
Abstract
Parkinson's disease (PD) is a neurodegenerative disorder marked primarily by motor symptoms such as rigidity, bradykinesia, postural instability and resting tremor associated with dopaminergic neuronal loss in the Substantia Nigra pars compacta (SNpc) and deficit of dopamine in the basal ganglia. These motor symptoms can be preceded by pre-motor symptoms whose recognition can be useful to apply different strategies to evaluate risk, early diagnosis and prevention of PD progression. Although clinical characteristics of PD are well defined, its pathogenesis is still not completely known, what makes discoveries of therapies capable of curing patients difficult to be reached. Several theories about the cause of idiopathic PD have been investigated and among them, the key role of inflammation, microglia and the inflammasome in the pathogenesis of PD has been considered. In this review, we describe the role and relation of both the inflammasome and microglial activation with the pathogenesis, symptoms, progression and the possibilities for new therapeutic strategies in PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In 2016, 6.1 million people worldwide were diagnosed with Parkinson's disease (PD), what represents 2.4 times more than the number of people diagnosed in 1990 (Dorsey et al. 2018a, b; Simon et al. 2019). In addition, it is estimated that the number of cases will exceed 12 million individuals in 2040. This growing number of cases is especially related to the increase in life expectancy, since PD is uncommon in individuals under 50 years, affecting predominantly individuals over 60 years of age and increasing dramatically after 75 years (Abdullah et al. 2015; Dorsey et al. 2018a, b). Age is the main risk factor for PD, but there is also an association with environmental factors related to industrialization, including pesticides, solvents and metals (Vlaar et al. 2018). The symptoms of PD were first described by James Parkinson in 1817 as a heterogeneous manifestation (Parkinson 1817). Nowadays, the symptoms of PD are well characterized, marked by motor symptoms such as bradykinesia, ataxia, postural stiffness and resting tremor associated with dopaminergic neuronal loss in the Substantia Nigra pars compacta (SNpc) and deficit of dopamine in the basal ganglia (Goldman and Postuma 2014; Obeso et al. 2017). However, it is believed that the pathogenic process begins in the pre-motor phase marked by sleep disturbance, olfactory deficit, anxiety and depression with pathogenic bases largely undefined (Schapira et al. 2017).
Most PD symptoms are associated with a slow and progressive degeneration of dopaminergic neurons in the SNpc with a subsequent dopamine depletion in the target areas (Obeso et al. 2017). The cause that leads to neuronal loss in PD is still unclear and has been object of continuous experimental studies in different systems (Cuenca et al. 2005; Herrero and Morelli 2017; Kalinderi et al. 2016; Kazlauskaite and Muqit 2015). However, even after many decades, the understanding of the mechanisms underlying the PD pathogenesis remains partially unknown. Several theories have been studied and some have been shown to indicate that the disease has multifactorial causes associated with genetic, environmental and aging changes that, when combined, confer a risk for the development of neuronal degeneration through molecular and cellular disorders, such as neurotoxicity by α-synuclein (Lau et al. 2020; Poewe et al. 2017) or product of dopamine oxidation (Segura-Aguilar 2017), oxidative stress (Puspita et al. 2017), reduction of endogenous neuroprotective molecules and mitochondrial dysfunction (Macdonald et al. 2018; Rani and Mondal 2020), dysfunction in protein degradation and autophagy system (Cheng, et al. 2020a; Hou et al. 2020; Lane et al. 2017; Menzies et al. 2017; Zhang et al. 2016b) and neuroinflammation (Arlehamn et al. 2020; Hirsch and Hunot 2009).
In particular, inflammation, a term that encompasses neuroinflammation and peripheral inflammatory responses, is documented in PD acting not only as a mere dysfunction that occurs in the disease process, but also as an important factor of PD pathogenesis (Glass et al. 2010; Salter and Stevens 2017; Schlachetzki et al. 2014). In the brain, continuous interactions between neurons, extracellular space and glial cells are determinant for the maintenance of neural homeostasis and/or for the emergence of neurological disorders, such as those occurring in PD (De Stefano and Herrero 2017; Heneka et al. 2010). Microglial activation is a typical pathological characteristic of neurodegenerative diseases. Emerging evidences indicate that sustained activation of the inflammatory response mediated by microglial activation in human and in animal models of PD plays an important role in explaining part of the cascade of events leading to dopaminergic degeneration in PD (Kim and Joh 2006).
Microglia is the main immunological cell of the Central Nervous System (CNS), responsible for its first line of defense, acting as a sensor that responds to physiological changes and pathological stimuli in the cerebral microenvironment (Aguzzi et al. 2013; Hanisch and Kettenmann 2007). These microglia changes, from a "quiescent state” to an activated phenotype, are characterized by a set of responses that may affect CNS function during the disease or injury, generating consequences ranging from the loss of synapses to progressive neurodegeneration (Bernier et al. 2020; Salter and Stevens 2017). During chronic brain damage, microglia release pro-inflammatory factors that are toxic to neurons (Cheng et al. 2020a; Wang et al. 2014). Among the released factors, the cytokine IL-1β is a product of inflammasome, a multiprotein complex present in the cytoplasm for the microglia responsible for the degradation of the pro-IL-1β zymogen in IL-1β. Several studies have shown the involvement of the NLRP3 type inflammasome in numerous human diseases in the CNS and found that the product of this molecule increases the rates of dopamine neuron degeneration in 6-Hydroxydopamine (6-OHDA) rat model (Chatterjee et al. 2020; Haque et al. 2020; Koprich et al. 2008; McGeer et al. 2002). In this review, we describe the role of microglial activation and inflammasome with clinical aspects, pathogenesis and therapeutic approaches in PD.
Symptoms in Parkinson’s Disease and Association with Neuroinflammation
This chronic and progressive neurodegenerative disease is mainly characterized by clinical motor manifestations that include bradykinesia, rigidity, postural instability and tremor at rest (Giráldez-Pérez et al. 2014; Das and Sharma 2016). Diagnosis of PD occurs primarily with the onset of motor symptoms that begins when 50–60% of the dopaminergic neurons are lost. On the other hand, these symptoms can be preceded by a pre-motor or prodromal phase that begins 20 years or more before the motor manifestations of the disease (Goldman and Postuma 2014; Kalia 2015).
Conditions associated with decreased olfaction, depression, disturbances in sleep behavior, anxiety and intestinal constipation are frequently reported by in patients with PD in retrospective and longitudinal studies and are recognized as the most common non-motor symptoms of this disease (Bhidayasiri and Martinez-Martin 2017; Reichmann 2017; Schapira et al. 2017). The progress of the disease involves other brain areas (thalamus, hypothalamus, brainstem, cortex) resulting in the increase in autonomic failures, sensory, cognitive and psychiatric disorders (Giráldez-Pérez et al. 2014). In a retrospective study, it was observed that in the years prior to the diagnosis, individuals complained to their primary care physicians about non-motor characteristics of PD, mainly for constipation, which was the most reported, neuropsychiatric disorders (depression, anxiety and memory problems), and disorder in sleep behavior (Schrag et al. 2015). In another retrospective case–control study, it was shown that 61.2% of the subjects with PD interviewed reported the presence of one or more pre-motor symptoms such as hyposmia, depression, anxiety, constipation and sleep disorders, with a significant relationship between the presence of symptoms and the risk of developing PD (Rodriguez-Violante et al. 2017). In fact, recognition of PD pre-motor symptoms is useful for the development of strategies to identify individuals at risk, to make early diagnosis and to prevent or stop the development and progression of the neurodegenerative process (Chaudhuri et al. 2006; Martinez-Martin et al. 2017).
The Braak hypothesis of PD development postulates that it begins in the periphery (enteric plexus and olfactory bulb) and works its way into the CNS in six neuropathological stages (Braak et al. 2004). It is an important support to provide evidence that inflammation is involved in the development of non-motor and motor symptoms in PD. The stage 1 can be associated with the activation of the immune system by Helicobacter pylori infection, which induces an autoimmune response targeting mitochondria and possibly leading to the deposition of α-synuclein, alterations in enteric nervous system that may manifest as gastrointestinal dysfunction (Barnum and Tansey 2012); the stage 2 is associated with an inflammatory transmission to the CNS, mainly expressed by high levels of TNF-α and IL-6 and a subsequent reduction in serotonin levels via an indoleamine-2, 3-dioxygenase (IDO) and kynurenine degradative pathway of tryptophan and degeneration of monoaminergic systems that results in low mood and sleep disturbances (Lim et al. 2017); the stages 3, 4, 5 and 6 are marked by widespread inflammation in the CNS that may contribute to cognitive decline, dementia, psychosis and motor symptoms (Barnum and Tansey 2012). The unidirectional spread of PD pathogenesis postulated by Braak and coworkers has been revised (Braak et al. 2004). Studies in monkeys reinforcing the involvement of alpha-synuclein in PD pathogenesis support the notion of the existence of a range of alpha-synuclein pathogenic structures with distinct toxic properties within the PD brain, and suggest a possible systemic mechanism in which the general circulation would act as a route for long-distance bidirectional transmission of endogenous α-synuclein between the enteric and the central nervous systems (Arotcarena et al. 2020; Bourdenx et al. 2020).
Animal models have contributed to the understanding of how neuroinflammation is involved in the development of pre-motor and motor symptoms. The injection of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is capable to induce reactive gliosis and dopaminergic degeneration in rodents and non-human primates (Annese et al. 2013, 2015; Barcia et al. 2013; Kastner et al. 1994). Studies showed that microglial activation starts in a distress phase that precedes neuronal death in MPTP animal model (Hirsch and Hunot 2009). Moreover, it is suggested that intranigral lipopolysaccharide (LPS) administration in Wistar rats can provide new insights about the role of neuroinflammation on simulating features of the pre-motor phase of PD, since it produces dopamine and glutathione impairment but not a reduction in locomotion frequency and rearing frequency in comparison with MPTP and 6-OHDA, nor did it induce an increase in immobility time frequency in comparison with 6-OHDA (Ariza et al. 2010). On the other hand, single systemic injection of lipopolysaccharide (5 mg/kg i.p.) in three-month-old male mice generated discrete, progressive neurodegeneration resembling the spatiotemporal pattern of neurodegeneration in PD. This LPS-induced neurodegeneration involves important brain regions associated with locomotor activities (substantia nigra and motor cortex), as well as areas associated with non-motor behavior activities, such as locus coeruleus (LC) and hippocampus (Song et al. 2019a). In addition, it is clear that the induction of non-motor symptoms including hyposmia, constipation, anxiety, sociability, exaggerated startle response and impaired learning, as well as motor symptoms including decreased rotarod activity, grip strength and gait disturbance in 9-week-old male mice with LPS intraperitoneal injection depends on a potentiation induced by the noradrenergic selective neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4), suggesting the association of noradrenergic dysfunctions and neuroinflammation in PD pathogeneses (Song et al. 2019b).
Microglial Functions in Homeostasis and in Neuroinflammation in Parkinson's Disease
Derived from early Erythroid Myeloid Precursors (eEMPs) from the yolk sac, the microglia represent 10–15% of the total glial cell population in the CNS (Tay et al. 2016) (Fig. 1). This cell was described morphologically a century ago by Del Río-Hortega (1919) and before the advent of immunological and molecular techniques, the morphological changes of the microglia were considered as the main characteristics of their activation and an indicator of pathology in the CNS, but currently we know that branched, hypertrophic and ameboid phenotypes are present in people without neurological diseases (Salamanca et al. 2019; Torres-Platas et al. 2014). The advancement in methodology tools using single-cell analysis allowed for the staggering in the identification of microglial types (branched, hypertrophic and ameboid) for the classification of subtypes and the demonstration of spatial heterogeneity of microglia in in vivo studies and postmortem brain tissue (Böttcher et al. 2019; Masuda et al. 2019; Silvin and Ginhoux 2018) (Fig. 1). The understanding of the mechanisms that regulate homeostasis and microglial function can provide means to manipulate these cells for therapeutic purposes. Studies have been advanced through the discovery of the microglial molecular diversity in a temporal and spatial way during embryogenesis, homeostasis, adulthood, aging and CNS disorders (Prinz et al. 2019).

Microglia are originated from the early Erythromyeloid Precursors (eEMPs) from the yolk sac embryonic. In the development, they migrate to the neural tube, where they proliferate, colonize the entire parenchyma and remain throughout the life of the organism. Neonatal microglia are characterized by an ameboid morphology with a high rate of proliferation and heterogeneity. In adult brain, microglia are represented by different phenotypes distributed in distinct regions of the CNS that can be identified through different morphologies and molecular markers. The satellite microglia, named due to its location near the neuron, have spherical morphology. These cells interact preferentially in the axon initial segment region. The microglia 1 are identified through the profile of markers: TMEM119+, P2RY12+, CX3CR1+, CD206lo. The microglia 2 are identified through the profile of markers: TMEM119+, P2RY12+, CX3CR1+, CD206lo. The microglia 3 are identified through the profile of markers: expresses TMEM119+, P2RY12+, CX3CR1+, CD11c+, CD68+. The microglia 4 are identified through the profile of markers: TMEM119lo, P2RY12lo, CX3CR1lo, SLC2A5lo, CCL2+, CCL4+, EGR2+, EGR3+. Figure created with BioRender.com
Microglia have long been erroneously considered as static observers in the healthy CNS with minimal functions in homeostasis. Nowadays, it is known that microglia are supremely agile, performing multitasking in the CNS during neurogenesis, adulthood and aging brain maintenance of homeostasis, neuronal survival, cell death and synaptic modulation (Colonna and Butovsky 2017). For example, microglia are required for synaptic pruning in neuronal development and provide support for neuronal networks functioning; they also phagocyte apoptotic cells during neurogenesis and may also support the formation of synapses associated with learning through the release of neurotrophic factors (Madore et al. 2020; Miyamoto et al. 2016; Paolicelli and Ferretti 2017). The microglia morphology in the healthy CNS is typically branched, where it maintains a steady state of constant surveillance. In this conditions, these cells are immobile, but their extensions can reach distances equivalent to ten times their size and are responsible for identifying changes in the cerebral microenvironment by making constant interactions with neurons and other glial cells, including other microglia, monitoring synapses and looking for any kind of breakdown of homeostasis (Arcuri et al. 2017; Savage et al. 2019). When there are small disturbances of homeostasis, the microglia change their morphology to hypertrophic. In large disorders, these cells acquire an amoeboid shape, with an increase in the phagocytic capacity and the expression of molecules associated with this profile, such as pro-inflammatory mediators and receptors for the antigen recognition (Anderson and Vetter 2019; Kirkley et al. 2017; Labzin et al. 2018; Sominsky et al. 2018) (Fig. 2).

Microglia acts on homeostasis and neurodegeneration. The microglia in homeostasis have important functions such as synaptic pruning, production of neurotrophic factors. For example, the brain-derived neurotrophic factor (BDNF) and the glia-derived neurotrophic factor (GDNF), both factors are essential for brain development. The microglia also support neuronal connections, phagocytosis of cellular debris and infection control. In neurodegenerative diseases, microglia become highly reactive, producing various neuroinflammatory molecules, such as IL-1β, IL-18, IL-6, TNF-α and chemokines, in addition to reactive species, such as nitric oxide, which are toxic to tissue and can damage neurons
Changes in the immune system of PD patients evidence continuous neuroinflammation. It is possible to observe in these individuals changes of lymphocyte population in cerebrospinal fluid and blood, increased synthesis of immunoglobulins, cytokines and acute phase proteins (Obeso et al. 2017). In addition, direct evidence of microgliosis can be provided in the CNS of PD patients by Positron Emission Tomography (PET) using the [18F]-radiolabeled prenoxyanilide ([18F]-FEPPA) radioligand, a biomarker known to interact with the translocating protein (TSPO) located in the microglia mitochondrial membrane (Koshimori et al. 2015; Roussakis and Piccini 2018). Furthermore, evidence of microgliosis shown in the SNpc of patients has revealed reactive microglia expressing complement receptor 3 (McGeer et al. 1988) and increase in the number of amoeboid immunoreactive microglia as detected by the expression of the ionized calcium-binding adaptor molecule 1 (Iba1) specific marker (Doorn et al. 2014). The microgliosis was also evidenced in animal models, such as MPTP-treated monkeys (Barcia et al. 2004, 2011) and Parkinsonian young and old mice (Gil-Martínez et al. 2019, 2018). In addition, studies show that blocking microglia activation and neuroinflammation with anti-inflammatory drugs, inhibitors of matrix metalloproteinase and inhibitors of activation of p21(ras) and Factor Nuclear kappa B (NF-κB) protect dopaminergic neurons in MPTP-treated young mice (Costa et al. 2020; Ghosh et al. 2009). MPTP is an exogenus neurotoxin that induces acute dopaminergic degeneration. On the other hand, aminochrome, an endogenous molecule derived from dopamine oxidation has been suggested as a neurotoxin capable to promote dysfunction in the dopaminergic system, slow dopaminergic degeneration in vivo, and microglia activation and neuroinflammation in vitro (Santos et al. 2017; de Araújo et al. 2018; to review see Segura-Aguilar et al. 2019).
Mechanism of Microglial Activation in Parkinson´s Disease
Microglia are endowed with Patterns-recognition receptors (PRRs) and their activation can be generated by the presence of Pathogen-Associated Molecular Pattern (PAMPs) highly conserved in microorganisms and/or by Damage-Associated Molecular Pattern (DAMPs), which can be generated by the presence of damaged cells and include poorly folded proteins, peptide aggregates and nucleic acids that are present in the neurodegenerative diseases (Wolf et al. 2017). An important family of PRRs is Toll-Like Receptor (TLR), which is composed of 13 highly conserved protein members. These proteins can be expressed in the cell membrane surface (TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10) or in intracellular vesicles, such as the endoplasmic reticulum, endosomes and lysosomes (TLR3, TLR7, TLR8 and TLR9) (Bayraktar et al. 2019). They play a key role in the activation of several signaling pathways and activation of transcription factors that induce the expression of important genes for the development of pro-inflammatory responses (Lu et al. 2018).
The structure of TLRs is composed of two domains: an extracellular one also known as ectodomain containing blocks of Leucine-Rich Repeats (LRR), and another with cysteine-rich coatings in the amino terminal and carboxy terminal domains. The C-terminal structure is connected to a transmembrane α-helix that attaches to the second domain of the protein, located in the cytoplasm known as the Toll/ interleukin-1 (TIR) receptor domain or TIR identity region that couples the transduction of the signal, activating the transcription cascade (Gay et al. 2006) (Fig. 3). TLRs are widely expressed in various CNS cells. Studies show that these receptors are present in neurons by activating different signaling pathways related to control of neuronal morphology, development and response to pathologies (Hung et al. 2018); in astrocytes, they are involved in several defensive mechanisms (Marinelli et al. 2015; Verkhratsky and Nedergaard 2018); in oligodendrocytes, the TLR7 is involved in the production of pro-inflammatory molecules such as Chemokine Ligand 2 (CCL2), Chemokine Ligand 8 (CXCL8) and Interleukin-6 (IL-6) (Parthasarathy and Philipp 2018); and in the microglia, which express all TLRs isoforms, these receptors are involved in the activation reported in several neurodegenerative diseases, such as PD and Alzheimer’s disease (Subhramanyam et al. 2019).

Structure of the Toll-like Receptor and signaling pathway responsible for stimulating the proliferation, survival and production of pro-inflammatory factors by the microglia through the activation of membrane TLRs. The presence of two domains, one extracellular responsible for the recognition of PRRs and the other intracellular responsible for signal transduction. α-Synuclein aggregates are recognized on the microglia surface by TLR type 1, 2 heterodimers or by a complex set of TLR 4 and Myeloid differentiation protein-2 (MD2). TLR stimulation recruits adapter proteins that include Myd88 and Mal/TIRAP. The next step is an Myd88-dependent signaling cascade leading to the formation and translocation of NFkB into the nucleus and transcription of cytokine and chemokine mRNA. MAPks activation is also observed as a consequent activation of the nuclear transcription factors JNK, ERK 1/2 and p38MAPK promoting the proliferation, survival and production of pro-inflammatory factors
In PD, endogenous molecules such as α-synuclein act as a DAMP leading to microglial activation through the TLR2, which induces a neuroinflammatory response with the production and release of Tumor Necrosis Factor-alpha (TNF-α), IL-6, Chemokine Ligand 1 (CX3CL1) and Chemokine Ligand 5 (CCL5) inflammatory mediators as a consequence of the activation of NF-κB and Mitogen-Activated Protein Kinases (MAPK) (Kim et al. 2018). The NFκB pathway is responsible for the production of TNF-α, Pro-Interleukin-1β (pro-IL-1β), and IL-6, Cyclooxygenase-2 (COX-2), Nitric Oxide (NO) and chemokines (CCL2, CXCL8, among others) (Dresselhaus and Meffert 2019; Taetzsch et al. 2015; Yan et al. 2017). On the other hand, the MAPKs, p38 MAPK, c-Jun NH2-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK 1/2) pathways, related to proliferation, survival and production of pro-inflammatory factors in microglia (Bohush et al. 2018; Tong et al. 2018) (Fig. 3). In addition, it was demonstrated in the microglia treated with α-synuclein that the formation of a heterodimer complex of TLR1 and TLR2 is involved in the increase of NF-κB nuclear translocation and consequently in the increase of TNF-α and IL-1β. Myeloid differentiation protein (MyD88), a molecular adapter critical for TLR, plays an important role in the increase of pro-inflammatory cytokine production, as it allows the dimerization of TLR1 and TLR2 receptors (Daniele et al. 2015). TLR4 is also expressed by astrocytes that act on its activation by α-synuclein; when this receptor is suppressed, astrocytes show a reduction in the pro-inflammatory response (Fellner et al. 2013).
Inflammasome Activation: Canonica and Non-canonica Pathway
TLR family is not the only receptors involved in the recognition of DAMPs and PAMPs. A second class of PRRs that is present in the intracellular compartments is also implicated in that function. This class includes the Absent in Melanoma 2 (AIM2), receptor-type AIM2-like (ALR) and the NOD-like receptors (NLRs) (Lamkanfi and Dixit 2014; Wang et al. 2020b). A subfamily of the NLRs is characterized by the presence of a central nucleotide-binding and oligomerization domain (NACHT), which is commonly flanked by C-terminal leucine-rich repeats (LRRs) and N-terminal caspase recruitment (CARD) or pyrin domains (PYD) (Yang et al. 2019a). The LRRs domain functions as a sensor that detects intracytoplasmic activation signals; the NACHT domain is present in all members of the NLRs family, being related to the activation of the complex through its oligomerization. The CARD or PYD domains mediate interactions between NLR and effector or adapter proteins, necessary for dowstream signaling (Schroder and Tschopp 2010). The NLRs containing a pyrin domain (NLRP1–NLRP14—to review see Table 1) has drawn attention due to its participation in the formation of inflammasome in the presence of activators (Platnich and Muruve 2019; Wang et al. 2017; Yang et al. 2019a).
The inflammasome is a multiprotein complex present in the microglia, other machophages, dendritic cells and some other immune cells. It controls the activation of the proteolytic enzyme caspase-1 and it can be subdivided into three components: a PRR as a sensing molecule, an adapter protein and an enzymatic component (Yang et al. 2019b). The most common inflammasome is NLRP3 due to its involvement in several human diseases, especially in PD (Haque et al. 2020; Lee et al. 2019). It has a domain for the recruitment of the caspase activating adapter protein (ASC) and an enzymatic component to caspase-1 (Guo et al. 2015; Wang et al. 2019). These three structures are assembled to react to infections or signs of endogenous danger through the production of IL-1β (Man and Kanneganti 2015). When ASC binds to NLRP3 through its pyrin domain, ASC induces the aggregation of pro-caspase-1 to initiate self-cleavage for activated caspase-1, which subsequently will carry out the zymogen cleavage of the pro-inflammatory cytokines IL-1β and IL-18 (He et al. 2016; Qiao et al. 2017). These cytokines are secreted and will activate other cells, amplifying the inflammatory response (Howrylak and Nakahira 2017).
In fact, the mechanism of the inflammatory activation of the NLRP3 involves two pathways: canonical and non-canonical. The canonical pathway inflammasome is dependent on caspase-1 and requires two signals for its function. The first signal, also known as priming, is responsible for sensitizing any receptor that activates the NFκB pathway by ligands for TLR, NLRs or IL-1R1, TNFR1 and TNFR2 cytokine receptors inducing the transcription and translation of Pro-IL-1β, pro-IL-18 and NLRP3 (Latz et al. 2013; Lin et al. 2014; Sutterwala et al. 2014). The production of pro-IL-1β, pro-IL-18 and NLRP3 is necessary because the basal levels of cytoplasmic NLRP3 are insufficient for the pathway activation and pro-IL-1β is not constitutively expressed (Vanaja et al. 2015). The second one is responsible for inflammasome activation mediated by Lys-63-specific deubiquitinase (BRCC3). This enzyme removes the ubiquitin bound to NLRP3 allowing the formation of the NLRP3-ASC, nucleated ASC sequentially recruits pro-caspase-1, which undergoes proximity-induced autocatalytic cleavage generating active subunits that will then cleave pro-IL-1β and pro-IL-18 in their active forms (Py et al. 2013; Xiang et al. 2020). Multiple danger signs can contribute to second signal NLRP3 inflammasome activation, including: ROS elevation (Tschopp and Schroder 2010), change in ion concentration (Hafner-Bratkovič and Pelegrín 2018) and mitochondrial dysfunction (Sarkar et al. 2017). The non-canonical pathway was evidenced for the first time by Kayagaki et al. (Kayagaki et al. 2011); in this study, it was observed that caspase-11 activated in mice performs the activation of caspase-1 and production of IL-1β. Functionally, caspase-11 has been identified as an LPS sensor in the cytoplasm of immune cells. It can induce a pyroptotic response and contribute to the assembly of the NLRP3 inflammasome in the non-canonical pathway (Sharma and Kanneganti 2016; Zheng et al. 2020).
NLRP3 Inflammasome Activation in Parkinson´s Disease
The main event that regulates the secretion of IL-1β by the microglia is the activation of inflammasome, a key function developed by the innate immune system in PD to sustain the neuroinflammatory process. This event marked by elevating IL-1β, IL18, caspase-1 and NLRP3 can be observed in a rodent study model of PD (Chen et al. 2019; Cheng et al. 2020a; Mao et al. 2017). In addition, studies in patients with this disease show an increase in IL-1β and IL-18 in the cerebrospinal fluid, cytokines that are generated by the action of inflammasome (Zhang et al. 2016a). These evidences demonstrate the key role of this multiprotein complex in the neuroinflammatory process.
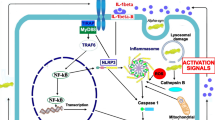
In PD, α-synuclein aggregates and DAMPs from damaged neurons can be released into the extracellular space and be recognized by TLR2 or other microglia TLRs. This recognition activates the canonical pathway followed by NFκB translocation for the production of Pro-IL-1β and NLRP3. It is important to note that the IL-1R and TNFR receptors can activate the signal priming when stimulated by their ligands (Chatterjee et al. 2020; Codolo et al. 2013; Javed et al. 2020; Lang et al. 2018; Sutterwala et al. 2014) (Fig. 4). The newly produced inflammasome NLRP3 is in a preactivated state, in which ubiquitination prevents its oligomerization with the ASC protein (Ren et al. 2019; Shim and Lee 2018). The second signal, generated by the presence of ROS and neurotoxic alpha-synuclein fibrils, stimulates NLRP3 deubiquitination mediated by BRCC3 deubiquitinase and activates the nucleation of the inflammasome with ASC forming the NLRP3-ASC-Caspase-1. This complex will form IL-1β and IL-18 from their zymogenes generated in the priming signal (Cheng et al. 2020b; Py et al. 2013; Sarkar et al. 2017) (Fig. 4). Another second signal, for example the increase in K+ efflux, increase in Ca+ influx, cathepsin B from lysosomes and mitochondrial DNA, can generate activation of NLRP3 inflammasome in PD (Haque et al. 2020). The secretion of IL-1β and IL-18 by the microglia occurs through the action of Gasdermin D; this protein is cleaved and activated by caspase-1, which after this process activates Gasdermin D translocates to the plasma membrane of the microglia forming pores through which IL-1β and IL-18 can be released into the extracellular space. This phenomenon will eventually induce pyroptosis, which is a pro-inflammatory form of cell death (Heneka et al. 2018; Shi et al. 2015).

Possible mechanism of inflammasome activation in PD through α-synuclein. Activation of the inflammasome requires two signals, the first one will generate the activation of NF-κB provided the production of NLRP3 and pro-IL-1β, and the second signal will provide the desubiquitination of NLRP3 making that molecule free to bond with ASC and caspase-1 and form the inflammasome complex that will cleave pro-IL-1β to IL-1β
Age plays an important role as a risk factor for the development of neurodegenerative diseases. In the elderly, an annual reduction in total brain volume between 0.5 and 1% can be seen in areas associated with cognition and memory (Scheiblich et al. 2020). Senescent cells of the elderly, and in in vivo study models, develop a secretome profile with high levels of pro-inflammatory markers, such as IL-1β and TNF-α, which has a summing effect for the progression of cellular dysfunction and tissue damage by impairing neuronal regeneration and growth, loss of synapses and reduction in the formation of synapses dependent on learning (Garré et al. 2017; Malaquin et al. 2016; Newman et al. 2016; Tsarouchas et al. 2018). The inflammation shown in the brain of these individuals promotes microglial activation with active participation of NLRP3 inflammasome in the production of IL-1β. Genetic or environmental risk factors can increase the risk of losing the age-associated inflammatory physiological control, which can result in sustained inflammatory exacerbation and development of neurodegenerative diseases such as PD (Scheiblich et al. 2020). Evidence also suggests that peripheral inflammasome activation in mice, through changes in the intestinal microbiota, can raise the levels of pro-inflammatory factors in the peripheral circulation, aggravating or promoting the inflammatory process at the CNS level with M1 reactivity of the microglial and consequent activation of the NLRP3 inflammasome that contributes to the development or aggravation of neurodegenerative diseases (Shen et al. 2020).
Inflammasome in Parkinson’s Disease: A Potential Target for New Therapies
A controlled and well-balanced inflammasome response is essential to maintain homeostasis, continuous and exacerbated activation of this complex can generate an inflammatory process harmful to the tissue. Regulatory feedback molecules that inactivate excessive inflammatory responses are essential to prevent tissue damage or even systemic inflammation. Understanding the effector mechanisms of these molecules can provide evidence of pharmacological targets helping to control the inappropriate inflammatory process(de Almeida et al. 2015). Immune cells naturally have endogenous molecules capable of making this regulatory feedback, and a group of proteins that can act on the inflammasome complex by inactivating its assembly are the PYRIN-only proteins (POPs) (Ratsimandresy et al. 2017). POP1 inhibits the assembly of the inflammasome; this protein is able to interact with the PYD of the ASC, its regulatory action is induced by IL-1β thus avoiding the nucleation of the ASC-NLRs and consequent perpertuation of the response (de Almeida et al. 2015). POP2 in addition to interacting with the PYD of the ASC also interacted with the PYD of other NLRPs and inhibited the activation of the NFkB. Thus, POP2 is able to simultaneously block the priming and the activation of the inflammasome (Ratsimandresy et al. 2017). POP3 does not bind to ASC, but interacts with AIM2, blocking the activation of the inflammasome and promoting the production of type I interferon (Khare et al. 2014).
The usual clinical treatment for PD is aimed at increasing dopamine levels in the brain using exogenous dopamine precursors (levodopa), monoamine oxidase B (MAO-B) inhibitors and dopamine receptor agonists (Goetz and Pal 2014). The dopamine precursor l-3,4-dihydroxyphenylalanine (l-dopa) produces important side effects, which usually appear several years after chronic use, such as motor fluctuations, dyskinesia and psychosis. On the other hand, despite its side effects, l-dopa remains as the best option for stiffness and akinesia, improving the patient's quality of life (Ramirez-Zamora and Molho 2014; Tarakad and Jankovic 2017). For four decades the main treatment for PD has been the use of l-dopa. However, these therapeutic approaches aimed at restoring dopamine levels in the CNS do not prevent or delay the neurodegenerative process in PD. As an alternative, neuroinflammation, which plays an important role in the development of the disease as discussed, has been investigated as a new therapeutic target for reducing the damage in dopaminergic neurons (Martinez et al. 2017; Tan et al. 2020).
Postmortem histological studies of PD patients revealed increased NLRP3 expression in mesencephalic neurons, highlighting that Human Embryonic Kidney 293 cells (HEK293) with NLRP3 rs7525979 polymorphism associated with protein instability, reduction in solubility and an increase in affinity for ubiquitination affect the progression of PD (von Herrmann et al. 2018). In NLRP3 (KO) mice treated with MPTP, a reduction in the progression of dopaminergic neurodegeneration has been shown in comparison with wild-type mice, suggesting a relation between inflammasome and PD (Yan et al. 2015). Moreover, Cx3Cr1CreER-microglia-based animals with specific expression of mutant NLRP3 presented exacerbated motor deficits and dopaminergic neuronal loss. It has also been shown that animals with NLRP3 deficits, when intoxicated with MPTP, present reduced motor deficit, neuronal loss, microglial recruitment, IL-1β production and caspase-1 activation (Lee et al. 2019). Not only the NLRP3 deficiency of inflammasome is able to reduce neuroinflammation in PD models, but some molecules are also able to induce their inhibition and reduction of inflammatory process (Yang et al. 2019b). For example, the tenuigenin, a mixture of saponins extracted from P. tenuifolia roots, was able to reduce the levels of NLRP3, caspase-1, pro-IL-1β and IL-1β in MPTP mouse acute model, and in BV2 microglia cells exposed to LPS (Fan et al. 2017).
The mechanisms underlying the pharmacological inhibition of NLRP3 inflammasome are diverse. Some agents such as glyburide present indirect action via ATP-sensitive K+ channels, while others such as VX-740, VX-765, parthenolide, CY-09 and MCC950 present direct action in one or more molecular target (NLRP3, Caspase 1, NF-κB, IKKβ) to inhibit NLRP3 inflammasome (Zahid et al. 2019). CY-09 is an molecule that directly binds to the ATP-binding motif of NLRP3 NACHT domain and inhibits NLRP3 ATPase activity, resulting in the suppression of NLRP3 inflammasome assembly and activation (Jiang et al. 2017), while MCC950 inhibition of NLRP3 inflammasome involves direct interaction with NLRP3 ATP hydrolysis motif within the NLRP3 NACHT domain, thereby blocking ATP hydrolysis and inhibiting canonical and non-canonical NLRP3 inflammasome activation (Shao et al. 2015).
It has been recently demonstrated that pharmacological inhibition of NLRP3 inflammasome activation with the oral treatment of MCC950, a small molecule derived from synthesis, prevents α-synuclein pathology and dopaminergic neurodegeneration in mice (Gordon et al. 2018). This is a promising drug for several inflammasome-related-diseases. However, in experimental autoimmune encephalomyelitis, a single-dose pharmacokinetic profile of MCC950 in C57Bl/6 mice via intravenous (3 mg/kg) and oral (20 mg/kg) administration resulted in a short half-life. This pharmacokinetic profile may be an obstacle to the success of the inhibitor in human clinical trials (Shao et al. 2015). Even so, the inhibitory effects of MCC950 and tenuigenin indicate NLRP3 inflammasome as a target for promising agents for alleviating dopaminergic degeneration in PD.
Additionally, there is information about the effect of some non-steroidal anti-inflammatory drugs (NSAIDs) in the NLRP3 inflammasome inhibition. The fenamate class is effective to inhibit IL-1β secretion from macrophages and selective inhibitors of the NLRP3 inflammasome via inhibition of the volume-regulated anion channel in macrophages, regardless of COX enzymes (Laliberte et al. 1994). The flufenamic acid and mefenamic acid therapeutic efficacy to inhibit NLRP3 inflammasome and induce neuroprotection in a model of amyloid beta induced memory loss, and in a transgenic mouse model of Alzheimer's disease, suggesting that fenamate NSAIDs could be repurposed as Alzheimer's disease therapeutics (Daniels et al. 2016).
Another important way for the regulation of inflammasome activation is the activation of autophagy, since it involves the degradation of damaged organelles and recycling of cellular metabolites that can active inflammassome; it can regulate inflammasome activation via a reduction of ROS production, degradation of ASC aggregates, and sequestration of pro-IL-1β (Harris et al. 2011; Jabir et al. 2015; Shi et al. 2012; Zhou et al. 2011). The involvement of autophagy in the neuroprotection in PD has been widely studied and associating the control of inflammasome as another mechanism of its neuroprotective action serves as a stimulus for the prospection of new molecules and investments for further studies in drugs with a potential inducer of autophagy.
Concluding Remarks
There are increased evidences that inflammatory reactions and changes in the immune system are always present in PD. Microglia, whose role is to orchestrate the immune responses in the CNS, can be activated when cerebral homeostasis breaks, releasing a series of pro-inflammatory cytokines and neurotoxic factors that induce neuronal death. In PD, production and release of α-synuclein will generate the activation of these cells with concomitant activation of the NLRP3 inflammasome that will stimulate the production of IL-1β, creating a toxic environment for neurons and potentiating the neurodegenerative process. Therefore, development of immunomodulatory therapeutic strategies could be beneficial for the survival of dopaminergic neurons and NLRP3 seems to be an important pharmacological target for the negative modulation of neuroinflammatory response in PD.
Abbreviations
- [18F]-FEPPA:
-
[18F]-Radiolabelled phenoxyanilide
- 6-OHDA:
-
6-Hydroxydopamine
- AIM2:
-
Absent in melanoma 2
- ALR:
-
AIM2-like receptor
- ASC:
-
Caspase activating adapter protein
- BDNF:
-
Brain-derived neurotrophic factor
- BRCC3:
-
Lys-63-specific deubiquitinase
- CARD:
-
N-terminal caspase recruitment
- CCL2:
-
Chemokine ligand 2
- CCL5:
-
Chemokine ligand 5
- CNS:
-
Central nervous system
- COX-2:
-
Cyclooxygenase-2
- CX3CL1:
-
Chemokine ligand 1
- CXCL8:
-
Chemokine ligand 8
- DAMPs:
-
Damage-associated molecular pattern
- PD:
-
Parkinson's disease
- DSP-4:
-
N-(2-Chloroethyl)-N-ethyl-2-bromobenzylamine
- EMPs:
-
Erythromyeloid precursors
- ERK 1/2:
-
Extracellular signal-regulated kinase
- FIND:
-
Function to find domain
- GDNF:
-
Glia-derived neurotrophic factor
- HEK293:
-
Human embryonic kidney 293 cells
- Iba1:
-
Ionized calcium-binding adaptor molecule 1
- IL-18:
-
Interleukin-18
- IL-1β :
-
Interleukin-1β
- IL-6:
-
Interleukin-6
- IL1-R1:
-
Interleukin-1 receptor 1
- IEA-NLRC4:
-
Infantile enterocolitis associated with NLRC4
- JNK:
-
C-Jun NH2-terminal kinase
- l-dopa:
-
l-3,4-Dihydroxyphenylalanine
- LPS:
-
Lipopolysaccharide
- LRR:
-
Leucine-rich repeats
- MAL/TIRAP:
-
MyD88 adaptor-like protein/TIR-containing adaptor protein
- MAS:
-
Macrophage activation syndrome
- MAO-B:
-
Monoamine oxidase B
- MAPK:
-
Mitogen-activated protein kinases
- MD2:
-
Myeloid differentiation protein-2
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MyD88:
-
Myeloid differentiation protein
- NACHT:
-
Nucleotide-binding and oligomerization
- NF-κB:
-
Factor nuclear kappa B
- NLRP3:
-
NOD-like receptor protein 3
- NLRs:
-
NOD-like receptors
- NO:
-
Nitric oxide
- PAMPs:
-
Pathogen-associated molecular pattern
- PET:
-
Positron emission tomography
- Pro-IL-1β :
-
Pro-interleukin-1β
- PRRs:
-
Pattern-recognition receptors
- PYD:
-
Pyrin domains
- ROS:
-
Reactive oxygen species
- SNpc:
-
Substantia nigra pars compacta
- TIR:
-
Toll/interleukin-1
- TNF-α :
-
Tumor necrosis factor-α
- TNFR1:
-
Tumor necrosis factor receptor 1
- TNFR12:
-
Tumor necrosis factor receptor 2
- TLR:
-
Toll-like receptor
- TSPO:
-
Translocating protein
References
Abdullah R, Basak I, Patil KS, Alves G, Larsen JP, Møller SG (2015) Parkinson’s disease and age: the obvious but largely unexplored link. Exp Gerontol 68:33–38
Abi Nahed R et al (2019) NLRP7 is increased in human idiopathic fetal growth restriction and plays a critical role in trophoblast differentiation. J Mol Med 97:355–367. https://doi.org/10.1007/s00109-018-01737-x
Aguilera M, Darby T, Melgar S (2014) The complex role of inflammasomes in the pathogenesis of inflammatory bowel diseases: lessons learned from experimental models vol 25. Cytokine Growth Factor Rev. https://doi.org/10.1016/j.cytogfr.2014.04.003
Aguzzi A, Barres BA, Bennett ML (2013) Microglia: scapegoat, saboteur, or something else? Science 339:156–161
Anderson SR, Vetter ML (2019) Developmental roles of microglia: a window into mechanisms of disease. Dev Dyn 248:98–117
Annese V et al (2013) Evidence of oligodendrogliosis in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Neuropathol Appl Neurobiol 39:132–143. https://doi.org/10.1111/j.1365-2990.2012.01271.x
Annese V et al (2015) Metalloproteinase-9 contributes to inflammatory glia activation and nigro-striatal pathway degeneration in both mouse and monkey models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Brain Struct Funct 220:703–727. https://doi.org/10.1007/s00429-014-0718-8
Arcuri C, Mecca C, Bianchi R, Giambanco I, Donato R (2017) The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front Mol Neurosci 10:191
Ariza D et al (2010) Intranigral LPS administration produces dopamine, glutathione but not behavioral impairment in comparison to MPTP and 6-OHDA neurotoxin models of Parkinson’s disease. Neurochem Res 35:1620–1627
Arlehamn CSL et al (2020) α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun 11:1–11
Arotcarena ML et al (2020) Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain 143:1462–1475. https://doi.org/10.1093/brain/awaa096
Barcia C et al (2004) Evidence of active microglia in substantia nigra pars compacta of parkinsonian monkeys 1 year after MPTP exposure. Glia 46:402–409
Barcia C et al (2011) IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis 2:e142–e142
Barcia C et al (2013) Persistent phagocytic characteristics of microglia in the substantia nigra of long-term Parkinsonian macaques. J Neuroimmunol 261:60–66. https://doi.org/10.1016/j.jneuroim.2013.05.001
Barnum CJ, Tansey MG (2012) Neuroinflammation and non-motor symptoms: the dark passenger of Parkinson’s disease? Curr Neurol Neurosci Rep 12:350–358. https://doi.org/10.1007/s11910-012-0283-6
Bayraktar R, Bertilaccio MTS, Calin GA (2019) The interaction between two worlds: microRNAs and Toll-like receptors. Front Immunol 10:1053
Bernier L-P, York EM, Kamyabi A, Choi HB, Weilinger NL, MacVicar BA (2020) Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat Commun 11(1):17
Bhidayasiri R, Martinez-Martin P (2017) Clinical assessments in Parkinson’s disease: scales and monitoring. International review of neurobiology, vol 132. Elsevier, New York, pp 129–182
Bohush A, Niewiadomska G, Filipek A (2018) Role of mitogen activated protein kinase signaling in Parkinson’s disease. Int J Mol Sci 19:2973
Bourdenx M et al (2020) Identification of distinct pathological signatures induced by patient-derived α-synuclein structures in nonhuman primates. Sci Adv. https://doi.org/10.1126/sciadv.aaz9165
Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134. https://doi.org/10.1007/s00441-004-0956-9
Bullon P, Navarro JM (2017) Inflammasome as a key pathogenic mechanism in endometriosis. Curr Drug Targets 18:997–1002. https://doi.org/10.2174/1389450117666160709013850
Böttcher C et al (2019) Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat Neurosci 22:78–90
Canna SW et al (2014) An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 46:1140–1146
Chatterjee K et al (2020) Inflammasome and α-synuclein in Parkinson’s disease: a cross-sectional study. J Neuroimmunol 338:577089
Chaudhuri KR et al (2006) International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson’s disease: the NMSQuest study. Mov Disord 21(916):923
Chavarría-Smith J, Vance RE (2015) The NLRP 1 inflammasomes. Immunol Rev 265:22–34
Chen L, Xue L, Zheng J, Tian X, Zhang Y, Tong Q (2019) PPARß/δ agonist alleviates NLRP3 inflammasome-mediated neuroinflammation in the MPTP mouse model of Parkinson’s disease. Behav Brain Res 356:483–489
Cheng J et al (2020a) Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy. https://doi.org/10.1080/15548627.2020.1719723
Cheng X, Xu S, Zhang C, Qin K, Yan J, Shao X (2020b) The BRCC3 regulated by Cdk5 promotes the activation of neuronal NLRP3 inflammasome in Parkinson’s disease models. Biochem Biophys Res Commun 522:647–654
Chu JQ et al (2016) Production of IL-1β and inflammasome with up-regulated expressions of NOD-like receptor related genes in Korean. J Parasitol 54:711–717. https://doi.org/10.3347/kjp.2016.54.6.711
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M (2013) Triggering of inflammasome by aggregated α–synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8:e55375
Colonna M, Butovsky O (2017) Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 35:441–468
Costa T, Fernandez-Villalba E, Izura V, Lucas-Ochoa AM, Menezes-Filho NJ, Santana RC, de Oliveira MD, Araújo FM, Estrada C, Silva V, Costa SL, Herrero MT (2020) Combined 1-deoxynojirimycin and ibuprofen treatment decreases microglial activation, phagocytosis and dopaminergic degeneration in MPTP-treated mice. J Neuroimmun Pharmacol. https://doi.org/10.1007/s11481-020-09925-8
Cuenca N et al (2005) Morphological impairments in retinal neurons of the scotopic visual pathway in a monkey model of Parkinson’s disease. J Comp Neurol 493:261–273
Cummings JR et al (2010) The genetics of NOD-like receptors in Crohn’s disease. Tissue Antigens 76:48–56. https://doi.org/10.1111/j.1399-0039.2010.01470.x
Dalbiès-Tran R, Papillier P, Pennetier S, Uzbekova S, Monget P (2005) Bovine mater-like NALP9 is an oocyte marker gene. Mol Reprod Dev 71:414–421. https://doi.org/10.1002/mrd.20298
Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA (2015) Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal 8:45
Daniels MJ et al (2016) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun 7:12504. https://doi.org/10.1038/ncomms12504
Das R, Sharma S (2016) Cognitive impairment associated with parkinson’s disease: role of mitochondria. Curr Neuropharmacol 14:584–592
Davis BK et al (2011) Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol 186:1333–1337. https://doi.org/10.4049/jimmunol.1003111
de Almeida L et al (2015) The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 43:264–276
De Stefano ME, Herrero MT (2017) The multifaceted role of metalloproteinases in physiological and pathological conditions in embryonic and adult brains. Prog Neurobiol 155:36–56
Del Rio-Hortega P (1919) El tercer elemento de los centros nerviosos. I. La microglia en estado nor-mal. II. Intervencion de la microglia en los procesos patologicos. III. Nat Prob Micro Boll Soc Esp Biol 9:69–120
Docherty LE et al (2015) Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans. Nat Commun 6:8086. https://doi.org/10.1038/ncomms9086
Doorn KJ, Moors T, Drukarch B, van de Berg WD, Lucassen PJ, van Dam A-M (2014) Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun 2:90
Dorsey ER et al (2018a) Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study. Lancet Neurol 17:939–953
Dorsey E, Sherer T, Okun MS, Bloem BR (2018b) The emerging evidence of the Parkinson pandemic. J Parkinson’s Dis 8(S3):S8
Dresselhaus EC, Meffert MK (2019) Cellular specificity of NF-κB function in the nervous system. Front Immunol 10(1043):1057
Duncan JA, Canna SW (2018) The NLRC 4 inflammasome. Immunol Rev 281:115–123
Eibl C et al (2012) Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry 51:7330–7341. https://doi.org/10.1021/bi3007059
Eisenbarth SC et al (2012) NLRP10 is a NOD-like receptor essential to initiate adaptive immunity by dendritic cells. Nature 484:510–513. https://doi.org/10.1038/nature11012
Fan Z, Liang Z, Yang H, Pan Y, Zheng Y, Wang X (2017) Tenuigenin protects dopaminergic neurons from inflammation via suppressing NLRP3 inflammasome activation in microglia. J Neuroinflamm 14:256
Fellner L et al (2013) Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 61:349–360
Fiorentino L, Stehlik C, Oliveira V, Ariza ME, Godzik A, Reed JC (2002) A novel PAAD-containing protein that modulates NF-kappa B induction by cytokines tumor necrosis factor-alpha and interleukin-1beta. J Biol Chem 277:35333–35340. https://doi.org/10.1074/jbc.M200446200
Frederick Lo C, Ning X, Gonzales C, Ozenberger BA (2008) Induced expression of death domain genes NALP1 and NALP5 following neuronal injury. Biochem Biophys Res Commun 366:664–669. https://doi.org/10.1016/j.bbrc.2007.11.174
Fu Y et al (2019) NLRC3 expression in dendritic cells attenuates CD4. EMBO J 38:e101397. https://doi.org/10.15252/embj.2018101397
Garré JM, Silva HM, Lafaille JJ, Yang G (2017) CX3CR1+ monocytes modulate learning and learning-dependent dendritic spine remodeling via TNF-α. Nat Med 23:714
Gay NJ, Gangloff M, Weber AN (2006) Toll-like receptors as molecular switches. Nat Rev Immunol 6:693–698
Ghimire L, Paudel S, Jin L, Jeyaseelan S (2020) The NLRP6 inflammasome in health and disease. Mucosal Immunol 13:388–398. https://doi.org/10.1038/s41385-020-0256-z
Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE, Pahan K (2009) Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci 29:13543–13556
Gil-Martínez A-L et al (2019) Local gastrointestinal injury exacerbates inflammation and dopaminergic cell death in parkinsonian mice. Neurotox Res 35:918–930
Gil-Martínez AL, Cuenca L, Estrada C, Sánchez-Rodrigo C, Fernández-Villalba E, Herrero MT (2018) Unexpected exacerbation of neuroinflammatory response after a combined therapy in old Parkinsonian mice. Front Cell Neurosci 12:451
Giráldez-Pérez RM, Antolín-Vallespín M, Muñoz MD, Sánchez-Capelo A (2014) Models of α-synuclein aggregation in Parkinson’s disease. Acta Neuropathol Commun 2:176
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140(918):934
Goetz CG, Pal G (2014) Initial management of Parkinson’s disease. BMJ. https://doi.org/10.1136/bmj.g6258
Goldman JG, Postuma R (2014) Premotor and non-motor features of Parkinson’s disease. Curr Opin Neurol 27:434
Gordon R et al (2018) Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Trans Med 10:4066
Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687
Ha HJ, Park HH (2020) Crystal structure of the human NLRP9 pyrin domain reveals a bent N-terminal loop that may regulate inflammasome assembly. FEBS Lett. https://doi.org/10.1002/1873-3468.13866
Hafner-Bratkovič I, Pelegrín P (2018) Ion homeostasis and ion channels in NLRP3 inflammasome activation and regulation. Curr Opin Immun 52:8–17
Hanisch U-K, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394
Haque ME, Akther M, Jakaria M, Kim IS, Azam S, Choi DK (2020) Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov Disord 35:20–33
Harris J et al (2011) Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 286:9587–9597. https://doi.org/10.1074/jbc.M110.202911
He Y, Hara H, Núñez G (2016) Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 41:1012–1021
Heneka MT, McManus RM, Latz E (2018) Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19:610–621
Heneka MT, Rodríguez JJ, Verkhratsky A (2010) Neuroglia in neurodegeneration. Brain Res Rev 63(189):211
Herrero MT, Morelli M (2017) Multiple mechanisms of neurodegeneration and progression. Prog Neurobiol 155:1–1
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8:382–397
Hou X, Watzlawik JO, Fiesel FC, Springer W (2020) Autophagy in Parkinson’s disease. J Mol Biol 432:2651–2672
Howrylak JA, Nakahira K (2017) Inflammasomes: key mediators of lung immunity. Ann Rev Physiol 79:471–494
Hung Y-F, Chen C-Y, Shih Y-C, Liu H-Y, Huang C-M, Hsueh Y-P (2018) Endosomal TLR3, TLR7, and TLR8 control neuronal morphology through different transcriptional programs. J Cell Biol 217:2727–2742
Imamura R et al (2010) Anti-inflammatory activity of PYNOD and its mechanism in humans and mice. J Immunol 184:5874–5884. https://doi.org/10.4049/jimmunol.0900779
Jabir MS et al (2015) Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11:166–182. https://doi.org/10.4161/15548627.2014.981915
Janowski AM, Sutterwala FS (2016) Atypical inflammasomes. Methods Mol Biol 1417(45):62. https://doi.org/10.1007/978-1-4939-3566-6_2
Javed H et al (2020) NLRP3 inflammasome and glia maturation factor coordinately regulate neuroinflammation and neuronal loss in MPTP mouse model of Parkinson’s disease. Int Immunopharmacol 83:106441
Jiang H et al (2017) Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med 214:3219–3238. https://doi.org/10.1084/jem.20171419
Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F (2011) NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol 186:1646–1655. https://doi.org/10.4049/jimmunol.1001654
Kalia L (2015) Lang A Parkinson’s disease. Lancet 386(9996):896–912
Kalinderi K, Bostantjopoulou S, Fidani L (2016) The genetic background of Parkinson’s disease: current progress and future prospects. Acta Neurol Scand 134:314–326
Karki R et al (2016) NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature 540:583–587. https://doi.org/10.1038/nature20597
Kastner A et al (1994) Decreased tyrosine hydroxylase content in the dopaminergic neurons of MPTP-intoxicated monkeys: effect of levodopa and GM1 ganglioside therapy. Ann Neurol 36:206–214. https://doi.org/10.1002/ana.410360213
Kayagaki N et al (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121
Kazlauskaite A, Muqit MM (2015) PINK1 and Parkin–mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J 282:215–223
Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20:3328
Khare S et al (2014) The PYRIN domain–only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat Immunol 15:343–353
Kim C et al (2018) Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating α-synuclein transmission and neuroinflammation. Mol Neurodegener 13:1–18
Kim YS, Joh TH (2006) Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp Mol Med 38:333–347
Kirkley KS, Popichak KA, Afzali MF, Legare ME, Tjalkens RB (2017) Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J Neuroinflamm 14:1–18
Koprich JB, Reske-Nielsen C, Mithal P, Isacson O (2008) Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J Neuroinflamm 5:8
Koshimori Y et al (2015) Imaging striatal microglial activation in patients with Parkinson’s disease. PLoS ONE 10:e0138721
Kuchmiy AA, D’Hont J, Hochepied T, Lamkanfi M (2016) NLRP2 controls age-associated maternal fertility. J Exp Med 213:2851–2860
Labzin LI, Heneka MT, Latz E (2018) Innate immunity and neurodegeneration. Annu Rev Med 69:437–449
Laliberte R, Perregaux D, Svensson L, Pazoles CJ, Gabel CA (1994) Tenidap modulates cytoplasmic pH and inhibits anion transport in vitro. II. Inhibition of IL-1 beta production from ATP-treated monocytes and macrophages. J Immunol 153:2168–2179
Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157(1013):1022
Lane JD, Korolchuk VI, Murray JT, Karabiyik C, Lee MJ, Rubinsztein DC (2017) Autophagy impairment in Parkinson’s disease. Essays Biochem 61(711):720
Lang Y, Chu F, Shen D, Zhang W, Zheng C, Zhu J, Cui L (2018) Role of inflammasomes in neuroimmune and neurodegenerative diseases: a systematic review. Mediat Inflamm. https://doi.org/10.1155/2018/1549549
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411
Lau A et al (2020) α-Synuclein strains target distinct brain regions and cell types. Nat Neurosci 23:21–31
Lee E et al (2019) MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ 26:213–228
Levy M et al (2015) Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163:1428–1443. https://doi.org/10.1016/j.cell.2015.10.048
Li R, Zhu S (2020) NLRP6 inflammasome. Mol Aspects Med. https://doi.org/10.1016/j.mam.2020.100859
Lim CK et al (2017) Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog Neurobiol 155:76–95. https://doi.org/10.1016/j.pneurobio.2015.12.009
Lin K-M et al (2014) IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci 111:775–780
Lu Y, Li X, Liu S, Zhang Y, Zhang D (2018) Toll-like receptors and inflammatory bowel disease. Front Immunol 9:72
Macdonald R, Barnes K, Hastings C, Mortiboys H (2018) Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: can mitochondria be targeted therapeutically? Biochem Soc Trans 46:891–909
Madore C, Yin Z, Leibowitz J, Butovsky O (2020) Microglia, lifestyle stress, and neurodegeneration. Immunity 52:222–240
Man SM, Kanneganti TD (2015) Regulation of inflammasome activation. Immunol Rev 265(6):21
Mangan MS, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E (2018) Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 17:588
Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H, Xue Z (2017) The NLRP3 inflammasome is involved in the pathogenesis of Parkinson’s disease in rats. Neurochem Res 42:1104–1115
Marinelli C et al (2015) Ligand engagement of Toll-like receptors regulates their expression in cortical microglia and astrocytes. J Neuroinflamm 12:1–20
Martinez EM et al (2017) Editor’s highlight: Nlrp3 is required for inflammatory changes and nigral cell loss resulting from chronic intragastric rotenone exposure in mice. Toxicol Sci 159:64–75
Martinez-Martin P, Rodriguez-Blazquez C, Forjaz MJ, Kurtis MM, Skorvanek M (2017) Measurement of nonmotor symptoms in clinical practice. International review of neurobiology, vol 133. Elsevier, New York, pp 291–345
Masuda T et al (2019) Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566:388–392
McGeer PL, Itagaki S, Akiyama H, McGeer EG (1988) Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol 24(574):576
McGeer PL, Yasojima K, McGeer EG (2002) Association of interleukin-1β polymorphisms with idiopathic Parkinson’s disease. Neurosci Lett 326:67–69
Menzies FM et al (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93:1015–1034
Mitchell PS, Sandstrom A, Vance RE (2019) The NLRP1 inflammasome: new mechanistic insights and unresolved mysteries. Curr Opin Immunol 60:37–45
Miyamoto A et al (2016) Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun 7(1):12
Mu J et al (2019) Mutations in NLRP2 and NLRP5 cause female infertility characterised by early embryonic arrest. J Med Genet 56:471–480
Newman AB, Sanders JL, Kizer JR, Boudreau RM, Odden MC, Zeki Al Hazzouri A, Arnold AM (2016) Trajectories of function and biomarkers with age: the CHS All Stars Study. Int J Epidemiol 45:1135–1145
Obeso J et al (2017) Past, present, and future of Parkinson’s disease: a special essay on the 200th anniversary of the shaking palsy. Mov Dis 32:1264–1310
Paolicelli RC, Ferretti MT (2017) Function and dysfunction of microglia during brain development: consequences for synapses and neural circuits. Front Synap Neurosci 9:9
Parkinson J (1817) An essay on the Shaking Palsy. Sherwood, Neely and Jones, London
Parthasarathy G, Philipp MT (2018) Intracellular TLR7 is activated in human oligodendrocytes in response to Borrelia burgdorferi exposure. Neurosci Lett 671:38–42
Peng H, Liu F, Li W, Zhang W (2015) Knockdown of NLRP5 arrests early embryogenesis in sows. Anim Reprod Sci 163:151–156. https://doi.org/10.1016/j.anireprosci.2015.11.004
Platnich JM, Muruve DA (2019) NOD-like receptors and inflammasomes: a review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys 670:4–14
Poewe W et al (2017) Parkinson disease. Nat Rev Dis Primers 3(1):21
Prinz M, Jung S, Priller J (2019) Microglia biology: one century of evolving concepts. Cell 179:292–311
Puspita L, Chung SY, Shim J-W (2017) Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain 10:53
Py BF, Kim M-S, Vakifahmetoglu-Norberg H, Yuan J (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49:331–338
Qiao C, Zhang L-X, Sun X-Y, Ding J-H, Lu M, Hu G (2017) Caspase-1 deficiency alleviates dopaminergic neuronal death via inhibiting caspase-7/AIF pathway in MPTP/p mouse model of Parkinson’s disease. Mol Neurob 54:4292–4302
Radian AD, de Almeida L, Dorfleutner A, Stehlik C (2013) NLRP7 and related inflammasome activating pattern recognition receptors and their function in host defense and disease. Microb Infect 15:630–639. https://doi.org/10.1016/j.micinf.2013.04.001
Ramirez-Zamora A, Molho E (2014) Treatment of motor fluctuations in Parkinson’s disease: recent developments and future directions. Expert Rev Neurother 14:93–103
Rani L, Mondal AC (2020) Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: pathogenic and therapeutic implications. Mitochondrion 50:25–34
Ratsimandresy RA et al (2017) The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat Commun 8:1–15
Reichmann H (2017) Premotor diagnosis of Parkinson’s disease. Neurosci Bull 33(526):534
Ren G et al (2019) ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J 38:e100376
Rodriguez-Violante M et al (2017) Premotor symptoms and the risk of Parkinson’s disease: a case-control study in Mexican population. Clin Neurol Neurosurg 160:46–49
Romberg N et al (2014) Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 46:1135–1139
Roussakis A-A, Piccini P (2018) Molecular Imaging of Neuroinflammation in Idiopathic Parkinson’s Disease. International review of neurobiology, vol 141. Elsevier, New York, pp 347–363
Salamanca L, Mechawar N, Murai KK, Balling R, Bouvier DS, Skupin A (2019) MIC-MAC: an automated pipeline for high-throughput characterization and classification of three-dimensional microglia morphologies in mouse and human postmortem brain samples. Glia 67:1496–1509
Salter MW, Stevens B (2017) Microglia emerge as central players in brain disease. Nat Med 23:1018
Saresella M et al (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener 11:23
Sarkar S et al (2017) Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinson’s Dis 3:1–15
Savage JC, Carrier M, Tremblay M-È (2019) Morphology of microglia across contexts of health and disease. microglia. Springer, New York, pp 13–26
Schapira AH, Chaudhuri KR, Jenner P (2017) Non-motor features of Parkinson disease. Nat Rev Neurosci 18:435
Scheiblich H, Trombly M, Ramirez A, Heneka MT (2020) Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol 41:300–312
Schlachetzki J, Marxreiter F, Regensburger M, Kulinich A, Winner B, Winkler J (2014) Increased tyrosine hydroxylase expression accompanied by glial changes within the non-lesioned hemisphere in the 6-hydroxydopamine model of Parkinson’s disease. Restor Neurol Neurosci 32:447–462
Schneider M et al (2012) The innate immune sensor NLRC3 attenuates toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol 13:823–831. https://doi.org/10.1038/ni.2378
Schrag A, Horsfall L, Walters K, Noyce A, Petersen I (2015) Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol 14:57–64
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140(821):832
Segura-Aguilar J (2017) On the role of endogenous neurotoxins and neuroprotection in Parkinson’s disease. Neural Regener Res 12:897
Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C (2015) NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol 6:262. https://doi.org/10.3389/fphar.2015.00262
Sharma D, Kanneganti T-D (2016) The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol 213:617–629
Shen H et al (2020) New mechanism of neuroinflammation in Alzheimer’s disease: the activation of NLRP3 inflammasome mediated by gut microbiota. Prog Neuro Psychopharmacol Biol Psychiatry 100:109884
Shi C-S et al (2012) Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263
Shi J et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665
Shim D-W, Lee K-H (2018) Posttranslational regulation of the NLR family pyrin domain-containing 3 inflammasome. Front Immunol 9:1054
Silvin A, Ginhoux F (2018) Microglia heterogeneity along a spatio–temporal axis: more questions than answers. Glia 66:2045–2057
Simon DK, Tanner CM, Brundin P (2019) Parkinson Disease epidemiology, pathology, genetics and pathophysiology. Clin Geriatr Med. https://doi.org/10.1016/j.cger.2019.08.002
Sominsky L, De Luca S, Spencer SJ (2018) Microglia: key players in neurodevelopment and neuronal plasticity. Int J Biochem Cell Biol 94:56–60
Song S et al (2019a) Loss of brain norepinephrine elicits neuroinflammation-mediated oxidative injury and selective caudo-rostral neurodegeneration. Mol Neurobiol 56:2653–2669. https://doi.org/10.1007/s12035-018-1235-1
Song S et al (2019b) Noradrenergic dysfunction accelerates LPS-elicited inflammation-related ascending sequential neurodegeneration and deficits in non-motor/motor functions. Brain Behav Immun 81:374–387
Su MY, Kuo CI, Chang CF, Chang CI (2013) Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PLoS ONE 8:e67843. https://doi.org/10.1371/journal.pone.0067843
Subhramanyam CS, Wang C, Hu Q, Dheen ST (2019) Microglia-mediated neuroinflammation in neurodegenerative diseases. Seminars in cell and developmental biology. Elsevier, New York, pp 112–120
Sutterwala FS, Haasken S, Cassel SL (2014) Mechanism of NLRP3 inflammasome activation. Ann NY Acad Sci 1319:82
Taetzsch T et al (2015) Redox regulation of NF-κB p50 and M1 polarization in microglia. Glia 63:423–440
Tan EK, Chao YX, West A, Chan LL, Poewe W, Jankovic J (2020) Parkinson disease and the immune system—associations, mechanisms and therapeutics. Nat Rev Neurol 16(6):303–318. https://doi.org/10.1038/s41582-020-0344-4
Tarakad A, Jankovic J (2017) Diagnosis and management of Parkinson’s disease. Seminars in neurology, vol 02. Thieme Medical Publishers, New York, pp 118–126
Tay TL, Hagemeyer N, Prinz M (2016) The force awakens: insights into the origin and formation of microglia. Curr Opin Neurobiol 39:30–37
Tian X, Pascal G, Monget P (2009) Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol Biol 9:202. https://doi.org/10.1186/1471-2148-9-202
Tilburgs T, Meissner TB, Ferreira LM, Mulder A, Musunuru K, Ye J, Strominger JL (2017) NLRP2 is a suppressor of NF-ƙB signaling and HLA-C expression in human trophoblasts. Biol Reprod 96:831–842
Tong H, Zhang X, Meng X, Lu L, Mai D, Qu S (2018) Simvastatin inhibits activation of NADPH oxidase/p38 MAPK pathway and enhances expression of antioxidant protein in Parkinson disease models. Front Mol Neurosci 11:165
Torres-Platas SG et al (2014) Morphometric characterization of microglial phenotypes in human cerebral cortex. J Neuroinflamm 11:12
Tsarouchas TM et al (2018) Dynamic control of proinflammatory cytokines Il-1β and Tnf-α by macrophages in zebrafish spinal cord regeneration. Nat Commun 9:1–17
Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10:210–215
Tuladhar S, Kanneganti TD (2020) NLRP12 in innate immunity and inflammation. Mol Aspects Med. https://doi.org/10.1016/j.mam.2020.100887
Vanaja SK, Rathinam VA, Fitzgerald KA (2015) Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 25:308–315
von Herrmann KM et al (2018) NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinson’s Dis 4:1–9
Verkhratsky A, Nedergaard M (2018) Physiology of astroglia. Physiol Rev 98(239):389
Vlaar T, Kab S, Schwaab Y, Fréry N, Elbaz A, Moisan F (2018) Association of Parkinson’s disease with industry sectors: a French nationwide incidence study. Eur J Epidemiol 33:1101–1111
Wang Y et al (2014) Interleukin-1β induces blood–brain barrier disruption by downregulating sonic hedgehog in astrocytes. PLoS ONE 9:e110024
Wang P et al (2015) Nlrp6 regulates intestinal antiviral innate immunity. Science 350:826–830
Wang L et al (2017) A comprehensive data mining study shows that most nuclear receptors act as newly proposed homeostasis-associated molecular pattern receptors. J Hematol Oncol 10:168
Wang Z et al (2020a) NLRP3 inflammasome and inflammatory diseases. Oxid Med Cell Longev. https://doi.org/10.1155/2020/4063562
Wang B, Bhattacharya M, Roy S, Tian Y, Yin Q (2020b) Immunobiology and structural biology of AIM2 inflammasome. Mol Aspects Med. https://doi.org/10.1016/j.mam.2020.100869
Wang Y, Hasegawa M, Imamura R, Kinoshita T, Kondo C, Konaka K, Suda T (2004) PYNOD, a novel Apaf-1/CED4-like protein is an inhibitor of ASC and caspase-1. Int Immunol 16:777–786. https://doi.org/10.1093/intimm/dxh081
Wang B, Tian Y, Yin Q (2019) AIM2 inflammasome assembly and signaling. Structural immunology. Springer, New York, pp 143–155
Westerveld GH, Korver CM, van Pelt AM, Leschot NJ, van der Veen F, Repping S, Lombardi MP (2006) Mutations in the testis-specific NALP14 gene in men suffering from spermatogenic failure. Hum Reprod 21:3178–3184. https://doi.org/10.1093/humrep/del293
Wlodarska M et al (2014) NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156:1045–1059
Wolf SA, Boddeke H, Kettenmann H (2017) Microglia in physiology and disease. Annu Rev Physiol 79:619–643
Xiang H, Zhu F, Xu Z, Xiong J (2020) Role of inflammasomes in kidney diseases via both canonical and non-canonical pathways. Front Cell Develop Biol 8:106
Yan X et al (2017) Vanillin protects dopaminergic neurons against inflammation-mediated cell death by inhibiting ERK1/2, p38 and the NF-κB signaling pathway. Int J Mol Sci 18:389
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R (2015) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160:62–73
Yang X, Lin G, Han Z, Chai J (2019a) Structural biology of NOD-like receptors. Structural immunology. Springer, New York, pp 119–141
Yang Y, Wang H, Kouadir M, Song H, Shi F (2019b) Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 10:1–11
Yao Y et al (2012) NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res 22:836–847. https://doi.org/10.1038/cr.2012.56
Yap JKY, Pickard BS, Chan EWL, Gan SY (2019) The role of neuronal NLRP1 inflammasome in Alzheimer’s disease: bringing neurons into the neuroinflammation game. Mol Neurobiol 56:7741–7753
Yin Y et al (2020) A noncanonical role of NOD-like receptor NLRP14 in PGCLC differentiation and spermatogenesis. Proc Natl Acad Sci USA 117:22237–22248. https://doi.org/10.1073/pnas.2005533117
Zahid A, Li B, Kombe AJK, Jin T, Tao J (2019) Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol 10:2538. https://doi.org/10.3389/fimmu.2019.02538
Zaki MH et al (2011) The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20:649–660. https://doi.org/10.1016/j.ccr.2011.10.022
Zeng Q, Hu C, Qi R, Lu D (2018) PYNOD reduces microglial inflammation and consequent neurotoxicity upon lipopolysaccharides stimulation. Exp Ther Med 15(5337):5343. https://doi.org/10.3892/etm.2018.6108
Zhang P et al (2016a) C dk5-Dependent Activation of Neuronal Inflammasomes in Parkinson’s Disease. Mov Disord 31:366–376
Zhang Q et al (2020) Kynurenine regulates NLRP2 inflammasome in astrocytes and its implications in depression. Brain Behav Immun. https://doi.org/10.1016/j.bbi.2020.04.016
Zhang C-W, Hang L, Yao T-P, Lim K-L (2016b) Parkin regulation and neurodegenerative disorders. Front Aging Neurosci 7:248
Zheng D, Liwinski T, Elinav E (2020) Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov 6:1–22
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225
Zhu S et al (2017) Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546:667–670. https://doi.org/10.1038/nature22967
Acknowledgements
F.M.A. thanks Coordenação de Apoio de Pessoal de Nível Superior (PDSE -47/2017) for its support; L.C.B. thanks the Spanish Ministry of Science, Innovation and Universities (FPU 18/02549) for its support; M.T.H. thanks the Federación Española de Parkinson (FIS PI13 01293) and the Fundación Séneca (19540/PI/14) for its support; V.D.A.S and S.L.C. thank Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ E. Universal/2018—429127/2018-9; and CNPQ—Research Fellowship) and Fundação de Amparo à Pesquisa do Estado da Bahia (JCB0057/2016) for its support.
Author information
Authors and Affiliations
Contributions
Both FMA, LC, and EF performed literature research and wrote the review. SLC, VDAS and MTH helped with the conceptualization of the review and performed literature research.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
de Araújo, F.M., Cuenca-Bermejo, L., Fernández-Villalba, E. et al. Role of Microgliosis and NLRP3 Inflammasome in Parkinson’s Disease Pathogenesis and Therapy. Cell Mol Neurobiol 42, 1283–1300 (2022). https://doi.org/10.1007/s10571-020-01027-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-020-01027-6