
Abstract
Ductal carcinoma in situ (DCIS) represents the earliest identifiable breast cancer lesion. Disruption of the myoepithelial cell layer and basement membrane is a prerequisite for DCIS to initiate invasion into the stroma. The majority of epithelial cells overlying a focally-disrupted myoepithelial cell layer are estrogen receptor-alpha negative (ER(−)); however, adjacent cells within the same duct confined by an intact myoepithelial cell layer express high levels of ER. These ER (+) and ER (−) cells were microdissected from the same ducts of breast cancer patients. Differential proteins expressed by ER(+) and ER(−) cells were identified using two-dimensional gel electrophoresis followed by mass spectrometry and Western blot analysis. ER(−) cells express lower levels of superoxide dismutase, RalA binding protein, galectin-1, uridine phosphorylase 2, cellular retinoic acid-binding protein 1, S100 calcium binding protein A11, and nucleoside diphosphate kinase A or non-metastasis protein 23-H1 (nm23-H1). The upregulated protein, Rho GDP-dissociation inhibitor 1 alpha, may induce chemotherapy resistance. The significant findings are that the microdissected ER(−) cells express 12.6 times less cellular retinoic acid-binding protein 1, a protein involved in cellular differentiation, and 4.1 times less nucleoside diphosphate kinase A or nm23-H1, a metastasis suppressor, and express fewer proteins than adjacent ER(+) cells. The collective role of the alterations of protein expression in ER(−) cells may be to promote a more malignant phenotype than adjacent ER(+) cells, including a decreased ability to undergo apoptosis and differentiation, and an increased potential to damage DNA, metastasize, and resist to chemotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human ductal carcinoma in situ (DCIS) is the earliest identifiable breast cancer lesion, in which tumor cells are confined to the duct and surrounded by a layer of myoepithelial (ME) cells and the basement membrane, with no evidence of invasion into the surrounding stroma [1]. While approximately half of DCIS cases may eventually progress to an invasive cancer [2], many are likely to remain non-threatening lesions, which complicates determination of the best treatment regimen for breast cancer patients. In human breast ducts, the ME cell layer and basement membrane form a physical barrier between the stroma and the luminal epithelium that serve to isolate in situ breast tumors. The ME cells themselves are joined by intercellular junctions and adhesion molecules, forming a continuous layer that encircles the entire duct system, and a discontinuous layer or a basket-like structure that covers a vast majority of lobules and terminal duct-lobular units [3–5]. The basement membrane is composed of laminins, type IV collagen, entactin, heparan sulfate proteoglycans, and glycosaminoglycans, forming a continuous lining surrounding and attaching to ME cells via hemidesmosomes and focal adhesion complexes [3–5]. These structural features confer two essential functions upon the ME cells and the basement membrane. First, as the epithelium is normally devoid of lymph and blood vessels, and is therefore dependent upon the stroma for its metabolic and survival needs, the ME cells and basement membrane function as gate-keepers, directly regulating the communication between these two compartments. Second, due to the physical interposition of the ME cell layer and basement membrane between the stroma and epithelium, ductal tumor cells must first break their way through the ME cell layer, then the basement membrane, to reach the stroma and initiate invasion and subsequent metastases [6].
Estrogens play a major role in both the development of sexual glands and in the reproductive cycle [7], and the biological effects of estrogen are mediated through the estrogen receptor (ER). ERα, cloned in 1986 [8, 9], was believed to be the sole form of this receptor until 1996, when a second ER, called ERβ, was also cloned [10, 11]. The exact roles of ERα and ERβ in breast cancer are not fully understood, though it has been reported that estrogens are involved in the promotion of human breast cancer, possibly by way of their mitogenic activity [12].
Immunohistochemical staining for ERβ in normal human breast tissues, DCIS, invasive cancers, and lymph node metastases has revealed a gradual reduction in ERβ expression during the transition from normal tissue to preinvasive lesions to invasive cancers, and ERβ was lost in 21% of the invasive cancers studied [13]. Another study utilizing similar techniques revealed that the percentage of cells positive for ERβ was high in normal mammary glands and in non-proliferative benign breast disease, but the percentage of positive cells was decreased significantly in proliferative benign breast disease and in carcinoma in situ [14]. These results are in agreement with results obtained when analyzing mRNA levels of ERβ in normal mammary, benign breast disease, breast cancer, and metastatic breast cancer in lymph nodes [15], where it was shown that ERβ mRNA expression was significantly decreased in breast cancer and lymph node metastatic tissues when compared with normal mammary and benign breast disease tissues. All of these results suggest that ERβ exerts a protective effect against the mitogenic activity of estrogens mediated by ERα, and may function as a tumor suppressor, as the loss of ERβ expression seems to correlate with the progression of breast carcinomas.
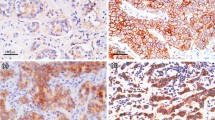
To identify potential precursors of invasive lesions, we have carried out studies focusing on the correlation between the structural integrity in ME cell layers and the immunohistochemical and genetic profiles in adjacent epithelial cells. In 5,698 duct cross-sections from 220 patients with ERα (+), non-invasive breast tumors, we detected a total of 405 focal ME cell layer disruptions, defined as the absence of ME cells resulting in a gap equal to or greater than the combined size of 3 ME or epithelial cells [16]. Of these focal disruptions, 350 (86.4%) were overlaid by epithelial cell clusters with no or substantially reduced ERα expression, in contrast to adjacent cells within the same duct, which expressed high levels of ERα and overlaid an intact ME cell layer [16]. Additional studies revealed that cell clusters overlying focally disrupted ME layers generally showed a substantially higher proliferation rate when compared to adjacent cells within the same duct, as assessed by immunohistochemical staining for Ki-67 [17, 18]. These features potentially signal the formation of a biologically more aggressive cell clone, with a protein expression profile that has yet to be fully examined, while focal breakdowns in the ME cell layer are likely to serve as portals for tumor progression and early invasion.
To identify new breast cancer biomarkers, the protein profiles of nipple aspirate fluids [19], or the serum of breast cancer patients [20], have been compared to that of healthy donors. Identifying biomarkers by these less invasive methods is of great importance, but questions remain as to what early differences in protein profiles of these samples might manifest to detectable levels, as the low abundance of proteins secreted in the serum and other biological fluids as a result of the disease is often masked by the high abundance of albumin and other highly abundant proteins [21]. The protein profiles of breast cancer cell lines have also been compared to the profiles of normal breast cell lines [22]. While tumor-derived cell lines can be useful for initial studies [23], each line might display a unique evolution that may not truly mimic real in vivo conditions. A proteomics study performed on prostate cells shows the discrepancies between microdissected prostate tissues and in vitro prostate lines [24]. Comparisons have also been made between microdissected breast tissues composed of either normal ductal epithelium or ductal epithelium containing DCIS lesions [25]. However, the samples collected for this study, composed of 50,000–100,000 cells, were unlikely to be have been homogeneous, and were likely composed of multiple cell types with divergent expression profiles.
Many diseases manifest themselves through severe changes in human pathophysiology, which forms the basis for clinical chemistry and provides its value in diagnoses and subsequent therapeutic interventions [26]. Proteomics includes the global analysis of proteins expressed by the genome of an organism, an organ, a tissue, or a group of cells with the typical aim being the evaluation of quantitative changes that occur as a function of disease, treatment, or environment [27]. Proteomics strategies are used to identify cancer-specific protein markers that could provide the basis for the development of new diagnostic methodologies, early disease detection, and monitoring the efficacy of treatments [27, 28]. Marker detection and correlation with tumor growth is less obstinate for advanced tumors, but the true value of these developing applications lies in early tumor detection and the prompt identification of invasive phenotypes [26].
To identify candidate markers specific to the initiation of invasion by DCIS cells, we here identify proteins that are differentially expressed by the ERα (−) cell clusters overlying focally disrupted ME cell layer when compared to adjacent ERα (+) cells overlying non-disrupted ME cell layer within the same duct (Fig. 1). Proteins extracted from ERα (+) and ERα (−) tissue samples were subjected to two-dimensional gel electrophoresis (2-DE) followed by MALDI-TOF-MS analysis of trypsin-digested spots. Differentially-expressed proteins that were identified by this method were investigated in ERα (+) and ERα (−) tissues microdissected from the ducts of the same patients (Fig. 1) using Western blot. We report the differential expression of proteins of ERα (−) cells overlying a focally disrupted ME layer compared to adjacent ERα (+) cells bound by an intact ME layer.

ERα expression status in cell clusters overlying focally-disrupted myoepithelial cell layers. Human breast tissue sections from DCIS (a), hyperplastic (b), and normal-appearing (c) ducts were double-immunostained for ERα (brown) and SMA (red). Arrows identify myoepithelial cell layers. Circles identify ERα-negative cell clusters overlying focal myoepithelial cell layer disruptions (300×) (Color figure online)
Materials and methods
Research involving human subjects was performed in accordance with the principles outlined in the declaration of Helsinki (1964). All the research projects involved in using human tissues and cells were approved by the Institutional Human Subject Review Board or Committees. Freshly-frozen DCIS breast tissue microdissected samples and corresponding slide-mounted, serial-sectioned paraffin-embedded tissues were provided by co-author Yan-Gao Man, M.D., Ph.D., Director of the Gynecologic and Breast Research Laboratory at the Armed Forces Institute of Pathology and American Registry of Pathology in Washington, D.C.
Tissue microdissection and protein extraction
Serial sections of 5 to 7 μm thickness were cut and placed on positively-charged microscopic slides and rehydrated by ethanol gradient. Sections were immunohistochemically stained for ERα and different proliferation markers. Nonspecific antibody binding was blocked with blocking buffer (0.2% Triton X-100, 5% normal goat serum, and 3% bovine serum albumin in Tris-buffered saline) for 1 h at room temperature prior to overnight incubation at 4°C with a monoclonal mouse anti-human ERα antibody (NCL-ER-6F11, Vector, Burlingame, CA) diluted into the same buffer (10 μg/mL). Samples were then incubated with an alkaline phosphatase-conjugated secondary antibody diluted 1:5000 in blocking buffer for 4 h at room temperature, and positive signals were detected using Fast-Red (Sigma). Clusters of cells overlying myoepithelial disruptions, composed primarily of ERα (–) cells, were excised by laser-capture microdissection from adjacent, unstained serial sections. Clusters of ERα (+) cells from the same duct, but bound by an intact ME layer, were similarly obtained. Microdissected tissues were then homogenized in a tissue lysis buffer composed of 10 mM Tris, 130 mM NaCl, 1% octylglucopyranoside (OG), 1% CHAPS, 10 mM sodium phosphate, and 10 mM sodium fluoride (pH 7.5). The ratio of the volume of tissue lysis buffer added to the weight of the tissue was 1/1 (v/v). This was followed by vortexing for 10 min and then by centrifugation at 17000 × g for 20 min. This resulted in the formation of 3 layers: The top layer was composed primarily of non-soluble lipids, the middle layer contained the soluble proteins, and the bottom layer was composed of cell debris. The middle layer was recovered and used for subsequent analyses.
Protein extraction from whole human breast tissue samples
ERα (+) and ERα (−) whole human breast tissue samples were minced using a scalpel, and proteins were extracted using the procedure outlined above.
Protein quantitation
Protein concentrations of the tissue lysates were determined using a bicinchoninic acid (BCA) Protein Assay Kit (Pierce, Rockford, IL) using bovine serum albumin (BSA) as a standard according to the manufacturer’s instructions. The concentrations of proteins in the tissue lysates from the microdissected cells and the tissue samples were determined using the standard curve generated by the absorbance of the standards at 562 nm in the Y axis versus the known concentration of the standards in the X axis.
Two-dimensional gel electrophoresis
2-DE was performed as described previously [29]. Briefly, proteins were reconstituted in a rehydration buffer composed of 5 M urea, 2 M thiourea, 1% OG, 0.25% v/v Biolyte 3-10 ampholyte, 12.5% water-saturated isobutanol, 10% isopropanol, 5% glycerol, and 50 mM dithiothreitol (DTT). The samples were rehydrated at 50 V for 16 h at 20°C using 11 cm Immobilized pH Gradient (IPG) strips, pH 4 to 7 (Bio-Rad). Proteins were focused at 250 V for 15 min, and then 8000 V was maintained for a total of 60,000 Vh per gel. The strips were then equilibrated for 10 min in 2.5 mL of a solution composed of 375 mM Tris–HCl pH 8.8, 6 M urea, 2% SDS, and 2% DTT. After this first equilibration, the strips were equilibrated for another 10 min in 2.5 mL of a second equilibration buffer composed of 375 mM Tris–HCl pH 8.8, 6 M urea, 2% SDS, and 2% iodoacetamide. The equilibrated IPG strips were washed with cathode buffer (0.1 M Tricine, 0.1 M Tris–HCl pH 8.2, and 0.1% SDS) and placed onto a 10% Tris–HCl polyacrylamide gel. The anode buffer consisted of 0.2 M Tris–HCl pH 8.9. Gels were electrophoresed at 50 V for 30 min, then at 100 mA/gel until the end of the separation. Gels were stained with SYPRO Ruby (Bio-Rad) according to the following procedure: Gels were fixed in 50% methanol (v/v) and 12% acetic acid (v/v) for 2 h, then incubated with SYPRO Ruby in the dark overnight. Gels were then washed with ddH2O for 3 h. Gels were scanned using Typhoon 9410 Scanning Systems (GE Healthcare). The excitation wavelength was 457 nm and the signals were detected at 610 nm. Spot detection and densitometry were performed using PDQuest 8.0 (Bio-Rad, Hercules, CA) and ImageJ (http://rsb.info.nih.gov/ij).
MALDI-TOF-MS
Proteins were identified utilizing Matrix-Assisted Laser Desorption-Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF–MS) [30–34] as described previously [35]. Briefly, after four iterations of 2-DE for each sample, protein spots were excised under UV light, combined, and incubated with 0.5 μg/mL TPCK-treated trypsin. Peptides generated from the trypsin digestion were then mixed with a matrix (α-cyano-hydroxycinnamic acid) [36, 37] at a 100–1000-fold molar excess over the analyte [38]. This mixture was then spotted onto a target plate and allowed to evaporate until crystals were formed. The MALDI-TOF used for this analysis was a Kratos PC Axima CFR+ (Shimadzu Biotech) that operated in the reflectron positive ion mode with an operating voltage of 20 kV, an extraction voltage of 19.98 kV, and a pulse voltage of 2000 V. Spectra were acquired over the mass range of 500 to 3000 Da. MASCOT software was used to identify the proteins. The search was performed against the SwissProt database for the Homo sapiens taxonomy. The following parameters were used: one missed cleavage, mass tolerance of 1.3 Da, oxidation of methionine (variable modification), and carbamidomethylation (fixed modification).
Western blotting
Proteins from microdissected ERα (+) and ERα (−) cells were reconstituted with SDS–PAGE buffer and 10 μg of each sample were loaded onto a 10% polyacrylamide gel. The gel was electrophoresed at 30 V for 30 min, then at 50 mA until the end of the separation. Proteins contained within the gel were then electroblotted onto a nitrocellulose membrane (50 V for 50 min). Western blot analyses were accomplished utilizing a 1 μg/mL dilution of primary antibody followed by incubation with a horseradish peroxidase-conjugated secondary antibody against the appropriate species. Visualization of the bands was then accomplished by the addition of a 1:1 ratio of Super Signal West Pico-Stable Peroxidase Solution and Luminol/Enhancer Solution (Pierce, Rockford, IL) and by developing the chemiluminescent signal in the dark using Kodak Scientific Imaging Film (Kodak cat. 1651496), Fixer and Replenisher/Developer and Replenisher (Kodak cat. 1901859) according to the manufacturer’s instructions.
Results and discussions
2-DE of microdissected ERα (+) and ERα (−) tissues
2-DE of proteins extracted from 150,000 microdissected ERα (+) and ERα (−) breast cancer cells shows that ERα (−) cells have reduced protein expression (Fig. 2a), although there are 3 proteins exclusively expressed by the ERα (−) cells (Fig. 2b Blue spots). PDQuest analysis of the images revealed the presence of 28 common proteins differentially-expressed between these two cell clusters (Fig. 2b Green spots). ERα (+) cells express 83 proteins that are not expressed by ERα (−) cells (Fig. 2 b Red spots).

a 2-DE of proteins extracted from 150,000 ERα (+) and ERα (−) cells, respectively, microdissected from human DCIS tissues originating from 3 patients. Proteins were rehydrated at 50 V for 16 h at 20°C using 11 cm Immobilized pH Gradient (IPG) strips, pH 4–7. A 10% polyacrylamide gel was used in the second dimension. Proteins were stained by SYPRO Ruby and visualized using an excitation wavelength of 457 nm. The signals were detected at 610 nm. b PDQuest imaging analysis of 2-DE representing proteins extracted from 150 thousand ERα (+) and ERα (−) cells microdissected from human DCIS tissues originating from 3 patients shows 28 differentially-expressed proteins
Differential protein expression between ERα (+) and ERα (−) human breast cancer tissue samples (whole tissue homogenates)
2-DE of ERα (+) invasive breast cancer tissue samples versus ERα (−) samples (Fig. 3a) followed by MALDI-TOF analysis of tryptic digests (Fig. 3b) revealed the differential expression of 247 proteins, 22 of which were identified (Table 1). Eight of these proteins were selected to be analyzed and validated in the microdissected ERα (+) and ERα (−) tissues by Western blotting: Superoxide Dismutase (Mr 16154; pI 5.7), RalA binding protein 1 variant (Mr 53893; pI 4.92), Rho GDP-Dissociation Inhibitor 1 (RhoGDIα) (Mr 23250; pI 5.02), Galectin-1 (Mr 15048; pI 5.34), Uridine Phosphorylase 2 (UPase2) (Mr 36245; pI 6.21), Cellular Retionic Acid-binding Protein 1 (CRABP1) (Mr 15727; pI 5.3), S100 calcium binding protein A11 (S100-A11) (Mr 11487; pI 6.56), and Nucleoside Diphosphate Kinase A (NDPK-A) (Mr 17309; pI 5.83). The selection was based primarily on the role these proteins play in cellular distribution, apoptosis, phosphorolysis, tumorigenesis, invasion, or metastasis, as well as the availability of commercial antibodies.

a 2-DE of proteins extracted from entire ERα (+) and ERα (−) tissue samples. Proteins were rehydrated at 50 V for 16 h at 20°C using 11 cm Immobilized pH Gradient (IPG) strips, pH 4–7. A 10% polyacrylamide gel was used in the second dimension. Proteins were stained by SYPRO Ruby and visualized using an excitation wavelength of 457 nm. The signals were detected at 610 nm. Proteins were identified using MALDI-TOF-MS. b MALDI-TOF mass spectra of tryptic digests obtained on a Kratos PC Axima CFR+ (Shimadzu Biotech) operated in the reflectron positive ion mode with an operating voltage of 20 kV, an extraction voltage of 19.98 kV, and a pulse voltage of 2000 V. Spectra were acquired over the mass range of 500 to 3000 Da
Microdissected tissues versus whole tissue samples
The low amount of proteins extracted from the ERα (+) and ERα (−) microdissected tissues did not allow for the identification of differentially-expressed proteins between these two cell clusters, and we therefore proceeded by analyzing differential protein expression from tissue samples composed exclusively of ERα (+) or ERα (−) invasive breast cancer cells as described in the pathology reports of these tissues and verified by Western blots. More than 1 mg of proteins was extracted from each tissue sample to allow for the protein identification using MALDI-TOF analysis on trypsin-digested spots. Western blotting was then performed to validate differential protein expression in 3 ERα (+) and ERα (−) pairs of tissue that were microdissected from the ducts of 3 patients.
Differential protein expression between microdissected ERα (+) and ERα (−) cells
Western Blotting was performed on lysates of the ERα (+) and ERα (−) tissues microdissected from the same duct of DCIS microinvasive cancer patients (Fig. 4). The expression levels of Superoxide Dismutase, RalA binding protein, Galectin-1, UPase, CRABP1, S100-A11, and NDPK-A were lower in ERα (−) cell clusters than their ERα (+) counterparts. Rho GDP-Dissociation Inhibitor 1 (RhoGDIα) was the only protein upregulated in the ERα (−) cells (Table 2).

Western Blots performed on microdissected ERα (+) and ERα (−) cells. Numbers 1–3 refer to the 3 pairs of samples microdissected from the ducts of 3 different patients that are either positive (+) or negative (−) for ERα expression. Individual rows include findings for Estrogen Receptor-α, superoxide dismutase, RalA binding protein 1, RhoGDIα, Galectin-1, Uridine Phosphorylase, cellular retinoic acid-binding protein 1, S100-A11, nucleoside diphosphate kinase A, and tubulin-α. Lanes represent 5 μg of total protein. Western blot analyses were accomplished utilizing a 1 μg/mL dilution of primary antibody followed by incubation with a horseradish peroxidase-conjugated secondary antibody against the appropriate species with positive signals detected by chemiluminescence
Superoxide dismutase plays a key anti-oxidant role in the cell by converting superoxide radicals to hydrogen peroxide and oxygen [39]. Its cellular role is to provide a defense against the potentially damaging activities of superoxide radicals; diminished superoxide dismutase enzyme activity leads to many of the observed properties in cancer cells such as damage to the mitochondrial and plasma membranes, modulating key enzyme activities by oxidizing sulfhydryl groups to disulfides, and causing DNA double-strand breakage and possibly mutation and genomic instability. Downregulation of this enzyme has been found in many cancers [40]. The superoxide dismutase protein expression level was 2.52 fold higher in ERα (+) than ERα (−) epithelial cells belonging to the same patient.
RalA protein is a Ras-related small GTPase. The Ral GTPase family of proteins plays a major role in intracellular membrane trafficking, as well as tumorigenesis, invasion, and metastasis [41, 42]. Because RalA binding protein interacts with RalA and binds directly to RalB [43], which is associated with cancer metastasis [44], and RalA binding protein activates the GTPase activity of Cdc42 and partially Rac1 [45], both proteins are involved in cell migration [46]. Furthermore, given the role of RalA binding protein in cancer cell chemoresistance [47], RalA binding protein may also be critical in tumor invasion and metastasis. RalA binding protein was found to be 4.5 times higher in ERα (+) than ERα (−).
RhoGDIα is a cellular protein that controls the cellular distribution and activity of RhoGTPases [48, 49]. Rho-GDIα is elevated in breast cancer cell lines and breast tumor tissues [50]. RhoGDIα has also been reported to promote the resistance of cancer cells to drug-induced toxicity, thus playing an anti-apoptotic role [51]. RhoGDIα was the only protein that was identified as elevated in ERα (−) cells when compared to its ERα (+) cells.
Galectin-1 can induce apoptosis in breast cancer cells by blocking the cell cycle at the S/G2 transition [52]. It has been shown to be involved in cancer invasion and metastasis by regulating cell adhesion and migration [53]. Galectin-1 is thought to be synthesized by the carcinoma cells and secreted into the stroma. A recent study demonstrated increased expression of Galectin-1 in human breast cancer cells and cancer-associated stroma and hypothesized that Galectin-1 plays a major role in tumor-stroma interaction in breast cancer [54]. The Galectin-1 protein level was 4.27 times higher in ERα (+) when compared to the ERα (−) expression. Uridine phosphorylase (UPase) catalyzes the phosphorolysis of uridine to uracil. It is also involved in fluoropyrimidine metabolism, playing a role in the intracellular activation of 5-fluorouracil [55].
UPase is elevated in various tumor tissues including breast cancer tissues. This elevation contributes to the therapeutic efficacy of fluoropyrimidines in cancer patients [56], and it has been demonstrated that elevated levels of UPase are correlated with favorable prognoses in cancer [57]. UPase was 5.1 times higher in ERα (+) than in ERα (−).
Cellular retinoic acid binding proteins are thought to play a role in shuttling retinoic acid (RA) from the cytoplasm to the nucleus, therefore playing a role in cell growth and differentiation regulation in fetal and adult tissues [58]. CRABP1 regulates RA activity by increasing its degradation rate by enhancing the production of RA-metabolizing enzymes [59]. CRABP1 levels were 12.6 times higher in ERα (+) than in neighboring ERα (−) cells.
S100-A11 is involved in tumorigenesis [60], although it has been shown to be upregulated in some cancers [61] and downregulated in others [62]. S100-A11 levels in ERα (+) samples were 4.48 more than that in ERα (−) samples.
Nucleoside diphosphate kinase-A (NDPK-A or nm23-H1) is a metastasis suppressor gene identified in 1988 [63]. Restoration of NDPK-A expression reduces metastasis of breast cancer [64]. The NDPK-A protein level was 4.1 times higher in ERα (+) cells than in nearby ERα (−) counterparts.
The loss of ERα expression, signaling the early signs of breast cancer invasion, was accompanied by the downregulation of superoxide dismutase that plays an anti-oxidant role, galectin-1 known to induce apoptosis, uridine phosphorylase that contributes to drug efficacy, cellular retinoic acid-binding protein 1 that plays a role in cell growth and differentiation, S100-A11 involvement in tumorigenesis, and a metastasis suppressor nucleoside diphosphate kinase A. RhoGDIα, known to induce the resistance of cancer cells to drug-induced toxicity, was the only protein that was elevated in ERα (−) cells when compared to adjacent ERα (+) cells. The level of RalA binding protein, involved in tumorigenesis and metastasis, was 4.5 times higher in the microdissected ERα (+) cells than in adjacent ERα (−) ones. The physiological and pathological roles of these differentially expressed proteins are complex, and may sometimes conflict with one another. Although the specific effects of the alteration of each of these identified proteins remains to be further investigated, their collective role may be to promote a malignant phenotype for ERα (−) cells, including a decreased ability to undergo programmed cell death and differentiate, an increased potential to damage DNA and generate genomic instability, a resistance to chemotherapy drugs, and ultimately to metastasize. Disruption of both the ME cell layer and the basement membrane is a prerequisite for tumorigenic DCIS cells to initiate invasion into the stroma. The majority of epithelial cells overlying focally-disrupted ME cell layers are ERα (−), while adjacent cells within the same duct confined by an intact ME cell layer express high levels of ERα. In this study, ERα (+) cells bound by an intact ME cell layer and ERα (−) cells overlying myoepithelial disruptions were microdissected from the same ducts of breast cancer patients, and proteomic analyses revealed great discrepancies in protein expression within the same breast cancer duct accompanied by non-expression of this hormonal receptor.
Conclusions
Our study shows that substantial alteration in protein expression was observed between clusters of adjacent ERα (+) and ERα (−) epithelial cells, although no estrogen-regulated genes were identified among the proteins exhibiting increased expression in ERα (+) cells. We propose two hypotheses to be further tested in the future. The first is that an ERα (−) cell cluster in DCIS with microinvasion may represent a clone from a genetically-altered cancer stem cell, explaining why many of the differentiation makers are not expressed or are expressed at a much lower level than those of adjacent ERα (+) cells. Second, an ERα (−) cell cluster may arise from dedifferentiated proliferating ERα (+) cells. The most significant facts are that the microdissected ERα (−) cells are pathologically more aggressive, overlay a focally-disrupted myoepithelial layer, express 12.6 times less cellular retinoic acid-binding protein 1, a protein involved in cellular differentiation, and 4.1 times less metastasis suppressor nucleoside diphosphate kinase A or nm23-H1, and express fewer proteins than adjacent ERα (+) cells in DCIS with microinvasion. Further research is required to test these two hypotheses and find out which one is the predominate case in the evolution of human breast DCIS to invasive carcinoma.
Abbreviations
- CRABP1:
-
Cellular retinoic acid-binding protein 1
- DCIS:
-
Ductal carcinoma in situ
- 2-DE:
-
Two-dimensional gel electrophoresis
- DTT:
-
Dithiothreitol
- ERα:
-
Estrogen receptor alpha
- IPG:
-
Immobilized pH gradient
- MALDI-TOF-MS:
-
Matrix-assisted laser desorption-ionization time-of-flight mass spectrometry
- ME cells:
-
Myoepithelial cells
- M r :
-
Molecular mass
- NDPK-A (or nm23-H1):
-
Nucleoside diphosphate kinase A (non-metastasis protein 23-H1)
- OG:
-
Octylglucopyranoside
- pI:
-
Isoelectric point
- RhoGDIα:
-
Rho GDP-dissociation inhibitor 1 alpha
- S100A11:
-
S100 calcium binding protein A11
- UPase:
-
Uridine phosphorylase
- SMA:
-
Smooth muscle actin
References
Skinner KA, Silverstein MJ (2001) The management of ductal carcinoma in situ of the breast. Endocr Relat Cancer 8(1):33–45
Jones JL (2006) Overdiagnosis and overtreatment of breast cancer: progression of ductal carcinoma in situ: the pathological perspective. Breast Cancer Res 8(2):204
Jolicoeur F, Seemayer TA, Gabbiani G et al (2002) Multifocal, nascent, and invasive myoepithelial carcinoma (Malignant myoepithelioma) of the breast: an immunohistochemical and ultrastructural study. Int J Surg Path 10(4):281–291
Nerlich AG, Haraida S, Hagedorn H et al (1995) Morphological aspects of basement-membranes and their receptors in benign and malignant neoplasms. Int J Oncol 6(6):1193–1202
Tsubura A, Shikata N, Inui T et al (1988) Immunohistochemical localization of myoepithelial cells and basement-membrane in normal, benign and malignant human-breast lesions. Virchows Archiv A Pathol Anat Histopathol 413(2):133–139
Man YG, Sang QXA (2004) The significance of focal myoepithelial cell layer disruptions in human breast tumor invasion: a paradigm shift from the “protease-centered” hypothesis. Exp Cell Res 301(2):103–118
Gustafsson JA (1999) Estrogen receptor beta—a new dimension in estrogen mechanism of action. J Endocrinol 163(3):379–383
Green S, Walter P, Kumar V et al (1986) Human estrogen-receptor cDNA—sequence, expression and homology to V-Erb-A. Nature 320(6058):134–139
Greene GL, Gilna P, Waterfield M et al (1986) Sequence and expression of human estrogen-receptor complementary-DNA. Science 231(4742):1150–1154
Kuiper G, Enmark E, Pelto-Huikko M et al (1996) Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA 93(12):5925–5930
Mosselman S, Polman J, Dijkema R (1996) ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett 392(1):49–53
Anderson E, Clarke R, Laidlaw I et al (1996) Experiments on proliferation of normal human breast tissue in nude mice do not show that progesterone does not stimulate breast cells: reply. Endocrinology 137(4):1505–1506
Skliris GP, Munot K, Bell SM et al (2003) Reduced expression of oestrogen receptor beta in invasive breast cancer and its re-expression using DNA methyl transferase inhibitors in a cell line model. J Pathol 201(2):213–220
Bardin A, Boulle N, Lazennec G et al (2004) Loss of ER beta expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer 11(3):537–551
Park BW, Kim KS, Heo MK et al (2003) Expression of estrogen receptor-beta in normal mammary and tumor tissues: is it protective in breast carcinogenesis? Breast Cancer Res Treat 80(1):79–85
Man YG, Tai L, Barner R et al (2003) Cell clusters overlying focally disrupted mammary myoepithelial cell layers and adjacent cells within the same duct display different immunohistochemical and genetic features: implications for tumor progression and invasion. Breast Cancer Res 5(6):R231–R241
Man YG, Shekitka KM, Bratthauer GL et al (2002) Immunohistochemical and genetic alterations in mammary epithelial cells immediately overlying focally disrupted myoepithelial cell layers. Breast Cancer Res Treat 76:S143–S143
Yousefi M, Mattu R, Gao CL et al (2005) Mammary ducts with and without focal myoepithelial cell layer disruptions show a different frequency of white blood cell infiltration and growth pattern: Implications for tumor progression and invasion. Appl Immunohistochem Mol Morphol 13(1):30–37
Sauter ER, Zhu W, Fan XJ et al (2002) Proteomic analysis of nipple aspirate fluid to detect biologic markers of breast cancer. Br J Cancer 86(9):1440–1443
Li JN, Zhang Z, Rosenzweig J et al (2002) Proteomics and bioinformatics approaches for identification of serum biomarkers to detect breast cancer. Clin Chem 48(8):1296–1304
Sahab ZJ, Semaan SM, Sang QXA (2007) Methodology and applications of disease biomarker identification in human serum. Biomark Insights 2:21–43
O’Neill KA, Miller FR, Barder TJ et al (2003) Profiling the progression of cancer: separation of microsomal proteins in MCF10 breast epithelial cell lines using nonporous chromatophoresis. Proteomics 3(7):1256–1269
Wu WG, Tang XM, Hu W et al (2002) Identification and validation of metastasis-associated proteins in head and neck cancer cell lines by two-dimensional electrophoresis and mass spectrometry. Clin Exp Metastasis 19(4):319–326
Ornstein DK, Gillespie JW, Paweletz CP et al (2000) Proteomic analysis of laser capture microdissected human prostate cancer and in vitro prostate cell lines. Electrophoresis 21(11):2235–2242
Wulfkuhle JD, Sgroi DC, Krutzsch H et al (2002) Proteomics of human breast ductal carcinoma in situ. Cancer Res 62(22):6740–6749
Bischoff R, Luider TM (2004) Methodological advances in the discovery of protein and peptide disease markers. J Chromatogr B Analyt Technol Biomed Life Sci 803(1):27–40
Somiari RI, Sullivan A, Russell S et al (2003) High-throughput proteomic analysis of human infiltrating ductal carcinoma of the breast. Proteomics 3(10):1863–1873
Hanash SM (2000) Biomedical applications of two-dimensional electrophoresis using immobilized pH gradients: current status. Electrophoresis 21(6):1202–1209
Sahab ZJ, Suh Y, Sang QXA (2005) Isoelectric point-based prefractionation of proteins from crude biological samples prior to two-dimensional gel electrophoresis. J Proteome Res 4(6):2266–2272
Binz PA, Hochstrasser DF, Appel RD (2003) Mass spectrometry-based proteomics: current status and potential use in clinical chemistry. Clin Chem Lab Med 41(12):1540–1551
Aebersold R, Mann M (2003) Mass spectrometry-based proteomics. Nature 422(6928):198–207
Ferguson PL, Smith RD (2003) Proteome analysis by mass spectrometry. Annu Rev Biophys Biomol Struct 32:399–424
Heck AJR, Krijgsveld J (2004) Mass spectrometry-based quantitative proteomics. Expert Rev Proteomics 1(3):317–326
Feuerstein I, Rainer M, Bernardo K et al (2005) Derivatized cellulose combined with MALDI-TOF MS: a new tool for serum protein profiling. J Proteome Res 4(6):2320–2326
Sahab ZJ, Iczkowski KA, Sang QXA (2007) Anion exchange fractionation of serum proteins versus albumin elimination. Anal Biochem 368(1):24–32
Karas M, Hillenkamp F (1988) Laser desorption ionization of proteins with molecular masses exceeding 10000 Daltons. Anal Chem 60(20):2299–2301
Tanaka K, Waki H, Ido Y et al (1988) Protein and polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 2(8):151–153
Schiller J, Arnhold J, Benard S et al (1999) Lipid analysis by matrix-assisted laser desorption and ionization mass spectrometry: a methodological approach. Anal Biochem 267(1):46–56
Briehl MM, Baker AF, Siemankowski LM et al (1997) Modulation of antioxidant defenses during apoptosis. Oncol Res 9(6–7):281–285
Oberley LW, Buettner GR (1979) Role of superoxide-dismutase in cancer—review. Cancer Res 39(4):1141–1149
Tchevkina E, Agapova L, Dyakova N et al (2005) The small G-protein RalA stimulates metastasis of transformed cells. Oncogene 24(3):329–335
Ward Y, Wang W, Woodhouse E et al (2001) Signal pathways which promote invasion and metastasis: critical and distinct contributions of extracellular signal-regulated kinase and Ral-specific guanine exchange factor pathways. Mol Cell Biol 21(17):5958–5969
Jullien-Flores V, Dorseuil O, Romero F et al (1995) Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J Biol Chem 270(38):22473–22477
Lim KH, O’Hayer K, Adam SJ et al (2006) Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol 16(24):2385–2394
Park SH, Weinberg RA (1995) A putative effector of Ral has homology to Rho/Rac GTPase activating proteins. Oncogene 11(11):2349–2355
Nobes CD, Hall A (1995) Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81(1):53–62
Awasthi S, Cheng J, Singhal SS (2000) Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry 39(31):9327–9334
Sasaki T, Takai Y (1998) The Rho small G protein family Rho GDI system as a temporal and spatial determinant for cytoskeletal control. Biochem Biophys Res Commun 245(3):641–645
Olofsson B (1999) Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal 11(8):545–554
Fritz G, Brachetti C, Bahlmann F et al (2002) Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br J Cancer 87(6):635–644
Zhang BL, Zhang YQ, Dagher MC et al (2005) Rho GDP dissociation inhibitor protects cancer cells against drug-induced apoptosis. Cancer Res 65(14):6054–6062
Wells V, Davies D, Mallucci L (1999) Cell cycle arrest and induction of apoptosis by beta galactoside binding protein (beta GBP) in human mammary cancer cells. A potential new approach to cancer control. Eur J Cancer 35(6):978–983
Rabinovich GA (2005) Galectin-1 as a potential cancer target. Br J Cancer 92(7):1188–1192
Jung EJ, Moon HG, Cho BI et al (2007) Galectin-1 expression in cancer-associated stromal cells correlates tumor invasiveness and tumor progression in breast cancer. Int J Cancer 120(11):2331–2338
Cao DL, Russell RL, Zhang DK et al (2002) Uridine phosphorylase (−/−) murine embryonic stem cells clarify the key role of this enzyme in the regulation of the pyrimidine salvage pathway and in the activation of fluoropyrimidines. Cancer Res 62(8):2313–2317
Yan R, Wan LX, Pizzorno G et al (2006) Uridine phosphorylase in breast cancer: a new prognostic factor? Front Biosci 11:2759–2766
Kawamura K, Takiguchi N, Wada A et al (2006) Up-regulated expression of the uridine phosphorylase gene in human gastric tumors is correlated with a favorable prognosis. Anticancer Res 26(6C):4647–4651
Gorry P, Lufkin T, Dierich A et al (1994) The cellular retinoic acid-binding protein-I is dispensable. Proc Natl Acad Sci USA 91(19):9032–9036
Dong D, Ruuska SE, Levinthal DJ et al (1999) Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J Biol Chem 274(34):23695–23698
Kondo A, Sakaguchi M, Makino E et al (2002) Localization of S100C immunoreactivity in various human tissues. Acta Med Okayama 56(1):31–34
Oue N, Hamai Y, Mitani Y et al (2004) Gene expression profile of gastric carcinoma: identification of genes and tags potentially involved in invasion, metastasis, and carcinogenesis by serial analysis of gene expression. Cancer Res 64(7):2397–2405
Memon AA, Sorensen BS, Meldgaard P et al (2005) Down-regulation of S100C is associated with bladder cancer progression and poor survival. Clin Cancer Res 11(2):606–611
Steeg PS, Bevilacqua G, Kopper L et al (1988) Evidence for a novel gene associated with low tumor metastatic potential. J Nat Cancer Inst 80(3):200–204
Leone A, Flatow U, King CR et al (1991) Reduced tumor-incidence, metastatic potential, and cytokine responsiveness of Nm23-transfected melanoma-cells. Cell 65(1):25–35
Acknowledgements
This work was supported in part by grant BCTR0504465 from the Susan G. Komen for the Cure Breast Cancer Foundation, grants from the Florida Breast Cancer Coalition Research Foundation, the Elsa U. Pardee Foundation, and the Florida State University to Dr. Q.-X. Sang, and a grant BCTR0706983 from the Susan G. Komen Breast Cancer Foundation, grant 05AA from the AFIP/ARP joint research initiative project, and grant 2006CB910505 from the Ministry of Chinese Science and Technology to Dr. Y.-G. Man.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sahab, Z.J., Man, YG., Semaan, S.M. et al. Alteration in protein expression in estrogen receptor alpha-negative human breast cancer tissues indicates a malignant and metastatic phenotype. Clin Exp Metastasis 27, 493–503 (2010). https://doi.org/10.1007/s10585-010-9338-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-010-9338-8