survive for significantly shorter times than those with metastasis only to bone [4]. Thus, while there is no cure for metastatic breast cancer, occasional prolonged periods of remission have been reported. For example, among patients who had a complete remission after anthracycline-based therapy, 17% (or 3% of all treated patients) remained disease-free for 5 years [42]. Thus, palliation of cancer-related symptoms and possible prolongation of life are primary goals of chemotherapy.
However, because they target DNA structure or the microtubules involved in spindle formation [43], chemotherapeutic agents also adversely affect rapidly proliferating cells of the gut, the skin, and the bone marrow. Furthermore, breast cancer cells can become resistant to anthracyclines and taxanes [43, 44]. Because breast cancer invasion is a very important, debilitating aspect of the metastatic phenotype [10], there is a need for an effective, targeted, systemic anti-invasive agent for breast cancer.
When unchecked by normal regulatory mechanisms, the regulatory pathways initiated by α5β1 integrin may give rise to the progressive growth and invasion observed in metastatic breast cancer. For example, the interaction of α5β1 with the PHSRN sequence of the fibronectin cell binding domain is sufficient to induce interstitial collagenase expression and basement membrane invasion by microvascular endothelial cells [6] and metastatic breast cancer cells [7], as well as prostate cancer cells [8, 9] and normal epithelial cells and fibroblasts [14]. Because fibronectin is found in all body fluids, proper regulation of α5β1-mediated invasion is very important. This is accomplished by another integrin fibronectin receptor, α4β1. When fibronectin is intact, α4β1 integrin interacts with the LDV sequence of the fibronectin connecting segment, LHGPEILDVPST, to repress α5β1-mediated interstitial collagenase expression [45]. Fragmentation of fibronectin by urokinase plasminogen activator, which also functions in clot dissolution (reviewed in [46]), gives rise to α5β1 mediated invasion during wound healing [47, 48]. Thus, an important attribute of α5β1-induced invasion in normal α5β1+, α4β1+ cells is its regulation by α4β1 integrin. Although still expressing abundant surface α5β1 many metastatic breast cancer cell lines have low levels of surface α4β1 relative to mammary epithelial cells [49]. Loss of surface α4β1, which can result from ErbB-2 oncogene overexpression in transformed mammary epithelial cells [49–51], has been shown to cause constitutive invasiveness in human metastatic breast and prostate cancer cell lines [7, 9] in the presence of the abundant pFn of blood, lymph, and interstitial fluid [52, 53].
Here, we show that attaching 8 PHSCN peptide moieties to a polylysine dendrimer results in a 60- to 305-fold increase in invasion-inhibitory potency for metastatic human breast cancer cell lines in vitro, and a significant increase in ability to inhibit extravasation and lung colony formation in vivo in athymic, nude mice. Because only highly elevated concentrations of PHSCN dendrimer affected clonogenic survival of SUM-149PT and MDA-MB-231 cells in vitro, inhibition of extravasation and colony formation by low PHSCN dendrimer concentrations was very unlikely to be due to reductions in survival.
The rationale for employing PHSCN-dendrimers as improved therapeutic agents is that their polyvalency will enhance the affinity of their interactions with their target protein, the α5β1 integrin. There are many examples of the ability of multimeric peptides to enhance binding affinity. For example, a peptide complementary to a specific region of endothelin exhibited a binding affinity that was 100-fold greater when it was synthesized as a MAP peptide [54]. Peptide dendrimers are also used as inhibitors of increased potency. For example, peptide dendrimers have been used to inhibit the entry of malaria parasites into hepatocytes [55]. In another example, a peptide dendrimer based on an 8 amino acid sequence from gp120, the HIV-1 surface envelope glycoprotein, has been shown to inhibit HIV-1 infection of both CD4+ and CD4− cells [56, 57]. In an application closely related to ours, the increased binding affinity of dendrimers, compared to monomeric peptides, has been used to design anti-metastatic drugs. For example, a dendrimer containing 16 copies of the YIGSR laminin peptide has been found to inhibit tumor metastasis to lung tissue, and slow tumor growth, when administered as a systemic agent in mice bearing B16F10 melanoma [58].
Thus, systemic therapy with a potent anti-invasive agent, such as the PHSCN–dendrimer, that effectively targets the activated α5β1 integrins of tumor cells, without affecting the unactivated α5β1 receptors of healthy tissues, could potentially allow patients with metastatic breast cancer to live for long periods of time with their disease, without many toxic side effects. Because of the PHSCN peptide’s efficacy and lack of toxicity, when used as a systemic monotherapy in Phase 1 clinical trial [17], more potent PHSCN peptide derivatives, such as the PHSCN-dendrimer, may also be useful additions to radiotherapy or more conventional chemotherapeutic regimens.
Abbreviations
- MAP:
-
Multiantigenic peptide
- SF:
-
Serum-free
- FBS:
-
Fetal bovine serum
- CI:
-
Combination Index
- DRI:
-
Dose reduction index
- Ova:
-
Ovalbumin
- EDC:
-
1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride
- HBSS:
-
Hanks buffered salt solution
- MALDI:
-
Matrix assisted laser desorption/ionization
- MMP-1:
-
Matrix metalloproteinase-1
- ELISA:
-
Enzyme-linked immunoabsorbant assay
- DiI:
-
1,1′-Dilinoleyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate
- MAb:
-
Monoclonal antibody
- SD:
-
Standard deviation
- SEM:
-
Standard error of the mean
- PECAM-1:
-
Platelet endothelial cell adhesion molecule-1
- OCT:
-
Optimal cutting temperature
- FITC:
-
Fluorescein isothiocyanate
References
Sanchez-Munoz A, Perez-Ruiz E, Ribelles N, Marquez A, Alba E (2008) Maintenance treatment in metastatic breast cancer. Expert Rev Anticancer Ther 8:1907–1912. doi:10.1586/14737140.8.12.1907
Perez EA (2009) Impact, mechanisms, and novel chemotherapy strategies for overcoming resistance to anthracyclines and taxanes in metastatic breast cancer. Breast Cancer Res Treat 114:195–201. doi:10.1007/s10549-008-0005-6
Chan A (2009) Antiangiogenic therapy for metastatic breast cancer: current status and future directions. Drugs 69:167–181. doi:10.2165/00003495-200969020-00003
Giordano SH, Buzdar AU, Smith TL, Kau SW, Yang Y, Hortobagyi GN (2004) Is breast cancer survival improving? Cancer 100:44–52. doi:10.1002/cncr.11859
Higgins MJ, Wolff AC (2008) Therapeutic options in the management of metastatic breast cancer. Oncology 22:614–623 (discussion 623, 627–629)
Zeng ZZ, Yao H, Staszewski ED, Rockwood KF, Markwart SM, Fay KS, Spalding AC, Livant DL (2009) Alpha(5)beta(1) integrin ligand PHSRN induces invasion and alpha(5) mRNA in endothelial cells to stimulate angiogenesis. Transl Oncol 2:8–20
Jia YF, Markwart SM, Rockwood KF, Woods-Ignatoski KM, Ethier SP, Livant DL (2004) Integrin fibronectin receptors in MMP-1 dependent invasion by breast cancer and mammary epithelial cells. Cancer Res 64:8674–8681
Livant DL, Brabec RK, Pienta KJ, Allen DL, Kurachi K, Markwart S, Upadhyaya A (2000) Anti-invasive, antitumorigenic, and antimetastatic activities of the PHSCN sequence in prostate carcinoma. Cancer Res 60:309–320
Zeng Z-Z, Jia YF, Hahn NJ, Markwart SM, Rockwood KF, Livant DL (2006) Role of focal adhesion kinase and phosphatidylinositol 3′-kinase in integrin fibronectin receptor-mediated, matrix metalloproteinase-1 dependent invasion by metastatic prostate cancer cells. Cancer Res 66:8091–8099
White DE, Muller WJ (2007) Multifaceted roles of integrins in breast cancer metastasis. J Mammary Gland Biol Neoplasia 12:135–142. doi:10.1007/s10911-007-9045-5
Miles FL, Pruitt FL, van Golen KL, Cooper CR (2008) Stepping out of the flow: capillary extravasation in cancer metastasis. Clin Exp Metastasis 25:305–324. doi:10.1007/s10585-007-9098-2
Guba M, Bosserhoff AK, Steinbauer M, Abels C, Anthuber M, Buettner R, Jauch KW (2000) Overexpression of melanoma inhibitory activity (MIA) enhances extravasation and metastasis of A-mel 3 melanoma cells in vivo. Br J Cancer 83:1216–1222. doi:10.1054/bjoc.2000.1424
Matsuura N, Puzon-McLaughlin W, Irie A, Morikawa Y, Kakudo K, Takada Y (1996) Induction of experimental bone metastasis in mice by transfection of integrin alpha 4 beta 1 into tumor cells. Am J Pathol 148:55–61
Livant DL, Kurachi K, Allen DL, Wu Y, Haaseth R, Andrews P, Ethier SP, Markwart S (2000) The PHSRN sequence induces extracellular matrix invasion and accelerates wound healing in obese diabetic mice. J Clin Invest 105:1537–1545
Aota S, Nagai T, Yamada KM (1991) Characterization of regions of fibronectin besides the arginine-glycine-aspartic acid sequence required for adhesive function of the cell-binding domain using site-directed mutagenesis. J Biol Chem 266:15938–15943
Mould AP, Askari JA, Aota SI, Yamada KM, Irie A, Takada Y, Mardon HJ, Humphries MJ (1997) Defining the topology of integrin alpha5beta1-fibronectin interactions using inhibitory anti-alpha5 and anti-beta1 monoclonal antibodies. Evidence that the synergy sequence of fibronectin is recognized by the amino-terminal repeats of the alpha5 subunit. J Biol Chem 272:17283–17292
Cianfrocca ME, Kimmel KA, Gallo J, Cardoso T, Brown MM, Hudes G, Lewis N, Weiner L, Lam GN, Brown SC, Shaw DE, Mazar AP, Cohen RB (2006) Phase I trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2) a beta integrin antagonist, in patients with solid tumours. Br J Cancer 94:1621–1626
Khalili P, Arakelian A, Chen G, Plunkett ML, Beck I, Parry GC, Donate F, Shaw DE, Mazar AP, Rabbani SA (2006) A non-RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol Cancer Ther 5:2271–2280
Stoeltzing O, Liu W, Reinmuth N, Fan F, Parry GC, Parikh AA, McCarty MF, Bucana CD, Mazar AP, Ellis LM (2003) Inhibition of integrin alpha5beta1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int J Cancer 104:496–503
van Golen KL, Bao LW, Brewer GJ, Pienta KJ, Kamradt JM, Livant DL, Merajver SD (2002) Suppression of tumor recurrence and metastasis by a combination of the PHSCN sequence and the antiangiogenic compound tetrathiomolybdate in prostate carcinoma. Neoplasia 4:373–379
Cailleau R, Olive M, Cruciger QV (1978) Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro 14:911–915
van Golen KL, Davies S, Wu ZF, Wang Y, Bucana CD, Root H, Chandrasekharappa S, Strawderman M, Ethier SP, Merajver SD (1999) A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res 5:2511–2519
Kaiser E, Colescott RL, Bossinger CD, Cook PI (1970) Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem 34:595–598
Remmer H, Fields G (2000) Chemical synthesis of peptides. In: Reid RE (ed) Peptide and protein drug analysis. Marcel Dekker, Inc., New York, pp 133–169
Grant GA (2002) Evaluation of the synthetic product. In: Grant GA (ed) Synthetic peptides: a user’s guide, 2nd edn. Oxford University Press, Oxford, pp 220–291
DeSilva NS, Ofek I, Crouch EC (2003) Interactions of surfactant protein D with fatty acids. Am J Respir Cell Mol Biol 29:757–770. doi:10.1165/rcmb.2003-0186OC
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55
Ren H, Tan X, Dong Y, Giese A, Chou TC, Rainov N, Yang B (2009) Differential effect of imatinib and synergism of combination treatment with chemotherapeutic agents in malignant glioma cells. Basic Clin Pharmacol Toxicol 104:241–252. doi:10.1111/j.1742-7843.2008.00371.x
Godement P, Vanselow J, Thanos S, Bonhoeffer F (1987) A study in developing visual systems with a new method of staining neurones and their processes in fixed tissue. Development 101:697–713
Heimer L, Záborszky L (1989) Neuroanatomical tract-tracing methods, 2: recent progress. Plenum, New York
Collazo A, Bronner-Fraser M, Fraser SE (1993) Vital dye labelling of Xenopus laevis trunk neural crest reveals multipotency and novel pathways of migration. Development 118:363–376
Kuffler DP (1990) Long-term survival and sprouting in culture by motoneurons isolated from the spinal cord of adult frogs. J Comp Neurol 302:729–738. doi:10.1002/cne.903020405
Peled A, Kollet O, Ponomaryov T, Petit I, Franitza S, Grabovsky V, Slav MM, Nagler A, Lider O, Alon R, Zipori D, Lapidot T (2000) The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 95:3289–3296
Yao H, Dashner EJ, van Golen CM, van Golen KL (2006) RhoC GTPase is required for PC-3 prostate cancer cell invasion but not motility. Oncogene 25:2285–2296
Baldwin HS, Shen HM, Yan HC, DeLisser HM, Chung A, Mickanin C, Trask T, Kirschbaum NE, Newman PJ, Albelda SM et al (1994) Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31): alternatively spliced, functionally distinct isoforms expressed during mammalian cardiovascular development. Development 120:2539–2553
Luo J, Guo P, Matsuda K, Truong N, Lee A, Chun C, Cheng SY, Korc M (2001) Pancreatic cancer cell-derived vascular endothelial growth factor is biologically active in vitro and enhances tumorigenicity in vivo. Int J Cancer 92:361–369. doi:10.1002/ijc.1202
Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, Gerald WL, Brogi E, Benezra R, Massague J (2007) ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci USA 104:19506–19511. doi:10.1073/pnas.0709185104
Rowland-Goldsmith MA, Maruyama H, Matsuda K, Idezawa T, Ralli M, Ralli S, Korc M (2002) Soluble type II transforming growth factor-beta receptor attenuates expression of metastasis-associated genes and suppresses pancreatic cancer cell metastasis. Mol Cancer Ther 1:161–167
Orr FW, Wang HH, Lafrenie RM, Scherbarth S, Nance DM (2000) Interactions between cancer cells and the endothelium in metastasis. J Pathol 190:310–329. doi:10.1002/(SICI)1096-9896(200002)190:3<310:AID-PATH525>3.0.CO;2-P
Lawrence TS, Davis MA, Maybaum J, Mukhopadhyay SK, Stetson PL, Normolle DP, McKeever PE, Ensminger WD (1992) The potential superiority of bromodeoxyuridine to iododeoxyuridine as a radiation sensitizer in the treatment of colorectal cancer. Cancer Res 52:3698–3704
Hortobagyi GN (1998) Treatment of breast cancer. N Engl J Med 339:974–984
Greenberg PA, Hortobagyi GN, Smith TL, Ziegler LD, Frye DK, Buzdar AU (1996) Long-term follow-up of patients with complete remission following combination chemotherapy for metastatic breast cancer. J Clin Oncol 14:2197–2205
McGrogan BT, Gilmartin B, Carney DN, McCann A (2008) Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta 1785:96–132. doi:10.1016/j.bbcan.2007.10.004
Chien AJ, Moasser MM (2008) Cellular mechanisms of resistance to anthracyclines and taxanes in cancer: intrinsic and acquired. Semin Oncol 35:S1–S14. doi:10.1053/j.seminoncol.2008.02.010
Huhtala P, Humphries MJ, McCarthy JB, Tremble PM, Werb Z, Damsky CH (1995) Cooperative signaling by alpha 5 beta 1 and alpha 4 beta 1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J Cell Biol 129:867–879
Livant DL (2005) Targeting invasion as a therapeutic strategy for the treatment of cancer. Curr Cancer Drug Targets 5:489–503
Greiling D, Clark RA (1997) Fibronectin provides a conduit for fibroblast transmigration from collagenous stroma into fibrin clot provisional matrix. J Cell Sci 110:861–870
Grinnell F, Zhu M (1994) Identification of neutrophil elastase as the proteinase in burn wound fluid responsible for degradation of fibronectin. J Invest Dermatol 103:155–161
Woods-Ignatoski KM, Grewal NK, Markwart SM, Livant DL, Ethier SP (2003) p38 MAPK induces cell surface α4 integrin down-regulation to facilitate erbB-2 mediated invasion. Neoplasia 5:128–134
Woods Ignatoski KM, Livant DL, Markwart S, Grewal NK, Ethier SP (2003) The role of phosphatidylinositol 3’-kinase and its downstream signals in erbB-2-mediated transformation. Mol Cancer Res 1:551–560
Woods-Ignatoski KM, Maehama T, Markwart SM, Dixon JE, LIvant DL, Ethier SP (2000) ERBB-2 overexpression confers P1 3′ kinase-dependent invasion capacity on human mammary epithelial cells. Br J Can 82:666–674
Mosher DF (1984) Physiology of fibronectin. Ann Rev Med 35:561–575
Ruoslahti E, Hayman EG, Pierschbacher M, Engvall E (1982) Fibronectin: purification, immunochemical properties, and biological activities. Methods Enzymol 82:803–831
Fassina G, Corti A, Cassani G (1992) Affinity enhancement of complementary peptide recognition. Int J Pept Protein Res 39:549–556
Sinnis P, Clavijo P, Fenyo D, Chait BT, Cerami C, Nussenzweig V (1994) Structural and functional properties of region II-plus of the malaria circumsporozoite protein. J Exp Med 180:297–306
Carlier E, Mabrouk K, Moulard M, Fajloun Z, Rochat H, De Waard M, Sabatier JM (2000) Ion channel activation by SPC3, a peptide derived from the HIV-1 gp120 V3 loop. J Pept Res 56:427–437
Yahi N, Sabatier JM, Baghdiguian S, Gonzalez-Scarano F, Fantini J (1995) Synthetic multimeric peptides derived from the principal neutralization domain (V3 loop) of human immunodeficiency virus type 1 (HIV-1) gp120 bind to galactosylceramide and block HIV-1 infection in a human CD4-negative mucosal epithelial cell line. J Virol 69:320–325
Nomizu M, Yamamura K, Kleinman HK, Yamada Y (1993) Multimeric forms of Tyr-Ile-Gly-Ser-Arg (YIGSR) peptide enhance the inhibition of tumor growth and metastasis. Cancer Res 53:3459–3461
Acknowledgments
The authors wish to thank Dr. Philip Andrews for his suggestion of utilizing MAP peptides. All peptides were synthesized by the University of Michigan Protein Structure Facility (Dr. Henriette A. Remmer). The masses of the MAP peptides were verified by Angela Walker, Ph.D of the Michigan Proteome Consortium (Dr. Philip C. Andrews). This research was supported by a Department of the Army IDEA award, W81XWH-06-1-0371, to DLL.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Yao, H., Veine, D.M., Fay, K.S. et al. The PHSCN dendrimer as a more potent inhibitor of human breast cancer cell invasion, extravasation, and lung colony formation. Breast Cancer Res Treat 125, 363–375 (2011). https://doi.org/10.1007/s10549-010-0826-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-010-0826-y