
Abstract
Since their discovery, a growing body of evidence has emerged demonstrating that transposable elements are important drivers of species diversity. These mobile elements exhibit a great variety in structure, size and mechanisms of transposition, making them important putative actors in organism evolution. The vertebrates represent a highly diverse and successful lineage that has adapted to a wide range of different environments. These animals also possess a rich repertoire of transposable elements, with highly diverse content between lineages and even between species. Here, we review how transposable elements are driving genomic diversity and lineage-specific innovation within vertebrates. We discuss the large differences in TE content between different vertebrate groups and then go on to look at how they affect organisms at a variety of levels: from the structure of chromosomes to their involvement in the regulation of gene expression, as well as in the formation and evolution of non-coding RNAs and protein-coding genes. In the process of doing this, we highlight how transposable elements have been involved in the evolution of some of the key innovations observed within the vertebrate lineage, driving the group’s diversity and success.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With approximately 65,000 species described, vertebrates represent a highly diverse taxon that has colonised a large range of biotopes, from all depths of the freezing oceans to arid deserts and snowy mountain ranges. Accompanying these ecological transitions, many different lineages have generated ingenious adaptations, such as gills, enlarged and highly complex brains, fur, the placenta, and immunity systems. How did these innovations arise, and where did the required genes and regulators come from? There are probably many interacting factors driving this evolution that have each played vital roles during the more than 500 million year (Myr) history of vertebrates.
One influential group of such factors are transposable elements (TEs). These represent genetic elements that are mostly selfish and capable of inserting themselves into novel locations in the genome, with generally no direct benefit (and occasionally deleterious effects) for their host. They are classified into two main categories: class I retroelements and class II DNA transposons (Fig. 1).

Different types of vertebrate transposable elements. HIV human immunodeficiency virus, HERV human endogenous retrovirus, SINE short interspersed element, MITE miniature inverted repeat transposable element, GAG group-specific antigen, PR protease, RT reverse transcriptase, RH RNAse H, IN integrase, ENV envelope, MT methyltransferase, TR tyrosine recombinase, APE apurinic/apyrimidic-like endonuclease, REL restriction-enzyme-like endonuclease, EN endonuclease, TNP transposase, RPA replication factor-A protein 1 transposase, Zf zinc finger, REP-HEL replication initiator and helicase, B-POL family B DNA polymerase. Filled arrows represent long terminal repeats (class I), empty arrows represent inverted terminal repeats (class II)
Class I retroelements (Fig. 1) propagate via an RNA intermediate that is then reverse transcribed into complementary DNA, using a “copy and paste” mechanism. These are populated by the long terminal repeat (LTR)-containing retroelements that include LTR retrotransposons but also endogenous retroviruses (ERVs). In addition, non-LTR containing retrotransposons characterised by the long interspersed nuclear element (LINE) elements and Penelope elements are also present in this class of TEs (Fig. 1).
Class II DNA transposons are characterised by the lack of an RNA transposition intermediate and generally use a direct “cut and paste” mechanism to move around the genomes (Fig. 1). Most of these elements are flanked by inverted repeats. Polintons (aka Mavericks) and Helitrons, which lack inverted repeats, are class II elements that present specific mechanisms of transposition. Polintons, also known as self-synthesising elements, are proposed to be excised as a single-strand DNA molecule that serves as a template for synthesis of its complement, the double-stranded DNA molecule being then inserted back into the genome (Kapitonov and Jurka 2006). Helitrons transpose through a so-called rolling-circle mechanism (Kapitonov and Jurka 2001). All the elements described so far are considered as “autonomous” as they contain functional sequence coding for the proteins required for their propagation, but non-autonomous, non-coding mobile elements also exist in both classes. They require the presence of an autonomous element in the host genome to provide in trans the proteins necessary for their transposition. By far, the most common class I non-autonomous elements are short interspersed nuclear elements (SINEs), with the classic example being the primate Alu elements, which are derived from the small cytoplasmic 7SL RNA, a component of the signal recognition particle ribonucleoprotein complex. In class II DNA transposons, the non-autonomous elements are known as “MITES” (miniature inverted repeat transposable elements), which are short sequences of 50–400 bp predominantly made up of two inverted repeats separated by short intervening DNA sequences (Fig. 1).
A role of TEs as drivers of diversity and speciation was initially proposed by Barbara McClintock (1956), but this idea was further expanded upon later (Coyne and Orr 1998; Kraaijeveld 2010). Subsequently, evidence has been growing in many areas of biology about the important roles that TEs play in lineage-specific diversification. There is no doubt on their ability to transpose and recombine to re-organise genomes and to be “co-opted” or “exapted” to form new exons and regulatory sequences and even new RNA and protein-coding genes. Here, we review the impact of TEs on genome evolution and their potential to contribute to the organismal diversification within the vertebrates. We start by looking at how genomes vary in just their TE content, but then go on to look at how TEs alter genome structure and gene regulation, and how TEs become incorporated into the expressed component of the genome in non-coding RNAs and protein-coding genes (Table 1).
TE diversity and genome plasticity
Lineage-specific diversity of TEs in vertebrates
The most immediate and simple way that TEs drive diversity in genomes is through the TE repertoire and copy number found in each species. The genomes of mammals and other vertebrates have been shown to be significantly repetitive, with a strong contribution of TEs to genome size and architecture (Kazazian 2004; Feschotte and Pritham 2007; Böhne et al. 2008; Chalopin et al. 2015). In the recent years, the number of papers studying TE diversity and evolution in vertebrate species has considerably increased, either in the context of genome sequencing projects or for particular goals such as the study of genome size evolution. For instance, LTR retrotransposon dynamics has been investigated in salamanders in order to highlight their potential role in genome gigantism (Sun et al. 2012). Similarly, non-LTR retrotransposon diversity and elimination were studied in several species such as stickleback (Blass et al. 2012), opossum (Gentles et al. 2007), chicken (Wicker et al. 2005), and lungfish (Metcalfe et al. 2012). Altogether, this information, as well as a recent large comparative analysis including 23 vertebrate species, have helped to infer a general overview of TE diversity in vertebrates that demonstrated the diverse range of TE repertoires present in each species (Chalopin et al. 2015).
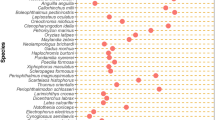
Almost all known types of eukaryotic TEs have been identified in vertebrates (Fig. 1). However, their composition, their copy number in the genome and their age can vary greatly both between and within major vertebrate lineages (Volff et al. 2003; Chalopin et al. 2015). Illustrating the difference of content, it was shown that mammals contain 10 times more TEs than birds. Within teleost fish, the zebrafish genome (55 % of TEs in the genome) is 10 times richer in TE content than that of the pufferfish Tetraodon (<6 %; Chalopin et al. 2015). Overall, TEs constitute a high proportion of the genome in mammals, squamates (comprising of lizards), turtles, sharks, lamprey and some fish genomes such as zebrafish, but are relatively poorly represented in birds and other fish genomes such as pufferfish or flatfish (Fig. 2, TE content). However, the contribution of TEs and other repeats to genome size is more important in fish than in other vertebrates. This suggests that variation in genome size in mammalian and other sarcopterygian genomes is more driven by non-repeated sequences, or possibly very divergent (i.e., old) repeated sequences (Chalopin et al. 2015).

Schematic comparison of TE diversity and content in vertebrates. The figure represents a non-exhaustive view of genome size, TE diversity and TE content in major vertebrate groups. For genome size: +++ bigger than 2.5 Gb; ++ from 1.5 Gb to 2.5 Gb; + less than 1.5 Gb. For TE content: +++ more than 25 % of the genome; ++ from 11 to 24 %; + less than 10 %. For all-TE diversity (without SINE superfamilies): +++ more than 18 superfamilies covering at least 0.001 % of the genome; ++ between 11 and 17; + less than 10
Regarding TE superfamilies present in the genome, a gradual decrease in TE diversity is observed from agnaths and cartilaginous fish to mammals and birds (Fig. 2, Chalopin et al. 2015). Indeed, mammals and birds present a reduced number of TE superfamilies (from 7 to 14), while turtles, squamates, crocodiles and amphibians harbour a higher diversity (from 15 to 21 superfamilies). Finally, the water-living vertebrates (coelacanth, teleost fish, cartilaginous fish and sea lamprey) show a much higher range of diversity (from 22 to 27 superfamilies). Some autonomous (e.g., ERVs, LINE1 retrotransposons, TcMariner or hAT DNA transposons) and non-autonomous (V-SINE, Piskurek and Jackson 2011) superfamilies are widespread in all vertebrates studied so far, suggesting their presence in ancestral vertebrate genomes. Conversely, other superfamilies were probably lost or are headed for extinction in some lineages, such as gypsy retrotransposons in birds and mammals (Volff et al. 2003) or L2 and Helitrons in birds (Chalopin et al. 2015).
At a finer scale, an additional level of variation in diversity can be observed within superfamilies. The LINE1 retrotransposon superfamily constitutes approximately 20 % of both human and mouse genomes with a single family, whereas zebrafish genome contains more than 30 different LINE1 families with much lower copy numbers (Furano et al. 2004). A different situation is observed for the Retroviridae: mammals and birds contain more ERV genera (gamma, epsilon, beta, HERVS/L in both groups, plus alpha only in birds, and lentiviruses only in mammals) than the teleosts, which harbour only epsilon and spuma retroviruses (Hayward et al. 2015).
The level of TE success and diversity, which has been compared between different lineages (i.e., mammals versus fish), can also be investigated within lineages (i.e., between mammalian species). For instance, LINE1 are widespread and thought to have remained active in all mammals except in megabats (Cantrell et al. 2008) and in one group of muroids (Grahn et al. 2005). In muroids, LINE1 extinction was shown to correlate with a massive invasion of ERV elements (Erickson et al. 2011). This highlights an important factor probably influencing TE success, namely the competition between TE families and superfamilies within genomes. It has indeed been proposed that the success of a particular TE superfamily can be associated with the loss of others (Le Rouzic and Capy 2006), since different families may not be able to coexist in the same host. Beside competition, other factors, such as rate of transposition, rate of DNA elimination, population size, mode of reproduction and variation in host-mediated defences may all play important roles in the observed TE diversity, making each lineage and each species unique in its TE content.
Horizontal transfers increase TE diversity
Horizontal transfers of TEs (HTT) are major events that can drive TE diversity between lineages. The invasion of a TE from a distant species by bypassing species barriers and entering into a new genome indeed constitutes an efficient way for TEs to spread. Following transfer, the newly acquired TEs may experience bursts of transposition, facilitated by the new host maybe lacking appropriate defence mechanisms. Due to the requirement of transfer vectors, which were proposed to be viruses, parasitoids or mites, HTT was considered for a long time to be a rare event in vertebrates (Wallau et al. 2012). However, the number of studies demonstrating cases of successful transfer increased in the past years, along with the interest for this phenomenon—evidenced by the establishment of an HTT database (Schaack et al. 2010; Ivancevic et al. 2013; Dotto et al. 2015).
Various classes of TEs have been shown to have been horizontally transferred between species. For example, DNA transposons, such as SPIN (for Space Invaders) elements have been transferred multiple times within mammals and other tetrapods (Gilbert et al. 2012). Occurrences of HTT events involving Helitrons have been reported in mammals, reptiles and fish (Thomas et al. 2010), and additional examples of HTT of DNA transposons include Merlin, TcMariner and OC1 (Feschotte 2004; De Boer et al. 2007; Gilbert et al. 2010). Transfers between vertebrates and invertebrates have been also observed, such as CACTA DNA transposons from insects to bats, possibly facilitated by a parasite-host interaction (Tang et al. 2015). Due to their mode of transposition, LINE retrotransposons are generally not considered as potential targets for HTT. However, the reported case of RTE BovB in Ruminantia (Kordiš and Gubensek 1998, 1999) suggests that HTT has probably happened more than originally thought. With the increase of genome sequencing projects, the number of identified HTT cases might grow considerably in the next few years.
Emergence of lineage-specific TE families
Some unique elements have emerged de novo and successfully invaded specific lineages, constituting punctuated events that might significantly contribute to genome divergence. As proposed for HTT, the newly emerged TEs can either be rapidly targeted by host defence and thus eliminated, or can experience a burst of transposition due to a lack of defences. Alu retrotransposons maybe constitute the best example of such successful lineage-specific elements in vertebrates. Alu sequences are 7SL RNA SINE elements specific to primates. They can be found in approximately 106 copies in the human genome, with different subfamilies present (Ullu and Tschudi 1984; Minghetti and Dugaiczyk 1993; Deininger 2011a). Other non-autonomous TEs even show a more restricted distribution, like the TX1 LTR retrotransposon, which is only found in poeciliid fish (Schartl et al. 1999).
Lineage-specific genome rearrangements
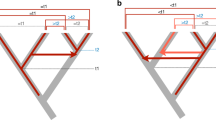
As a consequence of their mobility and high copy number, TEs can affect the structure of genomes by inducing different types of genomic rearrangements through insertion or ectopic recombination. First, the movement of TEs can alter gene structure or expression by inserting into or near exons, introns or regulatory regions. More drastically, TEs can also induce different types of rearrangements through homologous or illegitimate ectopic recombination that can modify or even delete genes (Fig. 3). Such TE-mediated rearrangements include deletions, duplications, inversions and translocations. All those processes can strongly affect the host at the individual scale, but also at the population or species levels.

Genomic innovations and re-arrangements mediated by transposable elements
At the individual scale, mutations may cause phenotypic changes. For example, TE insertions can lead to a number of human diseases including cancer (Deininger and Batzer 1999; Belancio et al. 2008). For instance, insertion of Alu SINEs into the BRCA1/2 genes is a well-described cause of breast cancer in women. Other diseases induced by TE insertions include ovarian carcinoma, haemophilia, colon cancer and Apert syndrome. In some cases, TE insertions can be beneficial: a mutant of the Xiphophorus fish, for example, presents a TX1 retrotransposon inserted in the Xmrk oncogene, which leads to the inability to form melanoma (Schartl et al. 1999). At the population or species level, some non-deleterious mutations can provide a source of phenotypic variation. For example, a Tol2 element (from the hAT DNA transposon family) in an inbred line of medaka fish is responsible for pigment variation. Depending on its homozygous presence, homozygous excision or heterozygous excision/presence, fish harbour albino, wild-type or new colouring phenotypes, respectively (Iida et al. 2005; Koga et al. 2006).
At a larger evolutionary scale, TE-mediated rearrangements can contribute to lineage divergence and speciation. A link between bursts of TE activity and species radiation has been proposed in apes, rodents and bats (Verneau et al. 1998; Chinwalla et al. 2002; Dobigny et al. 2004; Ray et al. 2008). Large rearrangements are thought to have played a fundamental role in hominid radiation during the last 5–20 Myr, some of them having been driven by ERVs, which account for 8 % of the human genome (Hughes and Coffin 2001, 2005). TE-mediated deletions were proposed to be involved in the process of rediploidization after genome duplication as well as in species diversification in salmonid fish (De Boer et al. 2007). Large inversions and deletions have also been observed in sex chromosome evolution. By mediating such rearrangements, TEs might participate in the differentiation of sex chromosomes from a pair of autosomes through the suppression of recombination, leading to non-recombining regions, as found between the X and Y chromosomes in mammals (Steinemann and Steinemann 2005). In some cases, lineage-specific TE-mediated rearrangements have been associated with functional differences. In humans, a 92-bp exon in the CMAH gene, encoding the cytidine monophosphate N-acetylneuraminic acid hydroxylase, was lost through recombination between two adjacent Alu elements (Hayakawa et al. 2001). This resulted in the CMAH gene being non-functional, with loss of synthesis of the sialic acid molecule Neu5Gc (N-glycolylneuraminic acid) and increase in expression of the precursor N-acetylneuraminic acid (Neu5Ac). Compared to non-human hominids, humans should consequently have more resistance to Neu5Gc-binding pathogens, but more risk to Neu5Ac-binding pathogens (Varki 2001, 2010).
TE activity as a reproductive barrier promoting speciation
In vertebrates, a significant increase in TE activity has been shown to coincide with some hybridization events between species. Similar to what has been observed in hybrid dysgenesis in Drosophila (Bingham et al. 1982), this might play a role in genomic instability, destabilisation and incompatibilities in the hybrid generation, and the concomitant TE activity may re-enforce reproductive barriers. Retroviral element amplification and chromosome remodelling associated with genome-wide under-methylation have been reported in a marsupial hybrid (O’Neill et al. 1998). In marsupial hybrids again, in a study focusing on centromeric instability and remodelling, the authors postulated that incompatibilities in hybrid genome involving small RNAs (such as siRNAs and piwi-RNAs), which are fundamental for restraining TE amplification, may result in TEs becoming activate, leading to changes in chromatin structure and to hybrid dysgenesis (Metcalfe et al. 2007). A 232-fold increase in TE activity has been found in malformed embryos of hybrid whitefish compared to wild type, suggesting that mobile sequences are key components of postzygotic isolation and thus drivers of speciation in lake whitefish (Dion-Cote et al. 2014). Alternatively, TE activation in hybrids might lead to beneficial new phenotypes and to hybrid speciation (Baack and Rieseberg 2007).
TE-mediated gene cis-regulation and regulatory network rewiring
TEs trigger lineage-specific regulatory diversity: a long-standing hypothesis
The idea that TEs could contribute to regulatory diversity and innovation is not recent, since they were initially called “controlling elements” by their discoverer, Barbara McClintock (1956). Indeed, by analysing maize transposons, she observed that these sequences could alter the expression of several loci in the genome. Some years later, Britten and Davidson suggested that the evolution of new structures or functions could be greatly accelerated by the co-option of TEs into regulatory elements (Britten and Davidson 1971). By such events, TEs would get involved in the activation of new groups of genes that were not co-regulated before in particular spatial and temporal conditions.
Some studies indeed support a global role of TEs in regulatory diversity and possibly, speciation in vertebrates. An analysis of open chromatin regions (as a proxy for regulatory regions) has shown that 63 % of the primate-specific regulatory regions are embedded within TEs, with particular involvement of ERVs (Jacques et al. 2013). Comparison of marmoset and anthropoid primate genomes showed that the vast majority of anthropoid-specific constrained regions are non-coding, and that >56 % correspond to TEs (Del Rosario et al. 2014). Interestingly, these anthropoid elements are particularly associated with genes involved in brain development, motor coordination, neurotransmission and vision. In African lakes, cichlid fish represent a highly diversified group (>1500 species) that rapidly expanded within a few million years. The sequencing and transcriptome analysis of five of these species allowed assessment of the genomic basis of this evolutionary radiation. TE insertions near 3ʹUTRs were shown to be significantly associated with increased gene expression in the majority of the tissues, suggesting a role for TEs in gene expression divergence, adaptation and possibly speciation (Brawand et al. 2014).
TE-derived host gene promoters and other regulatory sequences
A wealth of evidence has accumulated over the past 30 years that support the role of TEs in the evolution of the regulation of specific host genes within and outside of vertebrates (Böhne et al. 2008; Feschotte 2008; Bourque 2009; Rebollo et al. 2012; Cowley and Oakey 2013; De Souza et al. 2013; Gifford et al. 2013). Most of them implicate TE-derived sequences as particular promoters, enhancers, transcription terminators, silencers of genes and insulators (Table 1). Mobile sequences can additionally give birth to new splicing sites for genes, such events being coupled with the formation of a new exon (Fig. 3).
Many studies have described TE-derived sequences functioning as host gene promoters (Ting et al. 1992; Cristofano et al. 1995; Schulte and Wellstein 1998; Landry et al. 2002; Landry and Mager 2003; Bièche et al. 2003; Gombart et al. 2009; Thomson et al. 2009; Herpin et al. 2010; Emera and Wagner 2012a). The regulation of salivary amylase genes in primates constitutes a classical example of such an exaptation. Mammalian amylase genes are present in several copies that are derived from a single ancestral gene duplicated in tandem several times during mammalian evolution (Samuelson et al. 1990). While all mammals produce amylase in the pancreas, only primates, rodents and lagomorphs do so in saliva as well (Ting et al. 1992). Ting et al. demonstrated that the salivary expression of three of the human amylase genes is specifically conferred by an HERV-E retrovirus-derived sequence that inserted into the promoter of one of the ancestral genes prior to its subsequent duplication (Ting et al. 1992). The production of amylase in saliva probably improves the digestion of starchy food and thus increases the fitness of the species (Perry et al. 2007).
In fish, the downregulation of the male master sex-determining gene dmrt1bY of the medaka Oryzias latipes is exerted by a feedback loop involving a TE inserted into the proximal promoter region of the gene (Herpin et al. 2010). The emergence of this new regulatory feature is thought to have been crucial for the recruitment of dmrt1bY at the top of the male sexual development regulatory cascade of this fish. This example is particularly interesting since sex determination systems appear quite variable in fish (Volff et al. 2007) and illustrates how TEs constitute ideal driving factors in such fast-evolving pathways. Gombart et al. have shown how the insertion of an AluSx SINE element into the promoter of the primate CAMP gene (encoding a steroid hormone nuclear receptor) has placed it under the control of the vitamin D pathway (Gombart et al. 2009). They demonstrated the selection for this insertion in primates and suggested that it could counter the anti-inflammatory properties of vitamin D in these species compared to other mammals.
By inserting into regions more distal to the transcription start site, TEs can also give birth to new transcriptional enhancers that are lineage-specific. A number of them have been well characterised experimentally (Hambor et al. 1993; Bi et al. 1997; Pi et al. 2004; Santangelo et al. 2007; Sasaki et al. 2008; Franchini et al. 2011; Tashiro et al. 2011; Nakanishi et al. 2012; Santos et al. 2014). In mammals, expression of the fibroblast growth factor 8 (fgf8) gene is induced in the developing diencephalon by a mammalian-specific conserved element containing an AmnSINE1 sequence (Nakanishi et al. 2012). Reporter assays revealed that the AmnSINE1 part of the conserved element drives fgf8 expression in the ventral midline of the hypothalamus.
Many species of African cichlid fish use female mouth brooding of the eggs; males present conspicuous colour markings called “egg-spots” on their anal fin, which influence the behaviour of females during mating and facilitate egg fertilisation in their mouth. In these species, the occurrence of egg-spots has been linked to the integration of an AFC-SINE in the cis-regulatory region of the fhl2b pigmentation gene (Santos et al. 2014). This TE insertion was shown to be specific to egg-spot-bearing cichlids, and shows specific enhancer activities in pigment cells called iridophores.
Several studies have also suggested a role of TEs in the birth of lineage-specific polyadenylation (polyA) sites and thus in the evolution of novel 3ʹ-ends in genes. An analysis of the conservation of alternative polyA sites between human, mouse, rat and chicken revealed that non-conserved sites are much more associated with TEs than conserved ones (Lee et al. 2008). Ninety-four percent of human TE-associated polyA sites were non-conserved in the mouse, and conversely 93 % of mouse TE-associated polyA sites were not present in human. Many Alu sequences, which are present in primates but absent from rodents, can serve as polyA sites for host genes in humans, leading to divergences between both species (Chen et al. 2009). Some Alu-borne sites are intronic and probably lead to truncated transcripts. Strikingly, instead of constituting only weak alternative sites, they often represented a major polyA site for the gene.
TE-mediated rewiring of specific transcription factor regulatory networks
In the last years, genome-wide analyses, in particular using chromatin immunoprecipitation followed by sequencing (ChIP-seq), have suggested that TEs could control entire regulatory networks and rewire them by contributing, over a short evolutionary time frame, an important number of binding sites for specific transcription factors (Table 1; Mortazavi et al. 2006; Wang et al. 2007; Bourque et al. 2008; Mason et al. 2010; Cui et al. 2011; Micale et al. 2012; Schmidt et al. 2012; Cotney et al. 2013; Sundaram et al. 2014; Notwell et al. 2015) (for reviews, see Feschotte 2008; Rebollo et al. 2012).
Human embryonic stem cells (ESCs) constitute a good example illustrating how TEs might have shaped gene regulation during evolution. Comparison of binding profiles of the pluripotency factors NANOG, POU5F1 and CTCF in human and mouse ESCs has revealed that only 5 % of the binding regions are conserved between the two species for NANOG and POU5F1 (Kunarso et al. 2010). Strikingly, TEs have contributed up to 25 % of binding sites, possibly recruiting new genes to ESC. Notably, most of the recruited TE families are species-specific, with an important contribution of ERV1 elements. A more recent study focusing on human-specific regulatory loci binding NANOG, POU5F1 and CTCF indicated that 99.8 % of them are embedded within TEs, in particular, LTR7/HERV-H, LTR5_Hs and L1HS elements (Glinsky 2015). However, the drastic proportion observed might reflect the very stringent criteria used in this study to define human-specific loci: to be characterised as such, a region must have no orthologous (i.e., aligned) sequence in other species. The presented number thus does not take into account possible new regulatory loci that appeared by point mutations in more ancient sequences. Only 4.3 % of these regions could be retrieved in the genome of Neanderthal, leading to the conclusion that most of these putative regulatory sites derived from TEs arose in modern human.
In humans, more than one third of the pleiotropic tumour suppressor factor p53 binding sites overlap with ERV elements (Wang et al. 2007). These binding sites spread ∼40 million years ago (MYA) during the colonisation of the genome by these ERVs (Wang et al. 2007); they are thus primate-specific and not found in other mammals. The ERV progenitor was likely to contain a p53 motif in its LTR already. The functionality of the binding sites, detected by ChIP-seq experiment, was further demonstrated for five LTRs using a gene reporter assay (Wang et al. 2007). Focusing on a much smaller subset of binding sites (160 human binding sites with proven activity), Cui et al. (2011) observed that half of the repeat-associated p53 sites resided within Alu elements. LINE1 elements were also shown to play an important role in shaping the human p53 regulatory network (Harris et al. 2009), highlighting the fact that different TE families can encompass, or give birth to, multiple binding motifs for the same transcription factor. Binding sites from different TE origins probably harbour different sequence characteristics and therefore have different effects on the activity of the transcription factor and on the regulation it provides. Interestingly, a similar survey drawn from zebrafish detected the zebrafish-specific EnSpmN6_DR non-autonomous DNA transposon as a major contributor to p53 binding sites in this species (Micale et al. 2012). As several orthologous genes are controlled by p53 in both primates and teleost fish, these observations constitute a good example of convergent regulatory evolution driven by TEs.
Molecular specificities that could explain brain development in humans are the focus of many studies and provide more examples of lineage-specific enhancer regions provided by TEs. Notwell et al. have recently shown a significant enrichment of developing neocortex enhancers in MER130 repeats in the mouse (Notwell et al. 2015). MER130 is a non-autonomous TE that originated in the tetrapod or possibly Sarcopterygii ancestor. It presents putative binding sites for the Nfi and Neurod/g transcription factors, which are important for brain development. The functionality of MER130-containing enhancers was further demonstrated by luciferase reporter assays (Notwell et al. 2015). Additionally, six of the 22 validated MER130 enhancers are located near genes critical for neocortex development, such as Robo1 or Id4. Most MER130 instances identified in the mouse (96 %) were conserved in human, suggesting a possible ancestral exaptation of this element in the regulation of tetrapod brain development. MER130 sequences were found in several copies in the genomes up to the frog, but without signs of recent activity. Interestingly, a single and more divergent copy was found in the coelacanth; despite its low-conserved sequence, this MER130 instance could also drive significant activity in cortical neurons (Notwell et al. 2015).
Finally, by comparing the binding sites of 26 orthologous factors between human and mouse, Sundaram et al. evaluated that 20 % of sites were embedded within TEs (Sundaram et al. 2014). They also showed that most of these TE-derived binding sites were species-specific. For some transcription factors, the expansion of binding sites mediated by TEs even happened in only one of the two species analysed. This further sustains the Britten and Davidson model of TEs being major drivers of species-specific regulatory innovations.
Interestingly, a similar involvement of TEs has been shown to participate in the evolution of insulators. Insulators are boundary elements in the DNA that limit the action of enhancers within a particular region; their effect is thought to occur through a modification of 3D DNA structure mediated by proteins such as the transcriptional repressor CTCF. A number of CTCF sites have been provided by B1 or B2 SINEs in rodents (Bourque et al. 2008; Román et al. 2011).
While several genome-wide studies indeed suggest major roles for TEs in regulatory innovations, some of their conclusions must be taken cautiously, particularly concerning ChIP-seq experiments. As it was well synthesised in a previous review (De Souza et al. 2013), the transcription factor binding itself is not necessarily synonymous to functionality of the binding (i.e., effect on a target gene) neither to its physiological (effect at the organ scale) or its evolutionary impact (selected increase in fitness). A good proxy for functionality of a detected binding region could be its evolutionary conservation associated with an overlap with active histone marks (De Souza et al. 2013). What is more, the different transcription factors or tissues might not be equally susceptible to “TE-spread-sites.” Among the 26 transcription factors analysed by Sundaram et al. (2014), the proportion of sites located within TEs shows great variation, from 2 to 40 % of all the sites bound by each factor. Analysing enhancers involved in limb development in human, mouse and rhesus, Cotney et al. could not find any repeat enrichment in the 11 % of human enhancers that were specifically gained in humans (Cotney et al. 2013). Using a comparable approach, Villar et al. analysed the evolution of enhancers in the liver of 20 mammalian species (Villar et al. 2015). While they detected an important turnover of the regulatory sequences, only a minor proportion of the recently created enhancers corresponded to TEs. More work is needed to test if such differences are due to technical limitations of the studies, or if they reflect the diversity of TE contribution to regulatory network rewiring.
A major evolutionary innovation mediated by TE regulatory rewiring: the emergence of placental regulatory circuits
TEs might have played a decisive role in the birth of new traits by the new regulatory circuits they can mediate. A particularly well documented example is the emergence of placenta in mammals (for review, see Emera and Wagner 2012b). A role of TEs in such a major innovation was suggested by the analysis of a number of gene promoters triggering a placenta-specific expression. In primates, two enhancers located in a MER11 and an HERV elements allow placental expression of the leptin and insulin-like 4 protein (INSL4) genes, respectively (Bi et al. 1997; Bièche et al. 2003). INSL4 was previously shown to be upregulated during cytotrophoblast differentiation into syncytiotrophoblasts in human. Similarly, HERV-derived promoters control the EDNRB and mid1 genes in the human placenta (Landry et al. 2002; Landry and Mager 2003). In apes, the prolactin gene (Prl) is expressed in endometrium thanks to a strong promoter derived from a MER20 and a MER39 element (Emera and Wagner 2012a). Interestingly, the endometrial expression of Prl is not shared among all eutherian species (the gene is not expressed in placenta of rabbits, pigs, dogs or armadillos). In species expressing Prl in placenta, a striking case of evolutionary convergence is observed while the expression of Prl is controlled by the MER20/MER39 promoter in apes, the Prl promoter is derived from a MER77 element in mice and from a L1-2_LA element in elephant (Emera et al. 2012).
All these observations were recently extended to complete gene networks through genome-wide analyses. ERVs in particular were suggested to have participated in the emergence and diversification of placental structures among mammals (Chuong et al. 2013). ERVs were indeed shown to have spread a number of species-specific trophoblast cis-regulatory sequences that present binding motifs for key regulatory factors. In another study, about 1500 genes, ancestrally expressed in other non-placental tissues, were demonstrated to have gained expression in endometrial cells in placental mammals (Lynch et al. 2011). Interestingly, 13 % of these genes contained a MER20 element within 200 kb. These MER20 repeats exhibited enhancer signatures and were able to bind essential pregnancy factors linked to progesterone or cAMP signalling (Lynch et al. 2011). These observations thus suggested a major implication of MER20 TEs in foetus implantation and gestation. The same research group recently compared the uterine transcriptome among tetrapods, highlighting thousands of genes that acquired an expression in the uterus during mammal evolution (Lynch et al. 2015). Genes mediating decidualization and cell-type identity in decidualized stromal cells were found to be associated with numerous cis-regulatory elements derived from ancient TEs including MER20, most being eutherian-specific. This illustrates how TEs can rapidly rewire a network by putting previously non-co-regulated genes under new common specific regulations.
Impact of TEs on non-coding RNA structure and diversity
Almost three quarters (74.9 %) of the human genome is transcribed into primary RNA, and the vast majority of the resulting RNA does not code for proteins (Djebali et al. 2012). There are many types of non-coding RNAs, including ribosomal RNAs (rRNAs), microRNAs (miRNAs), long non-coding RNAs (lncRNAs), transfer RNAs (tRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), short interfering RNAs (siRNAs) and piwi-interacting RNAs (piRNAs) (Lukic and Chen 2011; Dozmorov et al. 2013; St Laurent et al. 2015). An emerging feature of many types of non-coding RNAs is their lineage specificity. In this section, we will focus on miRNAs and lncRNAs, as much recent work has shown a marked involvement of TEs in the origin and evolution of these sequences (Table 1).
TEs as drivers of the diversity of miRNA repertoires
Mature miRNAs are sequences of around 22 nucleotides; their main functions are mRNA degradation and translational regulation (Lee 1993; Bartel 2004). Their mature sequence arises from the processing of a longer sequence called “pri-miRNA,” which forms a self-folding hairpin structure. The pri-miRNA is then trimmed of the loop itself and the non-bound tails of the hairpin loop by complexes involving the proteins Drosha and Dicer (Ha and Kim 2014). This leaves only the overlapping RNA, containing the active part of the miRNA, which, when single stranded, acts as a guide to the protein Argonaute that represses translation of the target mRNA.
Over 3500 miRNA-producing loci have been identified in the human genome (Londin et al. 2015). More than half of the protein-coding genes in human are thought to be targets of miRNAs (Bartel 2004; Friedman et al. 2009). Evidence suggests that miRNAs are lineage-specific. For example, 56.7 % of human miRNAs were found to be species-specific, and 94.4 % of human miRNAs are restricted to the primate lineage (Londin et al. 2015). Furthermore, the target sites of miRNA have been shown to vary between human populations (Saunders et al. 2007).
Genome-wide studies have indicated that TEs are making significant contributions to miRNAs, but that their contribution can vary dramatically between species. Borchert et al. (2011) analysed the origin of 15,176 miRNAs across multiple species and found that >15 % contained TE-derived sequences, with DNA transposons and non-LTR retroelements accounting for over half of the TE-derived sequences. In humans, this number rises to over 20 % of miRNAs (Spengler et al. 2014; Qin et al. 2015). In contrast, no miRNAs containing TE-derived sequence have been detected in Xenopus, which has 25 % of its genome constituted by TEs (Hellsten et al. 2010; Qin et al. 2015). In the zebrafish (55 % of TEs in the genome), only 5 % of miRNAs contain TE-derived sequences (Howe et al. 2013; Qin et al. 2015; Chalopin et al. 2015). In chicken, 5 % of the genome is covered by TEs and almost 7 % of the miRNAs were derived from TEs (Hillier et al. 2004; Qin et al. 2015). These differences could be attributed to different types of TEs present between species and lineages, but also to the lack of fully and accurately characterised TEs and miRNAs in these species (Chalopin et al. 2015; Qin et al. 2015).
The data discussed earlier only pertain to whether TEs are present in the miRNAs, and not whether their presence is functionally relevant. The pivotal structure of the miRNA processing is the hairpin loop formed by imperfect binding of two inverted repeats. TEs can contribute to this in two major ways. First, two adjacent, inverted and diverged copies of the same element can form the basis of a hairpin loop. This was observed in 11.2 % of human TE-containing miRNAs, and often occurs with adjacent LINE elements (Qin et al. 2015). One such example is miR-28, which is a miRNA derived from the ends of two adjacent LINE2c insertions (Smalheiser and Torvik 2005; Gim et al. 2014).
A second-way TEs can contribute to miRNA hairpins is that a single TE forms the hairpin loop. For example, the terminal inverted repeats of DNA transposons, generally MITEs, can self-bind and form such hairpins. Examples include the MITE MADE1, and also members of the MER family (Piriyapongsa et al. 2007a; Qin et al. 2015). Additionally, some TEs have internal hairpin loops, such as the Alu elements (Deininger 2011b; Spengler et al. 2014; Qin et al. 2015). Such occurrences have been observed in over two thirds of TE-containing miRNAs. However, many TEs in human only overlap with a small portion of the miRNAs and constitute only part of the self-binding sequence. The exact functional role of the TEs in such cases is less clear.
The conservation of TE-derived miRNAs is generally much lower than for non-TE-derived miRNAs (Meunier et al. 2013). Interspecific comparisons indeed demonstrated high species specificity. No TE-derived miRNA was found to be common between zebrafish (346 TE-containing miRNAs) and mammals (615 to 1872 TE-containing miRNAs). Only 14 TE-containing miRNAs were shared among mammals, and 47 were common to primates (Qin et al. 2015). TE-derived miRNAs might contribute to lineage-specific functions: in mammals, the main recruitment of “young” miRNAs to exert regulatory functions in nervous tissues suggests their involvement in recent evolution (Meunier et al. 2013).
Some studies have provided clues on the important role of TEs in miRNA evolutionary dynamics. For example, the miR-1302 miRNA family has 11 members in the human genome and is derived from MER53, a DNA transposon with a short consensus sequence of 193 bp (Yuan et al. 2010). The exact function the miR-1302 family is unknown but targets include the male fertility-related gene CGA (Zhang et al. 2011). Homologs of miR-1302 are only observed in placental mammals and all are thought to be derived from MER53. Interestingly, across placental mammals, there is a high turnover of the miR-1302 family members. A repeated “birth and death” model has been proposed for these elements, with independent convergent recruitment of MER53 between lineages (Yuan et al. 2010).
TE contribution to the function and lineage-specific diversity of lncRNAs
Long non-coding RNAs (lncRNAs) represent a very interesting emerging class of ncRNAs. There are 10,000–18,000 lncRNAs in humans (Derrien et al. 2012; Hezroni et al. 2015), with a similar number in rhesus monkey and mouse (Hezroni et al. 2015). In other mammalian and vertebrate species, estimations of lncRNA numbers can greatly vary (only 1000 in the stickleback; Hezroni et al. 2015). Diversity is observed at the size level (from a few hundred bp to several kb in length), as well as the level of the processing: while some lncRNAs are only transcribed, others undergo post-transcriptional processing like mRNAs, including splicing, 5ʹ capping and poly-adenylation (Ruiz-Orera et al. 2014; Vance and Ponting 2014). Little is currently known about the role of the vast majority of lncRNAs; in humans, only 130 lncRNAs have been analysed at the functional level (Amaral et al. 2013). Originally thought to be mainly found in the nucleus and involved in gene regulation, lncRNAs have been detected in all cell compartments and vary greatly in their expression between tissues, suggesting a high diversity of functions (Ruiz-Orera et al. 2014; Vance and Ponting 2014).
Many lncRNAs are lineage-specific, but estimations might depend on the studies and/or on the types of lncRNAs analysed. Only 3 % of human lncRNAs have been reported to be conserved in non-primate species (Kutter et al. 2012). In another study specifically looking at a subclass of lncRNAs (lincRNAs), 60–70 % of sequences were shared between mice and humans (Kutter et al. 2012; Managadze et al. 2013). In a multi-species comparison, it was not possible to find orthologs for more than 30 % of lncRNAs between species having diverged more than 50 MYA (Hezroni et al. 2015). A better characterisation and classification of lncRNAs will help to elucidate their degree of conservation between species.
Genome-wide studies have demonstrated that the contribution of TEs to the sequence of lncRNAs is strong. In humans, between 69 and 83 % of lncRNAs contain TE-derived sequences, a proportion 10 times higher than that for protein-coding genes (Kelley and Rinn 2012; Kapusta et al. 2013; Kannan et al. 2015). Similar values have been reported in mouse (51–68 %) and zebrafish (67 %) (Kelley and Rinn 2012; Kapusta et al. 2013). In humans, 20 % of TE-containing lncRNAs have TEs making up more than 50 % of their sequence (Kapusta et al. 2013). However, TEs constitute less than 20 % of the sequence of most TE-containing lncRNAs in human (66 %) and mouse (78 %).
Several lines of evidence indicate that TE-containing lncRNAs are functional; they might even be under stronger functional constraints than non-TE containing lncRNAs (Kapusta et al. 2013). The types of TEs found in lncRNA sequences do not accurately reflect the TE composition of the genome, and thus, the TEs are probably not present by chance. For example, ERV/LTRs are over-represented in lncRNAs compared to background genomic levels in human (1.5x) and mouse (3x) (Kelley and Rinn 2012). As expected from their high copy number, the most common TEs found in lncRNAs are Alu and LINE elements in human. However, their contribution is lower than their representation in the genome (Kelley and Rinn 2012).
TE-containing lncRNAs have more stable secondary structures than non-TE containing lncRNAs (Kelley and Rinn 2012). The DNA transposon Angel is present in many lncRNAs in zebrafish, and its inverted repeats are hypothesised to form the basis of self-binding, leading to the formation of secondary structures. Compensatory substitutions have occurred to maintain the binding of the inverted repeats during evolution (Kelley and Rinn 2012). Inverted pairs of TEs can also enable binding and result in secondary structures.
Specific examples point towards the functional importance of TEs within lncRNAs and how they become incorporated (Santoni et al. 2012). The mature pluripotency-associated human lncRNA linc-ROR is mostly composed of TEs, with over 70 % of the sequence being derived predominantly from HERV-L, but also from LINE and SINE elements. This suggests a role of linc-ROR TE-derived sequences in pluripotency (Loewer et al. 2010; Santoni et al. 2012; Zhang et al. 2013; Johnson and Guigó 2014). A single nucleotide mutation localised in a LINE element within a lncRNA found in one intron of the SLC7A2 gene is associated with infantile encephalopathy (Cartault et al. 2012). This mutation might affect the secondary structure of the lncRNA. In humans, the lncRNA anti-sense noncoding RNA in the INK4 locus (ANRIL) binds to the polycomb protein and then form complexes with DNA to regulate expression of downstream target genes (He et al. 2013). The DNA binding of ANRIL is mediated by the Alu sequences present in the lncRNA (Holdt et al. 2013).
The origin of TE-containing lncRNAs is not always obvious to determine. TEs can be at the origin of the formation of the lncRNA, or can be incorporated subsequently (Kapusta et al. 2013; Kelley and Rinn 2012; Necsulea et al. 2014; Washietl et al. 2014). One of the best-studied lncRNAs is Xist, which is involved in X chromosome silencing and arose in the eutherian ancestor from the decayed protein-coding gene lnx3. Since its formation, Xist has gained several TE-based exons (Elisaphenko et al. 2008). Similarly, ANRIL is an lncRNA that in simians has become highly exonised, but not so in other mammal species (He et al. 2013). These exons are formed by both pre-existing TEs and the recruitment of TEs to the ANRIL sequence. The TE-based exon sequences are predicted to form important secondary structures for the lncRNAs (He et al. 2013). As for evidence describing lncRNAs being derived directly from TEs, Fort et al. (2014) performed deep transcriptome sequencing of stem cell lines in mouse and human, identifying 2372 and 639 novel LTR-associated lncRNAs, respectively, many of which appeared to have originated from TEs. The predominant TEs involved were ERVK and MaLR in mouse and ERV1 in humans. Knock-out of four of the LTR-based lncRNAs affected stem cell status, demonstrating a direct functional role (Fort et al. 2014). Similarly, Wang et al. showed that naive stem cells are characterised by a high expression level of HERV-H that leads to the production of hESC-specific chimerical transcripts, including a number of lncRNAs (Wang et al. 2014). These transcription events were triggered by binding sites found in ERV sequences that can recruit naive pluripotency transcription factors such as LP9. HERV-H-derived transcripts were demonstrated to be necessary for the self-renewal of the cells (Wang et al. 2014).
In summary, the studies described earlier are demonstrating the high level of involvement of TEs in lncRNAs, including potential functional roles (Kapusta et al. 2013; Kelley and Rinn 2012; Wang et al. 2014). Given the highly lineage-specific nature and the high turnover of lncRNAs (100 new lncRNA genes per Myr in rodents and primates; Kutter et al. 2012; Kapusta and Feschotte 2014), combined with the high contribution of TEs to their sequences, it seems clear that TEs will be a large contributing factor towards the lineage-specific nature of lncRNAs. The formation of lncRNAs directly from lineage-specific TEs (Fort et al. 2014; Wang et al. 2014) strongly indicates a role for TE-containing lncRNAs in vertebrate diversification. Currently, the exact structure and function of the vast majority of lncRNAs is not fully understood, but as a fuller understanding emerges, the extent and potential roles of TEs within lncRNAs will also become clear (Johnson and Guigó 2014).
TEs as a source of lineage-specific novel protein-coding sequence
TEs as a source of novel exons
The process of TE exonisation is when TEs contribute new exons within an existing host protein-coding gene, with incorporation of the TE-derived sequence into mature spliced mRNA. The insertion of a TE into a protein-coding gene can provide novel 3ʹ and 5ʹ splicing sites directly or after additional mutations (Fig. 3). If an open reading frame (ORF) is present in the inserted TE, then the exon can be included in the final coding sequence. In mammals, and particularly in humans, this is a common process, with over 2000 TE-derived exons being reported in humans (Piriyapongsa et al. 2007b; Sela et al. 2010). This is thought to be mainly due to the primate-specific Alu elements, which contain many 3ʹ splice sites in pyrimidine-rich tracts (Brow 2002). To become an exon, an Alu element would need to be present in the anti-sense orientation; this is indeed observed in 85 % of Alu-induced exons (Spengler et al. 2014).
Many examples of Alu exonisation have been reported (Singer et al. 2004; Krull et al. 2005; Schmitz and Brosius 2011). In the human tumour necrosis factor receptor gene type 2 (p75TNFR), an alternative first codon is contributed by an insertion of AluJ, which provides a novel N-terminal protein-coding domain (Singer et al. 2004). Alu integration and start codon formation occurred about 50 MYA in the common ancestor of anthropoid primates. Two additional single nucleotide mutations were required to provide a 5ʹ splice site and an ATG start codon, along with a 7-bp deletion to generate an ORF. These arose between 40 and 25 MYA in the Old World monkey lineage. Similarly, in the ribulose-5-phosphate-3-epimerase (RPE2-1, also known as phosphopentose epimerase), a novel exon has occurred caused by a truncated 75-bp AluJ element inserted between the second and third exons (Krull et al. 2005). The insertion took place 58–90 MYA, but is only active in hominids. Formation of a functional exon required the loss of an alternative distal 3ʹ splice site, a point mutation in a proximal 3ʹ splice site, and a 2-bp deletion that provided an ORF.
Alu elements are not the only TE found in the exonisation process (Piriyapongsa et al. 2007b; Krull et al. 2007; Lin et al. 2009; Bae et al. 2013). LTR retrotransposon-derived sequences have been found in as many as 1057 out of 18,241 genes in humans (Piriyapongsa et al. 2007b). For example, the MaLR element provided a novel exon to SUPT16H, a gene believed to be involved in the unpackaging of chromatin and DNA repair (Bae et al. 2013). The insertion occurred before the split of the New World monkeys and the promisians (40 MYA), between the second and third exons of the gene. The MaLR element provides the splicing sites; although the 5ʹ splice site is occasionally ignored and an alternative transcript that fuses with the third exon sometimes exists. Similarly, the DNA transposon family mammalian interspersed repeats (MIR) has caused many exonisation events (Lin et al. 2009). For example, in the gene encoding the zinc finger protein ZNF69, an inserted MIR is constitutively expressed and adds an extra 45 aa to the protein sequence (Krull et al. 2007). The MIR element provides a 3ʹ splice site half way through the element, but the 5ʹ splice site is taken from existing intronic sequence. It is present in all mammals, but not other vertebrates. The conservation and constitutive expression suggests that the extra 45 aa provide benefit, and the purifying selection that is observed on the exon supports this too.
Outside of humans, the number of detected TE exonisation events is generally a lot lower (Sela et al. 2007, 2010). In mice, 500 events have been detected, whereas only 70 have been identified in chicken and 53 in zebrafish (Sela et al. 2010). Outside of vertebrates, only 12 TE-based exonisation events were reported in Ciona intestinalis and none in Drosophila melanogaster and Caenorhabditis elegans. In primates, the Alu insertions occurred in younger genes (e.g., primate/human-specific) rather than older genes (e.g., mammalian- or vertebrate-specific; Shen et al. 2011). In addition, TE-based exons are generally not constitutively expressed, and often, their expression levels are low compared to alternative transcripts lacking the TE-derived exon (Zhang et al. 2013). Transcripts with older TE insertions are more likely to be expressed constitutively than those with younger insertions (Shen et al. 2011). A final interesting trend is that, in mammals, there is a strong preference for Alu-based exonisation events in zinc finger domain-containing proteins, which have undergone important expansion and diversification in primates (Emerson and Thomas 2009; Nowick et al. 2010; Shen et al. 2011).
The functional consequences of the TE-based exonisation events are not always clear and have been rarely tested so far (Lev-Maor et al. 2003; Shen et al. 2011). Any detected changes were marginal differences in binding activity or translational activity, but no direct connection to the function or the fitness of the host was observed (Lev-Maor et al. 2003; Shen et al. 2011).
TE-mediated retroposition and transduction
Transposable elements can generate novel coding sequence by partially or completely duplicating genes in the genome (Fig. 3). This can either be performed through retroposition (Vinckenbosch et al. 2006) or transduction (Xing et al. 2006). In retroposition, mRNA sequences of host genes are reverse transcribed into complementary DNA (cDNA) by reverse transcriptases encoded by autonomous retroelements. They are inserted into the genome as intronless coding sequences referred to as “retrocopies.” Generally, these new insertions do not recruit any promoter or regulatory sequences and degrade, but sometimes they evolve as functional genes and are termed “retrogenes.” For example, the glutamate dehydrogenase 2 (GLUD2) in human is a retrogene derived from GLUD1 about 18-25MYA (Marques et al. 2008). In the swordtail fish Xiphophorus helleri, there are four copies of the long wave-sensitive opsin gene LWSO, one of them being a functional retrogene that appeared somewhere in a common ancestor of guppies and swordtails (Watson et al. 2010). This provides a wider range of visual sensitivity, which is often tightly linked to adaptation and species diversification.
Gene duplication through retroposition has occurred in many species. In humans, there are estimated to be between 3500 and 17,000 retrocopies in the genome, 120–163 of them being bona fide functional retrogenes (Vinckenbosch et al. 2006; Marques et al. 2008; Henrichsen et al. 2009; Pan and Zhang 2009; Fu et al. 2010). It is estimated that primates gain one retrogene every 1 Myr (Marques et al. 2005). In other species, similar numbers of retrogenes have been detected, with the higher estimates seen in rats (226 retrogenes), opossum (232 retrogenes) and zebrafish (140 to 440 retrogenes) (Pan and Zhang 2009; Fu et al. 2010). The chicken genome contains only about 100 retrogenes, possibly because the CR1 LINE that is predominant in birds does not recognise polyA tails and therefore cannot easily retropose mRNAs (Haas et al. 1997). In mammals, many different retroposition events have occurred independently in a lineage-specific manner, sometimes in a convergent nature, with a slight emphasis on ribosome-associated genes (Pan and Zhang 2009). Retrogenes appear to have been important in the evolution of the mammalian X-chromosome, allowing gene copies to escape meiotic sex chromosome inactivation (Pang et al. 2009), but also in recruitment of genes to the X-chromosome (Potrzebowski et al. 2008, 2010). Retroposition mediated by LINE elements has been suggested to cause gene duplications involved in adaptation of Antarctic notohenioid fish to extreme cold (Chen et al. 2008).
Transduction can occur during the movement of LINE and SINE elements, when genomic sequences adjacent to the 3ʹ end of the element are transcribed together with the element and then inserted after reverse transcription into the genome in a new location (Xing et al. 2006). As with retroposition, the inserted sequence is usually non-functional, but functional examples exist, such as the acetyl malonyl condensing enzyme 1 (AMAC1). In the ancestor of the great African apes, an SVA SINE element inserted adjacent to the original AMAC1 gene on chromosome 17. Subsequently, retrotransposition of the SINE insertion together with the adjacent gene led to two extra copies on chromosomes 8 and 18 (Xing et al. 2006). Transduction associated with SVA elements accounts for 143 events and 53 kb of sequence in the human genome (Xing et al. 2006). Compared to retroposition, fewer confirmed examples of transduction exist, and often, examples are suggested rather than tested. For instance, duplications of the large lipid transfer protein superfamily (associated with yolk) are suggested to be due to LINE-associated transduction in zebrafish (Wu et al. 2013).
In vertebrates, retroposition and transduction events are predominantly associated with LTR, LINE and SINE elements but Helitrons can also play a role (Thomas et al. 2014). Helitron-mediated transduction of host coding sequences, which does not involve reverse transcription, has been reported in lepidopterans, fungi and plants (e.g., Cultrone et al. 2007; Hollister and Gaut 2007; Han et al. 2013) and more recently, in mammals, specifically in the bat Myotis lucifugus (Thomas et al. 2014). In this species, 110 out of 645 unique Helitrons contain sequences derived from 54 different genes. In a study of 36 copies of the TCF3 (E2A) fusion partner gene, two were found to be under purifying selection, suggesting functionality (Thomas et al. 2014).
Exaptation: TEs as a source of novel genes with new functions
TEs can act as a source of “ready to use” new protein-coding sequences that exaptated for the benefit of the host (a process sometimes referred to as “molecular domestication”) (Table 1). In the human genome, over 100 genes are believed to have originated from TE-coding sequences (Volff 2006; Kaessmann 2010; Alzohairy et al. 2013; Campillos et al. 2006). Some of these genes appear to have played critical roles in the evolution of mammals and other vertebrates. Many in-depth reviews exist, providing exhaustive lists of TE-derived genes (Volff 2006; Sinzelle et al. 2009; Alzohairy et al. 2013). Here, we look at the processes involved along with the emerging understanding of when these events happened and how they have affected vertebrate diversity.
Two main types of TE-derived protein-coding genes have been described: genes derived from a transposon sequence, and genes formed through the fusion of a TE sequence with a non-TE gene. Well-known examples of genes derived from an entire transposon are RAG1 and RAG2. These genes encode the recombinase catalysing the V(D)J recombination (or somatic recombination), which generate the highly diverse repertoire of antibodies/immunoglobulins and T cell receptors in vertebrates (Schatz and Swanson 2011). Both proteins have been formed from a Transib transposase over 500 MYA, and the recombination signal sequences they use might be derived from the original terminal inverted repeats of the ancestral transposon (Kapitonov and Jurka 2004; Kapitonov and Koonin 2015). Both RAG1 and RAG2 are crucial to the development of the vertebrate immune system and have probably played an important role in the emergence of the vertebrate lineage. Another example of transposase exaptation is CENP-B, the mammalian centromere-associated protein B, which is derived from a pogo-like transposase (Tomascik-Cheeseman et al. 2002; Casola et al. 2008). Interestingly, CENP-B-like genes have occurred through convergent exaptation events that arose independently in Drosophila, fungi and plants (Casola et al. 2008).
New protein-coding genes can also be formed from retrotransposon and retrovirus sequences. The best-studied examples are gag- and env-derived genes. The 85 human genes deriving from gag genes from retrotransposons of Ty3/Gypsy families are split into five main groups: the MART family (sushi/gypsy-derived gene family), the SCAN family (derived from Gmr1-like Gypsy), the paraneoplastic family (PNMA, also named Ma genes), the SASPase family and the ARC family (Campillos et al. 2006). The MART family contains 11 genes, and is derived from a sushi Ty3/Gypsy retrotransposon that is still functional in fish (Brandt et al. 2005). Expatation(s) and subsequent MART gene duplication events appear to have taken place in the ancestor of the eutherians. Eight out of 11 MART genes are expressed in the placenta, and several MART genes are involved in placental development (e.g., Peg10/Mart2, Peg11/rtl1; Ono et al. 2006; Sekita et al. 2008; Edwards et al. 2008; Kaneko-Ishino and Ishino 2012; Henke et al. 2015). The SCAN family of transcription factors originated from the C-terminal portion of the GAG protein from a gmr1-like retrotransposon in an early tetrapod ancestor, but has undergone a large expansion in mammals, with 60 and 40 SCAN proteins in human and mouse, respectively (Edelstein and Collins 2005). SCAN genes are frequently involved in development and cell differentiation (Li et al. 1999). The PNMA gene family is derived from a Gypsy12_DR-related GAG protein gene that is observed in zebrafish. A single exaptation event is believed to have occurred and then the gene family expanded from this point through gene duplications (Schüller et al. 2005; Kokošar and Kordiš 2013). No functional studies have been carried out on PNMA genes, but PNMA10 is a candidate for X-linked mental retardation (Cho et al. 2008a, 2011) and mouse forebrain development (Cho et al. 2008b). The SASPase gene is a single copy gene seen in all mammals, which is involved in skin development (Matsui et al. 2006, 2011; Barker et al. 2007). Finally, the ARC gene family is derived from a single-copy gene domesticated from the gag gene of a Gypsy-26-I_DR retrotransposon (Campillos et al. 2006). It is involved in neuronal functioning and memory development (Plath et al. 2006).
The exaptation of retroviral env genes produces one of the most intriguing examples of exaptation in the mammalian placenta (Emera and Wagner 2012b). Syncytin-1 (apes) and syncytin-2 (apes and monkeys) were identified to be derived from HERV-W and HERV-FRD env genes (Mi et al. 2000; Blaise et al. 2003). They are expressed in trophoblasts, which are cells constituting the intermediate layer between the mother and foetus in the placenta. Syncytin proteins were shown to be involved in cell-cell fusion and trophoblast differentiation. Similarly, syncytin-A and syncytin-B were discovered in mouse, and knock-out studies have shown them to be important for placental development (Dupressoir et al. 2005; Vernochet et al. 2014). Env-derived genes that are involved in cell fusion and placental function have been acquired independently in lagomorphs (Heidmann et al. 2009), carnivora (Cornelis et al. 2012), ruminants (Cornelis et al. 2013) and Afrotherian tenrecs (Cornelis et al. 2014). Expression of another independent env-derived syncytin gene has also been seen in the short-lived marsupial placenta (Cornelis et al. 2015). As with the other syncytin gene family members, the marsupial version has cell-cell fusogenic properties. This demonstrates an interesting pattern of convergent and repeated recruitments of TE genes to similar functions in a fundamental organ for the mammalian lineage, with a possible contribution of syncytin genes in lineage-specific variations in placental morphology.
A prominent example of fusion of a TE sequence with an existing host gene is the primate-specific setmar1 gene (Lee et al. 2005; Cordaux et al. 2006). SETMAR1 (Metnase) is a fusion of an N-terminal histone-lysine N-methyltransferase SET domain and a C-terminal transposase domain from the mariner-like Hsmar1 element, which appeared first in anthropoid primates. SETMAR1 is a non-homologous end-joining repair protein that regulates genomic integration of exogenous DNA (Lee et al. 2005). The mariner domain, with its DNA binding activity, might target the histone methylase domain to the multiple binding sites provided by copies of the Hsmar1 transposon in the human genome (Liu et al. 2007). Another example is the mammalian-specific GTF2IRD2 protein, which consists in a fusion of a Charlie8 transposase-like domain and the GTFI domain of the TFII-I transcription factor (Tipney et al. 2004). Deletion of GTF2IRD2 is observed in the Williams-Beuren syndrome, which is manifested by physical, neurological and behavioural disorders (Tipney et al. 2004).
Exaptation and subsequent differential evolution of TE-derived genes might be linked to diversification in the vertebrate lineage. A recent study surveyed 24 mammalian genes exaptated from TEs, mostly derived from GAG proteins, across 90 genomes, to identify when the exaptation events were taking place (Kokošar and Kordiš 2013). Few domesticated genes were found outside of the eutherians, only 10 in marsupials and three in monotremes. Outside of mammals, only two genes were found in reptiles (ARC and GIN1, of which only ARC is found in amphibians), and none of the studied genes are found in fish (Chalopin et al. 2012; Kokošar and Kordiš 2013). The authors suggested that the remnants of the rich TE repertoire found in the mammalian ancestor provided a rich resource of potential sequences for exaptation by the host genome. Indeed most of the exaptation events took place 90–100 MYA, correlating well with a drop in TE diversity in mammals (Kokošar and Kordiš 2013). This study only addresses a subset of domesticated genes and is human/mammalian centred. Little is known about exaptation events in other vertebrate groups apart from sporadic examples (e.g., Kobuta in Xenopus, Hikosaka et al. 2007). This represents a large gap in knowledge, although the increase in sequence data for all vertebrate species will soon rectify this situation.
In summary, exaptation of a large variety of TE genes has occurred in vertebrates. These exaptation events have occurred in many different ways and have proliferated in an almost idiosyncratic manner. There seem to be clear links with important organs and functions that have driven the diversification and success of different vertebrate lineages, but more studies across the group are required to fully understand the true evolutionary impact of these exaptation events.
Conclusions
Here, we have reviewed the potential roles and effects of TEs on genome and species diversification in vertebrates. The intrinsic properties of TEs (protein-binding, protein-coding, secondary structure formation etc.) make them a source of functional sequences that can be incorporated into a genome in a selectively advantageous manner. From a genomic point of view, it is abundantly clear that TE content and TE diversity dramatically vary between lineages. Lineage-specific mobilomes as well as lineage-specific rearrangements and innovations (summarised in Fig. 3) lead to variation in genome size and to genomic divergence and thus to the fact that each species harbours a unique genome, potentially favouring functional divergence and reproductive barriers.
In each vertebrate genome, lineage-specific TE families may play various roles. As an excellent model, the primate specific Alu element possesses many features that promote its recruitment for different functions. Alu has provided novel exonisation events, due to its possession of splicing sites; it can also self-fold to form hairpin loops and is capable of forming miRNAs and providing secondary structure to lncRNAs. Alu elements also provide many miRNA binding sites in the 3′ UTRs of mRNAs, further involving this SINE element in the regulation of the genome. The sheer success of Alu within the genome (106 copies in the human genome) increases not only its probability of being incorporated as a functional part of a gene or regulatory region, but also of generating deletions and genome rearrangements through recombination and thus participate to genome specificity.
From an organismal point of view, it is intriguing to see how TEs may be important for the regulation and maintenance of lineage-specific tissues. Indeed, through various examples, we highlighted that TEs may be linked, via different ways, to the evolution of organs and tissues. For instance, the placenta has recruited TE-derived genes on many occasions, as well as using TEs for novel binding sites in promoter regions. Similarly, TEs seem to be heavily involved in the regulation of embryonic stem cells, that vary in a highly lineage-specific manner, with TEs providing both novel promoter sites and novel lncRNAs that have been demonstrated to regulate pluripotency.
The various examples cited above demonstrate that TEs compose the major part of the genome either as active TE sequences, degenerated non-active TEs, exaptated genes, small TE-derived RNAs, transcription factor binding sites or other regulatory sequences. Furthermore, it is clear that TEs promote the evolution of their host genome in a very lineage-specific manner through the precise nature of the TE repertoire present, and the selection pressure experienced by the host (from the genomic to organismal level). All of this sustains the hypothesis that TEs are fundamental for genome evolution but also that they may account for a much bigger content of the genomes than was previously thought and may be involved in many various biological processes. The vast majority of the examples here are derived from mammals and frequently, primates. This is natural because of the high focus of research on, along with the availability of, the human genome. But we now reside firmly in a new age of genomics where sequence data is becoming rapidly available for traditionally non-model species, encompassing the full range of extant vertebrate species. Our laboratory and others have already taken a lot of this data to demonstrate the diverse range of TE repertoires present in vertebrates (Chalopin et al. 2015), but the next step is to understand exactly how these lineage-specific repertoires influence the evolution of their hosts.
Abbreviations
- ChIP-Seq:
-
Chromatin immunoprecipitation sequencing
- ERV:
-
Endogenous retrovirus
- ESC:
-
Embryonic stem cell
- HTT:
-
Horizontal transfer of TEs
- LINE (or SINE):
-
Long (or short) interspersed nuclear element
- lncRNA:
-
Long non-coding RNA
- LTR:
-
Long terminal repeat
- MYA:
-
Million years ago
- Myr:
-
Million years
- nt:
-
Nucleotides
- TE:
-
Transposable element
References
Alzohairy AM, Gyulai G, Jansen RK, Bahieldin A (2013) Transposable elements domesticated and neofunctionalized by eukaryotic genomes. Plasmid 69:1–15
Amaral PP, Dinger ME, Mattick JS (2013) Non-coding RNAs in homeostasis, disease and stress responses: an evolutionary perspective. Brief Funct Genomics 12:254–278
Baack EJ, Rieseberg LH (2007) A genomic view of introgression and hybrid speciation. Curr Opin Genet Dev 17:513–518
Bae MI, Kim YJ, Lee JR, Jung YD, Kim HS (2013) A new exon derived from a mammalian apparent LTR retrotransposon of the SUPT16H gene. Int J Genomics 2013:387594
Barker JN, Palmer CN, Zhao Y et al (2007) Null mutations in the filaggrin gene (FLG) determine major susceptibility to early-onset atopic dermatitis that persists into adulthood. J Invest Dermatol 127:564–567
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Belancio VP, Roy-Engel AM, Deininger P (2008) The impact of multiple splice sites in human L1 elements. Gene 411:38–45
Bi S, Gavrilova O, Gong DW, Mason MM, Reitman M (1997) Identification of a placental enhancer for the human leptin gene. J Biol Chem 272:30583–30588
Bièche I, Laurent A, Laurendeau I et al (2003) Placenta-specific INSL4 expression is mediated by a human endogenous retrovirus element. Biol Reprod 68:1422–1429
Bingham PM, Kidwell MG, Rubin GM (1982) The molecular basis of P-M hybrid dysgenesis: the role of the P element, a P-strain-specific transposon family. Cell 29:995–1004
Blaise S, de Parseval N, Bénit L, Heidmann T (2003) Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc Natl Acad Sci U S A 100:13013–13018
Blass E, Bell M, Boissinot S (2012) Accumulation and rapid decay of non-LTR retrotransposons in the genome of the three-spine stickleback. Genome Biol Evol 4:687–702
Böhne A, Brunet F, Galiana-Arnoux D, Schultheis C, Volff JN (2008) Transposable elements as drivers of genomic and biological diversity in vertebrates. Chromosome Res 16:203–215
Borchert GM, Holton NW, Williams JD et al (2011) Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob Genet Elem 1:8–17
Bourque G (2009) Transposable elements in gene regulation and in the evolution of vertebrate genomes. Curr Opin Genet Dev 19:607–612
Bourque G, Leong B, Vega VB et al (2008) Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res 18:1752–1762
Brandt J, Schrauth S, Veith AM et al (2005) Transposable elements as a source of genetic innovation: expression and evolution of a family of retrotransposon-derived neogenes in mammals. Gene 345:101–111
Brawand D, Wagner CE, Li YI et al (2014) The genomic substrate for adaptive radiation in African cichlid fish. Nature 513:375–381
Britten RJ, Davidson EH (1971) Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. Q Rev Biol 46:111–138
Brow DA (2002) Allosteric cascade of spliceosome activation. Annu Rev Genet 36:333–360
Campillos M, Doerks T, Shah PK, Bork P (2006) Computational characterization of multiple Gag-like human proteins. Trends Genet 22:585–589
Cantrell MA, Scott L, Brown CJ, Martinez AR, Wichman HA (2008) Loss of LINE-1 activity in the megabats. Genetics 178:393–404
Cartault F, Munier P, Benko E et al (2012) Mutation in a primate-conserved retrotransposon reveals a noncoding RNA as a mediator of infantile encephalopathy. Proc Natl Acad Sci U S A 109:4980–4985
Casola C, Hucks D, Feschotte C (2008) Convergent domestication of pogo-like transposases into centromere-binding proteins in fission yeast and mammals. Mol Biol Evol 25:29–41
Chalopin D, Galiana D, Volff JN (2012) Genetic innovation in vertebrates: gypsy integrase genes and other genes derived from transposable elements. Int J Evol Biol 2012:724519
Chalopin D, Naville M, Plard F, Galiana D, Volff JN (2015) Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol Evol 7:567–580
Chen Z, Cheng CH, Zhang J et al (2008) Transcriptomic and genomic evolution under constant cold in Antarctic notothenioid fish. Proc Natl Acad Sci U S A 105:12944–12949
Chen C, Ara T, Gautheret D (2009) Using Alu elements as polyadenylation sites: a case of retroposon exaptation. Mol Biol Evol 26:327–334
Chinwalla AT, Cook LL, Delehaunty KD et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562
Cho G, Bhat SS, Gao J et al (2008a) Evidence that SIZN1 is a candidate X-linked mental retardation gene. Am J Med Genet A 146A:2644–2650
Cho G, Lim Y, Zand D, Golden JA (2008b) Sizn1 is a novel protein that functions as a transcriptional coactivator of bone morphogenic protein signaling. Mol Cell Biol 28:1565–1572
Cho G, Lim Y, Golden JA (2011) XLMR candidate mouse gene, Zcchc12 (Sizn1) is a novel marker of Cajal-Retzius cells. Gene Expr Patterns 11:216–200
Chuong EB, Rumi MAK, Soares MJ, Baker JC (2013) Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat Genet 45:325–329
Cordaux R, Udit S, Batzer MA, Feschotte C (2006) Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc Natl Acad Sci U S A 103:8101–8106
Cornelis G, Heidmann O, Bernard-Stoecklin S et al (2012) Ancestral capture of syncytin-Car1, a fusogenic endogenous retroviral envelope gene involved in placentation and conserved in Carnivora. Proc Natl Acad Sci U S A 109:E432–E441
Cornelis G, Heidmann O, Degrelle SA et al (2013) Captured retroviral envelope syncytin gene associated with the unique placental structure of higher ruminants. Proc Natl Acad Sci U S A 110:E828–E837
Cornelis G, Vernochet C, Malicorne S et al (2014) Retroviral envelope syncytin capture in an ancestrally diverged mammalian clade for placentation in the primitive Afrotherian tenrecs. Proc Natl Acad Sci 111:E4332–E4341
Cornelis G, Vernochet C, Carradec Q et al (2015) Retroviral envelope gene captures and syncytin exaptation for placentation in marsupials. Proc Natl Acad Sci U S A 112:E487–E496
Cotney J, Leng J, Yin J et al (2013) The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154:185–196
Cowley M, Oakey RJ (2013) Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet 9:e1003234
Coyne JA, Orr HA (1998) The evolutionary genetics of speciation. Philos Trans R Soc Lond B Biol Sci 353:287–305
Cristofano AD, Strazzullo M, Longo L, Mantia GL (1995) Characterization and genomic mapping of the ZNF80 locus: expression of this zinc-finger gene is driven by a solitary LTR of ERV9 endogenous retrovrial family. Nucl Acids Res 23:2823–2830
Cui F, Sirotin MV, Zhurkin VB (2011) Impact of Alu repeats on the evolution of human p53 binding sites. Biol Direct 6:2
Cultrone A, Domínguez YR, Drevet C, Scazzocchio C, Fernández-Martín R (2007) The tightly regulated promoter of the xanA gene of Aspergillus nidulans is included in a helitron. Mol Microbiol 63:1577–1587
de Boer JG, Yazawa R, Davidson WS, Koop BF (2007) Bursts and horizontal evolution of DNA transposons in the speciation of pseudotetraploid salmonids. BMC Genomics 8:422
de Souza FSJ, Franchini LF, Rubinstein M (2013) Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Mol Biol Evol 30:1239–1251
Deininger P (2011a) Alu elements: know the SINEs. Genome Biol 12:236
Deininger P (2011) Alu elements. Genomic disorders: the genomic basis of disease 12:236
Deininger PL, Batzer MA (1999) Alu repeats and human disease. Mol Genet Metab 67:183–193
del Rosario RCH, Rayan NA, Prabhakar S (2014) Noncoding origins of anthropoid traits and a new null model of transposon functionalization. Genome Res 24:1469–1484
Derrien T, Johnson R, Bussotti G et al (2012) The GENCODE v7 catalogue of human long non-coding RNAs: analysis of their structure, evolution and expression. Genome Res 22:1775–1789
Dion-Cote AM, Renaut S, Normandeau E, Bernatchez L (2014) RNA-seq reveals transcriptomic shock involving transposable elements reactivation in hybrids of young lake whitefish species. Mol Biol Evol 31:1188–1199
Djebali S, Davis CA, Merkel A et al (2012) Landscape of transcription in human cells. Nature 489:101–108
Dobigny G, Ozouf-Costaz C, Waters PD, Bonillo C, Coutanceau JP, Volobouev V (2004) LINE-1 amplification accompanies explosive genome repatterning in rodents. Chromosome Res 12:787–793
Dotto BR, Carvalho EL, Silva AF et al (2015) HTT-DB: Horizontally transferred transposable elements database. Bioinformatics 31:2915–2917
Dozmorov MG, Giles CB, Koelsch KA, Wren JD (2013) Systematic classification of non-coding RNAs by epigenomic similarity. BMC Bioinf 14:S2
Dupressoir A, Marceau G, Vernochet C et al (2005) Syncytin-A and syncytin-B, two fusogenic placenta-specific murine envelope genes of retroviral origin conserved in Muridae. Proc Natl Acad Sci U S A 102:725–730
Edelstein LC, Collins T (2005) The SCAN domain family of zinc finger transcription factors. Gene 359:1–17
Edwards CA, Mungall AJ, Matthews L et al (2008) The evolution of the DLK1-DIO3 imprinted domain in mammals. PLoS Biol 6:1292–1305
Elisaphenko EA, Kolesnikov NN, Shevchenko AI et al (2008) A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS One 3:1–11
Emera D, Wagner GP (2012a) Transformation of a transposon into a derived prolactin promoter with function during human pregnancy. Proc Natl Acad Sci U S A 109:11246–11251
Emera D, Wagner GP (2012b) Transposable element recruitments in the mammalian placenta: impacts and mechanisms. Brief Funct Genomics 11:267–276
Emera D, Casola C, Lynch VJ, Wildman DE, Agnew D, Wagner GP (2012) Convergent evolution of endometrial prolactin expression in primates, mice, and elephants through the independent recruitment of transposable elements. Mol Biol Evol 29:239–247
Emerson RO, Thomas JH (2009) Adaptive evolution in zinc finger transcription factors. PLoS Genet 5:e1000325
Erickson IK, Cantrell MA, Scott L, Wichman HA (2011) Retrofitting the genome: L1 extinction follows endogenous retroviral expansion in a group of muroid rodents. J Virol 85:12315–12323
Feschotte C (2004) Merlin, a new superfamily of DNA transposons identified in diverse animal genomes and related to bacterial IS1016 insertion sequences. Mol Biol Evol 21:1769–1780
Feschotte C (2008) Transposable elements and the evolution of regulatory networks. Nat Rev Genet 9:397–405
Feschotte C, Pritham EJ (2007) DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet 41:331–368
Fort A, Hashimoto K, Yamada D et al (2014) Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet 46:558–566
Franchini LF, López-Leal R, Nasif S et al (2011) Convergent evolution of two mammalian neuronal enhancers by sequential exaptation of unrelated retroposons. Proc Natl Acad Sci U S A 108:15270–15275
Friedman RC, Farh KKH, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105
Fu B, Chen M, Zou M, Long M, He S (2010) The rapid generation of chimerical genes expanding protein diversity in zebrafish. BMC Genomics 11:657
Furano AV, Duvernell DD, Boissinot S (2004) L1 (LINE-1) retrotransposon diversity differs dramatically between mammals and fish. Trends Genet 20:9–14
Gentles AJ, Wakefield MJ, Kohany O et al (2007) Evolutionary dynamics of transposable elements in the short-tailed opossum Monodelphis domestica. Genome Res 17:992–1004
Gifford WD, Pfaff SL, Macfarlan TS (2013) Transposable elements as genetic regulatory substrates in early development. Trends Cell Biol 23:218–226
Gilbert C, Schaack S, Pace JK 2nd, Brindley PJ, Feschotte C (2010) A role for host-parasite interactions in the horizontal transfer of transposons across phyla. Nature 464:1347–1350
Gilbert C, Hernandez SS, Flores-Benabib J, Smith EN, Feschotte C (2012) Rampant horizontal transfer of SPIN transposons in squamate reptiles. Mol Biol Evol 29:503–515
Gim JA, Ha HS, Ahn K, Kim DS, Kim HS (2014) Genome-wide identification and classification of microRNAs derived from repetitive elements. Genomics Inform 12:261–267
Glinsky GV (2015) Transposable elements and DNA methylation create in embryonic stem cells human-specific regulatory sequences associated with distal enhancers and noncoding RNAs. Genome Biol Evol 7:1432–1454
Gombart AF, Saito T, Koeffler HP (2009) Exaptation of an ancient Alu short interspersed element provides a highly conserved vitamin D-mediated innate immune response in humans and primates. BMC Genomics 10:321
Grahn RA, Rinehart TA, Cantrell MA, Wichman HA (2005) Extinction of LINE-1 activity coincident with a major mammalian radiation in rodents. Cytogenet Genome Res 110:407–415
Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15:509–524
Haas NB, Grabowski JM, Sivitz AB, Burch JBE (1997) Chicken repeat 1 (CR1) elements, which define an ancient family of vertebrate non-LTR retrotransposons, contain two closely spaced open reading frames. Gene 197:305–309
Hambor JE, Mennone J, Coon ME, Hanke JH, Kavathas P (1993) Identification and characterization of an Alu-containing, T-cell-specific enhancer located in the last intron of the human CD8 alpha gene. Mol Cell Biol 13:7056–7070
Han MJ, Shen YH, Xu MS, Liang HY, Zhang HH, Zhang Z (2013) Identification and evolution of the silkworm helitrons and their contribution to transcripts. DNA Res 20:471–484
Harris CR, Dewan A, Zupnick A et al (2009) p53 responsive elements in human retrotransposons. Oncogene 28:3857–3865
Hayakawa T, Satta Y, Gagneux P, Varki A, Takahata N (2001) Alu-mediated inactivation of the human CMP- N-acetylneuraminic acid hydroxylase gene. Proc Natl Acad Sci U S A 98:11399–11404
Hayward A, Cornwallis CK, Jern P (2015) Pan-vertebrate comparative genomics unmasks retrovirus macroevolution. Proc Natl Acad Sci U S A 112:464–469
He S, Gu W, Li Y, Zhu H (2013) ANRIL/CDKN2B-AS shows two-stage clade-specific evolution and becomes conserved after transposon insertions in simians. BMC Evol Biol 13:247
Heidmann O, Vernochet C, Dupressoir A, Heidmann T (2009) Identification of an endogenous retroviral envelope gene with fusogenic activity and placenta-specific expression in the rabbit: a new “syncytin” in a third order of mammals. Retrovirology 6:107
Hellsten U, Harland RM, Gilchrist MJ et al (2010) The genome of the Western clawed frog Xenopus tropicalis. Science 328:633–636
Henke C, Strissel PL, Schubert MT et al (2015) Selective expression of sense and antisense transcripts of the sushi-ichi-related retrotransposon—derived family during mouse placentogenesis. Retrovirology 12:9
Henrichsen CN, Vinckenbosch N, Zöllner S et al (2009) Segmental copy number variation shapes tissue transcriptomes. Nat Genet 41:424–429
Herpin A, Braasch I, Kraeussling M et al (2010) Transcriptional rewiring of the sex determining dmrt1 gene duplicate by transposable elements. PLoS Genet 6:e1000844
Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I (2015) Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep 11:1110–1122
Hikosaka A, Kobayashi T, Saito Y, Kawahara A (2007) Evolution of the Xenopus piggyBac transposon family TxpB: domesticated and untamed strategies of transposon subfamilies. Mol Biol Evol 24:2648–2656
Hillier L, Miller W, Birney E et al (2004) Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432:695–716
Holdt LM, Hoffmann S, Sass K et al (2013) Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet 9:1–12
Hollister JD, Gaut BS (2007) Population and evolutionary dynamics of helitron transposable elements in Arabidopsis thaliana. Mol Biol Evol 24:2515–2524
Howe K, Clark MD, Torroja CF et al (2013) The zebrafish reference genome sequence and its relationship to the human genome. Nature 496:498–503
Hughes JF, Coffin JM (2001) Evidence for genomic rearrangements mediated by human endogenous retroviruses during primate evolution. Nat Genet 29:487–489
Hughes JF, Coffin JM (2005) Human endogenous retroviral elements as indicators of ectopic recombination events in the primate genome. Genetics 171:1183–1194
Iida A, Takamatsu N, Hori H et al (2005) Reversion mutation of ib oculocutaneous albinism to wild-type pigmentation in medaka fish. Pigment Cell Res 18:382–384
Ivancevic AM, Walsh AM, Kortschak RD, Adelson DL (2013) Jumping the fine LINE between species: horizontal transfer of transposable elements in animals catalyses genome evolution. BioEssays 35:1071–1082
Jacques PÉ, Jeyakani J, Bourque G (2013) The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet 9:e1003504
Johnson R, Guigó R (2014) The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA 20:959–976
Kaessmann H (2010) Origins, evolution, and phenotypic impact of new genes. Genome Res 20:1313–1326
Kaneko-Ishino T, Ishino F (2012) The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front Microbiol 3:1–10
Kannan S, Chernikova D, Rogozin IB et al (2015) Transposable element insertions in long intergenic non-coding RNA genes. Front Bioeng Biotechnol 3:1–9
Kapitonov VV, Jurka J (2001) Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci U S A 98:8714–8719
Kapitonov VV, Jurka J (2004) Harbinger transposons and an ancient HARBI1 gene derived from a transposase. DNA Cell Biol 23:311–324
Kapitonov VV, Jurka J (2006) Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci U S A 103:4540–4545
Kapitonov VV, Koonin EV (2015) Evolution of the RAG1-RAG2 locus: both proteins came from the same transposon. Biol Direct 10:1–8
Kapusta A, Feschotte C (2014) Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications. Trends Genet 30:439–452
Kapusta A, Kronenberg Z, Lynch VJ et al (2013) Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet 9:e1003470
Kazazian HH Jr (2004) Mobile elements: drivers of genome evolution. Science 303:1626–1632
Kelley D, Rinn J (2012) Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 13:R107
Koga A, Iida A, Hori H, Shimada A, Shima A (2006) Vertebrate DNA transposon as a natural mutator: the medaka fish Tol2 element contributes to genetic variation without recognizable traces. Mol Biol Evol 23:1414–1419
Kokošar J, Kordiš D (2013) Genesis and regulatory wiring of retroelement-derived domesticated genes: a phylogenomic perspective. Mol Biol Evol 30:1015–1031
Kordiš D, Gubensek F (1998) The Bov-B lines found in Vipera ammodytes toxic PLA2 genes are widespread in snake genomes. Toxicon 36:1585–1590
Kordiš D, Gubensek F (1999) Molecular evolution of Bov-B LINEs in vertebrates. Gene 238:171–178
Kraaijeveld K (2010) Genome size and species diversification. Evol Biol 37:227–233
Krull M, Brosius J, Schmitz J (2005) Alu-SINE exonization: en route to protein-coding function. Mol Biol Evol 22:1702–1711
Krull M, Petrusma M, Makalowski W, Brosius J, Schmitz J (2007) Functional persistence of exonized mammalian-wide interspersed repeat elements (MIRs). Genome Res 17:1139–1145
Kunarso G, Chia NY, Jeyakani J et al (2010) Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42:631–634
Kutter C, Watt S, Stefflova K et al (2012) Rapid turnover of long noncoding RNAs and the evolution of gene expression. PLoS Genet 8:e1002841
Landry JR, Mager DL (2003) Functional analysis of the endogenous retroviral promoter of the human endothelin B receptor gene. J Virol 77:7459–7466
Landry JR, Rouhi A, Medstrand P, Mager DL (2002) The Opitz syndrome gene mid1 is transcribed from a human endogenous retroviral promoter. Mol Biol Evol 19:1934–1942
Le Rouzic A, Capy P (2006) Population genetics models of competition between transposable element subfamilies. Genetics 174:785–793
Lee RC (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Lee SH, Oshige M, Durant ST et al (2005) The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc Natl Acad Sci U S A 102:18075–18080
Lee JY, Ji Z, Tian B (2008) Phylogenetic analysis of mRNA polyadenylation sites reveals a role of transposable elements in evolution of the 3′-end of genes. Nucl Acids Res 36:5581–5590
Lev-Maor G, Sorek R, Shomron N, Ast G (2003) The birth of an alternatively spliced exon: 3ʹ splice-site selection in Alu exons. Science 300:1288–1291
Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA (1999) Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science 284:330–333
Lin L, Jiang P, Shen S, Sato S, Davidson BL, Xing Y (2009) Large-scale analysis of exonized mammalian-wide interspersed repeats in primate genomes. Hum Mol Genet 18:2204–2214
Liu D, Bischerour J, Siddique A, Buisine N, Bigot Y, Chalmers R (2007) The human SETMAR protein preserves most of the activities of the ancestral Hsmar1 transposase. Mol Cell Biol 27:1125–1132
Loewer S, Cabili MN, Guttman M et al (2010) Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 42:1113–1117
Londin E, Loher P, Telonis AG et al (2015) Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc Natl Acad Sci U S A 112:E1106–E1115
Lukic S, Chen K (2011) Human piRNAs are under selection in Africans and repress transposable elements. Mol Biol Evol 28:3061–3067
Lynch VJ, Leclerc RD, May G, Wagner GP (2011) Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat Genet 43:1154–1159
Lynch VJ, Nnamani MC, Kapusta A et al (2015) Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep 10:551–561
Managadze D, Lobkovsky AE, Wolf YI, Shabalina SA, Rogozin IB, Koonin EV (2013) The vast, conserved mammalian lincRNome. PLoS Comput Biol 9:e1002917
Marques AC, Dupanloup I, Vinckenbosch N, Reymond A, Kaessmann H (2005) Emergence of young human genes after a burst of retroposition in primates. PLoS Biol 3:1970–1979
Marques AC, Vinckenbosch N, Brawand D, Kaessmann H (2008) Functional diversification of duplicate genes through subcellular adaptation of encoded proteins. Genome Biol 9:R54
Mason CE, Shu FJ, Wang C et al (2010) Location analysis for the estrogen receptor-α reveals binding to diverse ERE sequences and widespread binding within repetitive DNA elements. Nucl Acids Res 38:2355–2368
Matsui T, Kinoshita-Ida Y, Hayashi-Kisumi F et al (2006) Mouse homologue of skin-specific retroviral-like aspartic protease involved in wrinkle formation. J Biol Chem 281:27512–27525
Matsui T, Miyamoto K, Kubo A et al (2011) SASPase regulates stratum corneum hydration through profilaggrin-to-filaggrin processing. EMBO Mol Med 3:320–333
McClintock B (1956) Controlling elements and the gene. Cold Spring Harb Symp Quant Biol 21:197–216
Metcalfe CJ, Bulazel KV, Ferreri GC et al (2007) Genomic instability within centromeres of interspecific marsupial hybrids. Genetics 177:2507–2517
Metcalfe CJ, Filee J, Germon I, Joss J, Casane D (2012) Evolution of the Australian lungfish (Neoceratodus forsteri) genome: a major role for CR1 and L2 LINE elements. Mol Biol Evol 29:3529–3539
Meunier J, Lemoine F, Soumillon M et al (2013) Birth and expression evolution of mammalian microRNA genes. Genome Res 23:34–45
Mi S, Lee X, Li X et al (2000) Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403:785–789
Micale L, Loviglio MN, Manzoni M et al (2012) A fish-specific transposable element shapes the repertoire of p53 target genes in zebrafish. PLoS One 7:e46642
Minghetti PP, Dugaiczyk A (1993) The emergence of new DNA repeats and the divergence of primates. Proc Natl Acad Sci U S A 90:1872–1876
Mortazavi A, Leeper Thompson EC, Garcia ST, Myers RM, Wold B (2006) Comparative genomics modeling of the NRSF/REST repressor network: from single conserved sites to genome-wide repertoire. Genome Res 16:1208–1221
Nakanishi A, Kobayashi N, Suzuki-Hirano A et al (2012) A SINE-derived element constitutes a unique modular enhancer for mammalian diencephalic Fgf8. PLoS One 7:e43785
Necsulea A, Soumillon M, Warnefors M et al (2014) The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505:635–640
Notwell JH, Chung T, Heavner W, Bejerano G (2015) A family of transposable elements co-opted into developmental enhancers in the mouse neocortex. Nat Commun 6:6644
Nowick K, Hamilton AT, Zhang H, Stubbs L (2010) Rapid sequence and expression divergence suggest selection for novel function in primate-specific KRAB-ZNF genes. Mol Biol Evol 27:2606–2617
O’Neill RJ, O’Neill MJ, Graves JA (1998) Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393:68–72
Ono R, Nakamura K, Inoue K et al (2006) Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat Genet 38:101–106
Pan D, Zhang L (2009) Burst of young retrogenes and independent retrogene formation in mammals. PLoS One 4:e5040
Pang ALY, Peacock S, Johnson W, Bear DH, Rennert OM, Chan WY (2009) Cloning, characterization, and expression analysis of the novel acetyltransferase retrogene Ard1b in the mouse. Biol Reprod 81:302–309
Perry GH, Dominy NJ, Claw KG et al (2007) Diet and the evolution of human amylase gene copy number variation. Nat Genet 39:1256–1260
Pi W, Yang Z, Wang J et al (2004) The LTR enhancer of ERV-9 human endogenous retrovirus is active in oocytes and progenitor cells in transgenic zebrafish and humans. Proc Natl Acad Sci U S A 101:805–810
Piriyapongsa J, Jordan IK (2007) A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2:e203
Piriyapongsa J, Mariño-Ramírez L, Jordan IK (2007a) Origin and evolution of human microRNAs from transposable elements. Genetics 176:1323–1337
Piriyapongsa J, Polavarapu N, Borodovsky M, McDonald J (2007b) Exonization of the LTR transposable elements in human genome. BMC Genomics 8:291
Piskurek O, Jackson DJ (2011) Tracking the ancestry of a deeply conserved eumetazoan SINE domain. Mol Biol Evol 28:2727–2730
Plath N, Ohana O, Dammermann B et al (2006) Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52:437–444
Potrzebowski L, Vinckenbosch N, Marques AC, Chalmel F, Jégou B, Kaessmann H (2008) Chromosomal gene movements reflect the recent origin and biology of therian sex chromosomes. PLoS Biol 6:709–716
Potrzebowski L, Vinckenbosch N, Kaessmann H (2010) The emergence of new genes on the young therian X. Trends Genet 26:1–4
Qin S, Jin P, Zhou X, Chen L, Ma F (2015) The role of transposable elements in the origin and evolution of microRNAs in human. Plos One 10:e0131365
Ray DA, Feschotte C, Pagan HJT et al (2008) Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res 18:717–728
Rebollo R, Romanish MT, Mager DL (2012) Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu Rev Genet 46:21–42
Román AC, González-Rico FJ, Moltó E et al (2011) Dioxin receptor and SLUG transcription factors regulate the insulator activity of B1 SINE retrotransposons via an RNA polymerase switch. Genome Res 21:422–432
Ruiz-Orera J, Messeguer X, Subirana JA, Alba MM (2014) Long non-coding RNAs as a source of new peptides. Elife e03523
Samuelson LC, Wiebauer K, Snow CM, Meisler MH (1990) Retroviral and pseudogene insertion sites reveal the lineage of human salivary and pancreatic amylase genes from a single gene during primate evolution. Mol Cell Biol 10:2513–2520
Santangelo AM, de Souza FSJ, Franchini LF, Bumaschny VF, Low MJ, Rubinstein M (2007) Ancient exaptation of a CORE-SINE retroposon into a highly conserved mammalian neuronal enhancer of the proopiomelanocortin gene. PLoS Genet 3:1813–1826
Santoni FA, Guerra J, Luban J (2012) HERV-H RNA is abundant in human embryonic stem cells and a precise marker for pluripotency. Retrovirology 9:111
Santos ME, Braasch I, Boileau N et al (2014) The evolution of cichlid fish egg-spots is linked with a cis-regulatory change. Nat Commun 5:5149
Sasaki T, Nishihara H, Hirakawa M et al (2008) Possible involvement of SINEs in mammalian-specific brain formation. Proc Natl Acad Sci U S A 105:4220–4225
Saunders MA, Liang H, Li WH (2007) Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci U S A 104:3300–3305
Schaack S, Gilbert C, Feschotte C (2010) Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol 25:537–546
Schartl M, Hornung U, Gutbrod H, Volff JN, Wittbrodt J (1999) Melanoma loss-of-function mutants in Xiphophorus caused by Xmrk-oncogene deletion and gene disruption by a transposable element. Genetics 153:1385–1394
Schatz DG, Swanson PC (2011) V(D)J recombination: mechanisms of initiation. Annu Rev Genet 45:167–202
Schmidt D, Schwalie PC, Wilson MD et al (2012) Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148:335–348
Schmitz J, Brosius J (2011) Exonization of transposed elements: a challenge and opportunity for evolution. Biochimie 93:1928–1934
Schüller M, Jenne D, Voltz R (2005) The human PNMA family: novel neuronal proteins implicated in paraneoplastic neurological disease. J Neuroimmunol 169:172–176
Schulte AM, Wellstein A (1998) Structure and phylogenetic analysis of an endogenous retrovirus inserted into the human growth factor gene pleiotrophin. J Virol 72:6065–6072
Sekita Y, Wagatsuma H, Nakamura K et al (2008) Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet 40:243–248
Sela N, Mersch B, Gal-Mark N, Lev-Maor G, Hotz-Wagenblatt A, Ast G (2007) Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu’s unique role in shaping the human transcriptome. Genome Biol 8:R127
Sela N, Mersch B, Hotz-Wagenblatt A, Ast G (2010) Characteristics of transposable element exonization within human and mouse. PLoS One 5:e10907
Shen S, Lin L, Cai JJ et al (2011) Widespread establishment and regulatory impact of Alu exons in human genes. Proc Natl Acad Sci U S A 108:2837–2842
Singer SS, Männel DN, Hehlgans T, Brosius J, Schmitz J (2004) From “junk” to gene: curriculum vitae of a primate receptor isoform gene. J Mol Biol 341:883–886
Sinzelle L, Izsvák Z, Ivics Z (2009) Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell Mol Life Sci 66:1073–1093
Smalheiser NR, Torvik VI (2005) Mammalian microRNAs derived from genomic repeats. Trends Genet 21:322–326
Spengler RM, Oakley CK, Davidson BL (2014) Functional microRNAs and target sites are created by lineage-specific transposition. Hum Mol Genet 23:1783–1793
St. Laurent G, Wahlestedt C, Kapranov P (2015) The landscape of long noncoding RNA classification. Trends Genet 31:239–251
Steinemann S, Steinemann M (2005) Y chromosomes: born to be destroyed. BioEssays 27:1076–1083
Sun C, Shepard DB, Chong RA et al (2012) LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders. Genome Biol Evol 4:168–183
Sundaram V, Cheng Y, Ma Z et al (2014) Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res 24:1963–1976
Tang Z, Zhang HH, Huang K, Zhang XG, Han MJ, Zhang Z (2015) Repeated horizontal transfers of four DNA transposons in invertebrates and bats. Mob DNA 6:3
Tashiro K, Teissier A, Kobayashi N et al (2011) A mammalian conserved element derived from SINE displays enhancer properties recapitulating satb2 expression in early-born callosal projection neurons. PLoS One 6:e28497
Thomas J, Schaack S, Pritham EJ (2010) Pervasive horizontal transfer of rolling-circle transposons among animals. Genome Biol Evol 2:656–664
Thomas J, Phillips CD, Baker RJ, Pritham EJ (2014) Rolling-circle transposons catalyze genomic innovation in a mammalian lineage. Genome Biol Evol 6:2595–2610
Thomson SJP, Goh FG, Banks H et al (2009) The role of transposable elements in the regulation of IFN-λ1 gene expression. Proc Natl Acad Sci U S A 106:11564–11569
Ting CN, Rosenberg MP, Snow CM, Samuelson LC, Meisler MH (1992) Endogenous retroviral sequences are required for tissue-specific expression of a human salivary amylase gene. Genes Dev 6:1457–1465
Tipney HJ, Hinsley TA, Brass A, Metcalfe K, Donnai D, Tassabehji M (2004) Isolation and characterisation of GTF2IRD2, a novel fusion gene and member of the TFII-I family of transcription factors, deleted in Williams-Beuren syndrome. Eur J Hum Genet 12:551–560
Tomascik-Cheeseman L, Marchetti F, Lowe X et al (2002) CENP-B is not critical for meiotic chromosome segregation in male mice. Mutat Res 513:197–203
Ullu E, Tschudi C (1984) Alu sequences are processed 7SL RNA genes. Nature 312:171–172
Vance KW, Ponting CP (2014) Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet 30:348–355
Varki A (2001) Loss of N-glycolylneuraminic acid in humans: mechanisms, consequences, and implications for hominid evolution. Am J Phys Anthropol Suppl 33:54–69
Varki A (2010) Colloquium paper: uniquely human evolution of sialic acid genetics and biology. Proc Natl Acad Sci U S A 107(Suppl):8939–8946
Verneau O, Catzeflis F, Furano AV (1998) Determining and dating recent rodent speciation events by using L1 (LINE-1) retrotransposons. Proc Natl Acad Sci U S A 95:11284–11289
Vernochet C, Redelsperger F, Harper F et al (2014) The captured retroviral envelope syncytin-A and syncytin-B genes are conserved in the Spalacidae together with hemotrichorial placentation. Biol Reprod 91:148–148
Villar D, Berthelot C, Aldridge S et al (2015) Enhancer evolution across 20 mammalian species. Cell 160:554–566
Vinckenbosch N, Dupanloup I, Kaessmann H (2006) Evolutionary fate of retroposed gene copies in the human genome. Proc Natl Acad Sci U S A 103:3220–3225
Volff JN (2006) Turning junk into gold: domestication of transposable elements and the creation of new genes in eukaryotes. BioEssays 28:913–922
Volff JN, Bouneau L, Ozouf-Costaz C, Fischer C (2003) Diversity of retrotransposable elements in compact pufferfish genomes. Trends Genet 19:674–678
Volff JN, Nanda I, Schmid M, Schartl M (2007) Governing sex determination in fish: regulatory putsches and ephemeral dictators. Sex Dev 1:85–99
Wallau GL, Ortiz MF, Loreto EL (2012) Horizontal transposon transfer in eukarya: detection, bias, and perspectives. Genome Biol Evol 4:689–699
Wang T, Zeng J, Lowe CB et al (2007) Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci U S A 104:18613–18618
Wang J, Xie G, Singh M et al (2014) Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 516:405–409
Washietl S, Kellis M, Garber M (2014) Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res 24:616–628
Watson CT, Lubieniecki KP, Loew E, Davidson WS, Breden F (2010) Genomic organization of duplicated short wave-sensitive and long wave-sensitive opsin genes in the green swordtail, Xiphophorus helleri. BMC Evol Biol 10:87
Wicker T, Robertson JS, Schulze SR et al (2005) The repetitive landscape of the chicken genome. Genome Res 15:126–136
Wu LT, Hui JHL, Chu KH (2013) Origin and evolution of yolk proteins: expansion and functional diversification of large lipid transfer protein superfamily. Biol Reprod 88:102
Xing J, Wang H, Belancio VP, Cordaux R, Deininger PL, Batzer MA (2006) Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc Natl Acad Sci U S A 103:17608–17613
Yuan Z, Sun X, Jiang D et al (2010) Origin and evolution of a placental-specific microRNA family in the human genome. BMC Evol Biol 10:346
Zhang H, Liu Y, Su D et al (2011) A single nucleotide polymorphism in a miR-1302 binding site in CGA increases the risk of idiopathic male infertility. Fertil Steril 96:34–39
Zhang W, Edwards A, Fan W, Fang Z, Deininger P, Zhang K (2013) Inferring the expression variability of human transposable element-derived exons by linear model analysis of deep RNA sequencing data. BMC Genomics 14:584
Conflict of interest
The authors declare that they have no competing interest.
Compliance with ethical standards
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editors: Maria Assunta Biscotti, Pat Heslop-Harrison and Ettore Olmo
Ian Warren, Magali Naville and Domitille Chalopin contributed equally to this work.
Rights and permissions
About this article

Cite this article
Warren, I.A., Naville, M., Chalopin, D. et al. Evolutionary impact of transposable elements on genomic diversity and lineage-specific innovation in vertebrates. Chromosome Res 23, 505–531 (2015). https://doi.org/10.1007/s10577-015-9493-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-015-9493-5