
Abstract
It is estimated that nearly a third of people who abuse drugs started with prescription opioid medicines. Approximately, 11.5 million Americans used prescription drugs recreationally in 2016, and in 2018, 46,802 Americans died as the result of an opioid overdose, including prescription opioids, heroin, and illicitly manufactured fentanyl (National Institutes on Drug Abuse (2020) Opioid Overdose Crisis. https://www.drugabuse.gov/drugs-abuse/opioids/opioid-overdose-crisis. Accessed 06 June 2020). Yet physicians will continue to prescribe oral opioids for moderate-to-severe pain in the absence of alternative therapeutics, underscoring the importance in understanding how drug choice can influence detrimental outcomes. One of the opioid prescription medications that led to this crisis is oxycodone, where misuse of this drug has been rampant. Being one of the most highly prescribed opioid medications for treating moderate-to-severe pain as reflected in the skyrocketed increase in retail sales of 866% between 1997 and 2007, oxycodone was initially suggested to be less addictive than morphine. The false-claimed non-addictive formulation of oxycodone, OxyContin, further contributed to the opioid crisis. Abuse was often carried out by crushing the pills for immediate burst release, typically by nasal insufflation, or by liquefying the pills for intravenous injection. Here, we review oxycodone pharmacology and abuse liability as well as present the hypothesis that oxycodone may exhibit a unique pharmacology that contributes to its high likability and abuse susceptibility. We will discuss various mechanisms that likely contribute to the high abuse rate of oxycodone including clinical drug likability, pharmacokinetics, pharmacodynamics, differences in its actions within mesolimbic reward circuity compared to other opioids, and the possibility of differential molecular and cellular receptor interactions that contribute to its selective effects. We will also discuss marketing strategies and drug difference that likely contributes to the oxycodone opioid use disorders and addiction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oxycodone belongs to the family of one of the most powerful analgesic compounds available for treating moderate-to-severe pain. Chemically similar to morphine, oxycodone was first synthesized in 1916 in Germany and is classified as a semi-synthetic opioid, because it is created by a chemical modification of the opium poppy alkaloid, thebaine (Fig. 1). Oxycodone was approved by the US Food and Drug Administration (FDA) as a schedule II narcotic analgesic; the FDA is tasked with protecting the public health by ensuring the safety, efficacy, and security of human and veterinary drug. The first formulation containing oxycodone produced by Merck in 1939 was combined with scopolamine and ephedrine, but the company discontinued it in 1987 (Defalque et al. 2003). Today (2020), oxycodone is marketed alone or as controlled/extended release (ER) (OxyContin®) and immediate release formulations (OxyIR®, OxyFast®), or in combination with other nonnarcotic analgesics such as aspirin (Percodan®) or acetaminophen (Percocet®). Many pharmaceutical companies have made attempts to formulate abuse-deterrent drugs in an effort to prevent opioid abuse. Prior to declaring bankruptcy, Purdue Pharma incorporated physical and chemical barriers in several of their opioid formulations, including OxyContin® OP and Hysingla™ ER. However, despite strong efforts to prevent substance abuse through various designs, abusers have found ways to circumvent the deterrents, such as microwaving the pills to melt them, or freezing them, followed by shaving with a razor (Cicero and Ellis 2015). In addition, most of the heroin-using subjects transitioned from oxycodone to heroin because of the difficulties in procuring extended release oxycodone, both in respect to its availability and cost (Cicero et al. 2013; Remillard et al. 2019).

Metabolism of thebaine and oxycodone. Oxycodone is derived from thebaine and is metabolized in humans by hepatic cytochrome P450 (CYP) isoenzymes into three major metabolites. Oxycodone is either N-demethylated by the cytochrome P450 CYP-3A4/5 to noroxycodone or O-demethylated by CYP-2D6 to oxymorphone. Then, oxymorphone is converted into noroxymorphone by CYP-3A4/5 (Lemberg et al. 2008)
Oxycodone is often prescribed to treat pain associated with acute traumatic injuries (e.g., broken bone), post-operative pain, and cancer pain, including bone cancer pain (Zech et al. 1995; King et al. 2011; Bercovitch and Adunsky 2006; Moradi et al. 2012). In contrast to morphine, oxycodone gained popularity in clinical practice because of its ability to improve the quality of life of patients under constant chronic pain,Footnote 1 because of less overall side effects compared to morphine (Kalso and Vainio 1990; Riley et al. 2008; Hamunen et al. 2009; Moradi et al. 2012; Caraceni et al. 2013). Indeed, it has been increasingly prescribed for alleviating certain chronic neuropathic non-cancer pain syndromes such as those caused by post-herpetic neuralgia (Pappagallo et al. 1994; Watson and Babul 1998; Kalso 2005; Gavin et al. 2017), diabetic neuropathy (Watson et al. 2003; Kalso 2005; Minami et al. 2009), back pain, or osteoarthritis (Taylor et al. 2012). However, there is a lack of evidence regarding the long-term effectiveness of oxycodone (or any opioid treatment) in diminishing chronic non-cancer pain, and long-term oxycodone therapy involves serious health and dependency risks (Ballantyne 2015).Footnote 2 The preference of oxycodone over morphine as an analgesic in pain management is reflected in its consumer sales (See Footnote 1).Footnote 3 It is likely that marketing and promotion contributed to this preference, rather than it being solely driven by consumer choice.
Oxycodone-Related Deaths and Driving Factors That Got Us Here
Morbidity and mortality specifically involving oxycodone are undisputed. Oxycodone is the third leading opioid (behind heroin and fentanyl) that has contributed to overdose deaths in America (See Footnote 3).Footnote 4 From 2011 to 2016, there was a total of 33,154 overdose deaths (4967–6199 annually) in America4 involving oxycodone overdose,Footnote 5 although many deaths involve synergy with other drugs such as alcohol, benzodiazepines, or other sedatives. In terms of morbidity, an estimate of 49,609 emergency department visits per year involving non-medical use of oxycodone products was suggested by NEISS-CADES data. One of the reasons for such high morbidity and mortality rates is attributed to the fact that oxycodone is often diverted and abused by street users to alleviate or prevent the onset of opiate withdrawal (Evans and Cahill 2016). According to a DEA report published in July 2019,Footnote 6 oxycodone products such as the 40-mg OxyContin® tablet became the most frequently encountered pharmaceutical drug by law enforcement in 2009 and are still among the most identified pharmaceutical drugs by federal, state, and local forensic laboratories in the USA (See Footnote 5). Likewise, data on the consumption and sale of oxycodone reflect the major role of oxycodone in growth of the opioid epidemic. It was the second most prescribed opioid in clinics after hydrocodone in 2016 according to a FDA analysis published on May 1st, 2018.Footnote 7 In 2018, the total worldwide consumption of oxycodone (prescribed and illegal) reached an all-time high of 45,717 defined daily doses for statistical purposes, placing oxycodone as the second most consumed opioid after fentanyl. While the use of oxycodone increased, its total quantities manufactured reduced in 2018. The difference between the total quantities of oxycodone produced and those consumed was suggested to be due to large manufacturing losses during the processing of some semi-synthetic opioids.Footnote 8 The analgesic effectiveness and the rewarding and euphoric properties of oxycodone as the driving force for abuse, diversion, and addiction of oxycodone are among the scopes of our review. Nonetheless, it is important to mention that these intrinsic values of oxycodone are not the only factors responsible for the increased morbidity and mortality associated with the opioid.
Misleading marketing presented oxycodone as being non-addictive to physicians and consumers. This generated the high rates of prescription, then abuse and diversion of the drug (Van Zee 2009; Severino et al. 2018). It all started when Purdue Pharma reformulated and manufactured the extended release version of oxycodone, OxyContin®, whose distribution and release was approved by the FDA in 1995 in the USA.Footnote 9 Purdue Pharma based its marketing strategy on the claim that a decrease dosing interval (made available through an extended release formulation) would reduce its abuse liability. Indeed, the original label read the following: “Delayed absorption as provided by OxyContin tablets, is believed to reduce the abuse liability of a drug”.Footnote 10 Purdue Pharma not only used this claim to persuade physicians to prescribe OxyContin® for many yearsFootnote 11 but also organized more than 40 national pain management and speaker training conferences from 1996 to 2001 to recruit more physicians to prescribe OxyContin®Footnote 12 (Orlowski and Wateska 1992; Van Zee 2009). Misguided dosing recommendations also contributed to the higher abuse liability of OxyContin®. OxyContin® was marketed as a drug to be dosed every 12 h; however, the pharmacokinetics did not support this dosing regimen. An FDA post-market analysis identified that the actual half-life of serum drug levels was as little as 4.5 h,Footnote 13 which resulted in misuse of the prescription opioid, where patients started self-medicating by taking it earlier than the prescribed dosing schedule to either treat break-through pain or circumvent withdrawal symptoms (e.g., nausea, diarrhea, vomiting, lacrimation, and rhinorrhea as well as mood changes such as anxiety and depression). The lack of adequate dosing had the potential for patients to go through repeated withdrawal, which is now recognized as an underlying factor in opioid-induced adaptive changes in brain structures important in decision-making, reward, and motivation (Lee et al 2018; Lefevre et al 2020; Cahill 2020). Another important cornerstone of Purdue Pharma’s marketing plan was to target health care professionals with specifically high rates of opioid prescriptions. By building a database on the prescription habits of physicians who were prescribing opioids for treating chronic pain patients, the pharmaceutic company increased the number of sales representative visits to these high opioid prescribers (See Footnote 12) (Orlowski and Wateska 1992; Van Zee 2009) and provided promotional items such as brochures, OxyContin® fishing hats, stuffed plush toys, and music compact disksFootnote 14 (Van Zee 2009). Hence, in addition to the aggressive physician-directed marketing, another foundation of Purdue’s marketing was the direct-to-consumer pharmaceutical advertising, which not only includes brochures distribution from physicians’ offices, but also television, print (magazines, newspapers), radio, the Internet, and other forms of mass media, such as billboards and direct mailings (Abel et al. 2006; Connors 2009; Ventola 2011; Severino et al. 2018). Both direct-to-consumer pharmaceutical advertising, which was only allowed in the USA and New Zealand, combined with influencing the prescribing practice of physicians, contributed to mass consumer awareness of the availability of drugs, such as oxycodone (Ventola 2011; Fain and Alexander 2014; Severino et al. 2018). In the end, Purdue’s marketing blitz was successful, and OxyContin® became one of the top prescribed opioids in the USA.12 Sales grew from $48 million in 1996 to approximately $1.1 billion in 2000 (Van Zee 2009). Unfortunately, by the early 2000s (from 1999 to 2002), there was a dramatic rise in opioid overdoses and deaths from prescription opioids, especially related to OxyContin® (Paulozzi et al. 2006). The number of death certificate poisoning occurrences related to prescription opioids increased by 57.4% concurrently with oxycodone sales, which were the highest (130.3%) among prescription opioids (Paulozzi et al. 2006). The increase in mortality triggered the FDA to respond, where from 2003 to 2017, it issued several warnings to the manufacturer about misleading advertising, lack of warning statements regarding the addictive nature of the drug, and the overstated efficacy of the drug. Purdue ultimately faced criminal and civil charges by the US Department of Justice for false marketing claims that Oxycontin® was less addictive than other medications.Footnote 15,Footnote 16,Footnote 17 On May 10, 2007, Purdue Frederick Company Inc, a holding of Purdue Pharma, along with 3 company executives, pled guilty to misbranding OxyContin® and agreed to pay $634 million in finesFootnote 18 (Van Zee 2009; Severino et al. 2018). In the report of the US case court, the USA vs. The Purdue Frederick Company (See Footnote 15), it was revealed that Purdue Frederick Co. had conducted a study that showed that a drug abuser could extract nearly 68% of oxycodone from a 10-mg OxyContin® tablet by crushing the tablet, stirring it in water, and drawing the solution through cotton into a syringe. This motivated the development, in 2010, of a new formulation of Oxycontin®, an abuse-deterrent formulation (ADF) that is harder to crush and, thus, supposedly with less addictive and abuse potential as demonstrated by several studies (Cone et al. 2013; Harris et al. 2014). Unfortunately, the new formulation could still be abused. Patients take intact tablets orally or transition to other addictive drugs such as heroinFootnote 19,Footnote 20 (Harris et al. 2014; Dart et al. 2015; Cicero and Ellis 2015), leading to the current wave of the unprecedented opioid-related overdose death toll. All the efforts concentrated on designing improved ADF of Oxycontin® to diminish the potential of diversion after extraction (e.g., chewing/crushing) do not address the high abuse liability of oxycodone associated to the intrinsic “feel-good effect” or “like effect” property of the drug. Indeed, studies have suggested that oxycodone has now become a highly likable analgesic by drug abusers (Wightman et al. 2012). While morphine and oxycodone appear to be equally effective and equipotent in some types of pain (e.g., post-operative pain and cancer pain) (Ruan et al. 2017; Guo et al. 2018), a plethora of clinical reports in the literature demonstrating the undeniable drug abusers’ preference for oxycodone in comparison with other medical and non-medical opioids (Siegal et al. 2003; Grau et al. 2007; Zancy and Gutierrez 2003, 2008, 2009; Rosenblum et al. 2007; Comer et al. 2008; Katz et al. 2008; Zacny and Lichtor 2008; Pollini et al. 2011; Cicero et al. 2012; Osgood et al. 2012; Wightman et al. 2012; Young and Havens 2012; Atluri et al. 2014; Mars et al. 2014; Setnik et al. 2015; Carlson et al. 2016; Remillard et al. 2019). These studies were conducted in non-opioid abusing subjects (Zancy and Gutierrez 2003, 2008, 2009; Zacny et al. 2003; Zacny and Lichtor 2008), heroin-dependent individuals (Comer et al. 2008), prescription opioid-dependent patients or abusers (Cicero et al. 2010, 2013), recreational opioid users (Setnik et al. 2015), and methadone-treated patients (Rosenblum et al. 2007). Two systematic reviews search in EMBASE and MEDLINE databases by Wightman and collaborators and by Remillard and associates and compiled articles that investigated the comparative likability and/or abuse potential of the most commonly prescribed opioids, including oxycodone (Wightman et al. 2012; Remillard et al. 2019). The authors highlighted that oxycodone has a significantly elevated abuse liability profile due to high likability scores combined with fewer negative subjective side effects compared to morphine and hydrocodone (Wightman et al. 2012; Remillard et al. 2019). For example, eight heroin-dependent individuals (five men, three women) maintained on 120 mg/day of oral morphine rated the intravenous oxycodone as being the most ‘potent’ compared with morphine due to it producing positive subjective dose-related experience (i.e., positive subjective ratings such as ‘I feel a good drug effect’, ‘I like the drug’, ‘I want to take the drug again’) over (in descending order) heroin, buprenorphine, and fentanyl (Comer et al. 2008). Interestingly, oxycodone had the least aversive drug side effects compared to the other opioids (i.e., ‘I feel a bad drug effect’). The larger gap between the pleasurable effect of oxycodone (i.e., positive subjective ratings) and the lack of bad emotional effects (i.e., ‘feel bad’) is the reason why oxycodone comes first in term of preferability and propensity for abuse.
The reports in recreational and opioid-dependent individuals is similar to that reported by patients prescribed opioids for pain treatment. Patients taking oxycodone for alleviating pain report common opioid-related adverse effects (e.g., constipation, nausea, dizziness, etc.); however, the use of oxycodone in many types of pain treatments causes fewer side effects than several other opioids. For instance, patients with severe cancer pain experienced less nausea and vomiting with controlled release oxycodone than with controlled release morphine (Kalso and Vainio 1990; Heiskanen and Kalso 1997) or immediate release (IR) oxycodone/acetaminophen (Caldwell et al. 1999) as well as fewer hallucinations than either controlled release morphine (Kalso and Vainio 1990; Maddocks et al. 1996; Mucci-LoRusso et al. 1998; Lauretti et al. 2003) or controlled release hydromorphone (Hagen and Babul 1997), and less itching compared to controlled release morphine (Mucci-LoRusso et al. 1998). In post-operative patients with major abdominal surgery, intravenous oxycodone caused less sedation than morphine (Kalso et al. 1991; Riley et al 2008; Lenz et al. 2009; Raff et al. 2019). Oxycodone is also better tolerated than tramadol in post-operative pain surgery (Wirz et al. 2005; Riley et al. 2008). Thus, the better analgesic efficacy of oxycodone combined with less deleterious physical side effects appears to explain why oxycodone became the top choice in moderate-to-severe clinical pain management.
The preferential use of oxycodone to treat many types of pain increases the availability of the drug to consumers, which is important to consider regarding the propensity of oxycodone to be abused. The absence or low level of negative effects of oxycodone in pain patients is a positive feature where it could contribute to greater compliance; however, this same preferred side effect profile has contributed to the higher abuse associated with this drug. This is an important consideration given that a number of clinical trials show better outcomes with perioperative oxycodone compared to other opioids such as remifentanil and fentanyl. For example, patient satisfaction scores, pain ratings and rescue medications were significantly lower in patients that received oxycodone compared to those patients that were given remifentanil for a percutaneous radiofrequency ablation used to treat hepatic cancer (Wu et al. 2019). Similarly, in a randomized double-blind study on patients undergoing total hip replacement, patients with oxycodone administered 20 min prior to the end of surgery had lower pain ratings and required less cumulative opioid requirement post-surgery compared to those that received fentanyl (Kim et al. 2018).
High Likability (‘Liking’), Incentive Salience (‘Wanting’) and Abuse Liability of Oxycodone
High Likability and Incentive Salience of Oxycodone Assessed by Behavioral Studies in Humans
Before discussing likability and incentive salience, it is important to understand the pharmacokinetic and analgesic profile of oxycodone compared to other opioids. Most morphine analgesic equivalent charts report equianalgesic dosing between oxycodone and morphine for parenteral administration, but oxycodone is 1.5 times more potent than morphine following oral administrationFootnote 21 (Bruera et al. 1998; Hanks et al. 2001; Zacny and Lichtor 2008). Oral oxycodone has better bioavailability (about 60–90%) than morphine and with the same unbound concentrations of oxycodone and morphine in the blood, unbound oxycodone in the brain is as much as six times higher than morphine (Boström et al. 2008). Preclinical research reports differences in potency, where oxycodone was 2 to 4 times more potent than morphine after subcutaneous and intraperitoneal injection in rats (Pöyhiä and Kalso 1992). Interestingly, oxycodone is 10 times less potent than morphine when administered epidurally (Backlund et al. 1997; Plummer et al. 1990; Kalso 2005), suggesting that the primary difference that accounts for the oxycodone enhanced analgesia is due to activity in the brain, rather than altering pain transmission at the spinal level. This is also born out in rodents (Lemberg et al. 2008) where intrathecal oxycodone has about a 50th the analgesic potency of morphine and the oxycodone metabolites oxymorphone and noroxymorphone (Fig. 1), yet when given subcutaneously oxycodone is a more potent than all these other opioid chemical entities. The unique analgesic effectiveness of oxycodone compared to other opioid medications could be an important component underlying the abuse potential of oxycodone. The enhanced analgesic efficacy of systemic administration of oxycodone, over other opioids, was reported in many clinical studies. For example, a systematic literature search of PubMed, Cochrane Library, and EMBASE databases for randomized controlled trials published from 2008 through 2017 revealed that post-operative intravenous oxycodone provides higher analgesic efficacy than fentanyl and sufentanil, and comparable efficacy to morphine, with less adverse events such as sedation (Raff et al. 2019).
As stated by Wightman and colleagues, the term ‘likability’ or ‘liking’ generally represents the positive psychoactive component of a drug's subjective effects (Wightman et al. 2012; Kuypers et al. 2018). Berridge defines ‘liking’ as the hedonic impact of sensory pleasures or the pleasure generated from a stimulus/reward (Nesse and Berridge 1997; Berridge et al. 2009; Berridge and Kringelbach 2015; Anselme and Robinson 2016; Berridge and Robinson 2016; Castro and Berridge 2017; Berridge 2018). Terms that are synonyms of positive psychoactive effects of a drug are euphoria, feel-good, feeling-high, dreamy, elated, carefree, coasting, mellow, social, stimulated, pleasant thoughts, invincibility, and pleasurable physical sensations (Wightman et al. 2012; Remillard et al. 2019). Abuse propensity is also intricately linked to the presence or absence of negative subjective effects. Thus, the abuse liability of a drug has been assumed to depend on the balance between net positive and negative subjective effects. The more a substance induces enjoyable subjective emotional experience without uncomfortable negative effects, the more the substance is liked. Drug ‘liking’ (and ‘wanting’) will also have more salience in subjects where oxycodone blocks negative affect (anxiety, depression or pain) or withdrawal symptoms. Drug likability, attractiveness, or euphoria in humans can be evaluated by asking participants to complete drug rating questionnaires (MacKillop and de Wit 2013; Butler et al. 2010; Morean et al. 2013). For example, one of the most commonly used evaluation forms is the Drug Effects Questionnaire (DEQ), which measures two critical aspects of subjective experience: the strength of the substance effects and the desirability of substance effects (Fraser et al. 1961; de Wit and Phillips 2012; Morean et al. 2013). In the DEQ, where the format can vary across studies, the questions are, “do you feel any substance effect(s)?” (FEEL), “do you like the effects that you feel?” (LIKE), “do you feel high?” (HIGH), “do you dislike the effects?” (DISLIKE), and “do you want more of the substance?” (MORE or TAKE AGAIN) (MacKillop and de Wit 2013; Morean et al. 2013). The desire to “TAKE AGAIN” is considered to reflect the reinforcing effect of a drug, that is the ‘wanting’ or incentive salience (motivation, desire, attractiveness or craving for a reward) for a substance (Wightman et al. 2012; Berridge and Robinson 2016). The DEQ was used to evaluate the ‘liking’ and ‘wanting’ of many opioids, including oxycodone (Webster et al. 2012), morphine (Webster et al. 2011), and heroin (Comer et al. 2005).
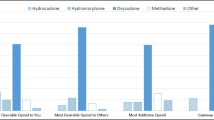
Clinicians and researchers proposed that the preference for oxycodone over other opioid drugs may not be due to its specific analgesic properties but to its’ positive subjective effects (and its reduced negative effects) (Wightman et al. 2012). That is to say, drug consumers appear to feel a particularly enjoyable “smooth” high experience with oxycodone that they qualify as the ‘Rolls Royce’ of opioidsFootnote 22 (Comer et al. 2008). For example, the very much cited health survey established by Cicero et al. in 2010 reported that extended release oxycodone was the most liked and desired (‘wanted’) drug by approximately 50% of [1818] prescription opioid-dependent patients to produce euphoria (both in healthcare and non-healthcare environments); an effect confirmed with a larger cohort [3520] of opioid-dependent subjects (Cicero et al. 2013). Following oxycodone, the most liked and desired opioid drugs were heroin and hydrocodone, with 20–25% of those using these drugs to get high (Cicero et al. 2010). The most recent clinical study and mini review on prescription opioids ‘liking’, ‘wanting’ and addiction liability was published in 2019 by Remillard et al. (2019). In their survey conducted between 2013 and 2018, 113 opioid-dependent subjects from a pain management practice in south-central Alaska were presented with a list of commonly prescribed oral prescription opioids including hydrocodone, hydromorphone, methadone, morphine, oxycodone, oxymorphone, tapentadol. They were asked to identify the prescription opioid which was the (1) most desirable to them, (2) most desirable among drug-using associates or community, (3) the most addictive to them, and (4) the one that is used as gateway to heroin. They had 4 main conclusions: (1) The highest number of respondents, 60.2% of subjects rated oxycodone as most ‘desirable’ over the other opioids they had experience with. Hydrocodone followed in the second place with 16.9% of subjects rating it as most desirable. (2) Oxycodone was claimed by the largest number of participants, 75.4%, to be the most ‘desirable’ among the general drug-consuming population. (3) 59.4% of respondents additionally stated that oxycodone was the most ‘addictive’ prescription opioid, followed distantly by methadone, hydrocodone, and hydromorphone. (4) Among heroin users, oxycodone was also rated as their gateway to heroin (Remillard et al. 2019).
It is important to highlight that drug likability or drug ‘liking’ is not equivalent to incentive salience or ‘wanting’. Despite the fact that the indices of likability or ‘liking’ often correlate with addiction, ‘liking’ seems to not be a determining factor in the abuse liability, as compared to ‘wanting’ (Robinson and Berridge 1993; Berridge and Robinson 2016). Indeed, the neural circuits responsible for likability of a particular reward are different from the network that mediates the incentive salience of the same reward. ‘Liking’ is mediated by small and short-lived hedonic hotspot systems, and is not dependent on dopamine (DA), whereas incentive salience or ‘wanting’ is generated by larger and more robust neural systems that include mesolimbic DA circuits (Berridge and Robinson 2016). The hedonic hotspots underlying ‘liking’ are dispersed and interconnected areas in the brain that act together so that stimulating one hotspot with a drug microinjection causes other hotspots to be recruited into neurobiological activation (Smith and Berridge 2007; Castro and Berridge 2014, 2017; Berridge 2018). These areas are located in the limbic prefrontal cortex such as the orbitofrontal cortex and the insula cortex, and in subcortical structures such as the nucleus accumbens (NAc), ventral pallidum, and the brainstem pons (Small et al. 2001; Kringelbach et al. 2003; Smith and Berridge 2007; Kringelbach, 2010; Castro and Berridge 2017; Berridge 2018). In the NAc and specifically in the rostrodorsal medial shell, the hedonic hotspot measures only one cubic millimeter in a rat brain, and probably about a cubic centimeter in humans (Peciña and Berridge 2000, 2005; Smith and Berridge 2007; Berridge and Robinson 2016). The hotspot was estimated to represent 10% of total NAc volume. The remaining 90% of the NAc cannot enhance ‘liking’, though still strongly generates intense ‘wanting’ (Peciña and Berridge 2000, 2005; Berridge and Robinson 2016). Indeed, mu opioid receptor (MOR) stimulation in the rostrodorsal medial shell of the NAc by microinjections of an opioid such as DAMGO, increased ‘liking’ reactions measured by hedonic orofacial expressions elicited by sucrose taste (e.g., tongue protrusions) (Berridge 2000; Steiner et al. 2001; Peciña and Berridge 2000, 2005; Smith and Berridge 2005; Castro and Berridge 2012; Castro and Berridge 2017). In contrast, microinjections of the same drug in other areas of the NAc outside of the hedonic hotspot enhanced motivational ‘wanting’ for food, but not ‘liking’ reactions to taste (Zhang et al. 1998; Peciña and Berridge 2000, 2005; Smith and Berridge 2005; Shimura et al. 2006; Woolley et al. 2006; Berridge 2018). Moreover, ‘liking’ reactions are not thought to be dependent on dopamine, as dopamine depletion via neurochemical lesions in the whole brain (Berridge et al. 1989) and dopamine stimulation in hedonic hotspots (Smith et al. 2011; Berridge and Kringelbach 2015) do not affect hedonic orofacial expressions elicited by sugar taste. Unlike ‘liking’, ‘wanting’ requires dopamine as shown by suppression of motivation to seek food reward after dopamine lesions in rats (Berridge et al. 1989). In humans, suppressing dopamine neurotransmission did not reduce ‘liking’ ratings of drug rewards, such as cocaine or amphetamine, but did lower the desire to consume more drug (Brauer and De Wit 1997; Leyton 2010). Similarly, implanted electrodes to stimulate the mesolimbic systems and elevate extracellular dopamine enhanced ‘wanting’ but not ‘liking’ (Berridge and Valenstein 1991; Berridge and Kringelbach 2015). Finally, despite the clear evidences showing that dopamine mediates incentive salience but not ‘liking’, the circuitry underlying ‘wanting’ is more complex than what we describe.
One of the main characteristics of the addiction state is the underlying long-lasting changes in the dopaminergic neurotransmission in mesolimbic circuits. This dopaminergic system mediates ‘wanting’ responses to drugs and drug-related cues following prolonged drug use (Koob and Le Moal 1997; Wise 1985; Robinson and Berridge 1993; Berridge and Robinson 2016), but is not involved in ‘liking’. Thus, as the incentive sensitization theory postulates, the essence of drug addiction is the excessive amplification specifically of psychological ‘wanting’, without necessarily an amplification of ‘liking’ (Berridge and Robinson 2016). Incentive sensitization signifies that the dopamine system becomes hyper-reactive to cues and contexts associated with a drug, thus conferring more intense incentive salience on those cues or contexts (Berridge and Robinson 2016). When the mesolimbic dopamine system becomes sensitized and hyper-reactive to the incentive motivational properties of drug cues, drug-seeking and taking become compulsive (Childress et al. 2008; Ostlund et al. 2014; Zhou et al. 2012; Witteman et al. 2015; Berridge and Robinson 2016). ‘Take again’ ratings were consistently higher for oxycodone (Zacny and Gutierrez 2003; Zacny et al. 2003; Comer et al. 2008; Remillard et al. 2019) suggesting that oxycodone precipitates ‘wanting’ and reinforcing characteristics, which infer higher abuse liability. However, we need to take in consideration behavioral studies to confirm that oxycodone may have greater ‘wanting’ or reinforcing effects both in humans and animals. ‘Wanting’ is evaluated via the increase in effort to obtain a reward upon presentation of a cue that was previously associated with the same reward (Pool et al. 2016). Likability is assessed differently, by hedonic pleasure displayed during reward consumption, such as prototypical orofacial expressions elicited by sweet taste in human and animals (Berridge 2000; Berridge et al. 2009; Pool et al. 2016). The relationship between oxycodone administration rating and oxycodone taking behavior in humans has been investigated. Comer et al. in 2008, asked heroin- and morphine-dependent subjects to choose between a monetary reward and an oxycodone dose that they had received during the sample session in a self-administration task. Oxycodone exhibited strong reinforcing effects, similar to those of morphine and heroin, suggesting equivalent abuse liability among these opioids. This latter study raised concerns over the potential for oxycodone abuse, because it presented some of highest positive subjective ratings and no increases in ratings of bad effects compared to the other opioids (Comer et al. 2008). Nevertheless, the experimental paradigm did not allow differentiation between fentanyl, oxycodone, morphine, and heroin. To more closely examine the relative abuse liabilities of oxycodone and morphine using a drug versus drug self-administration procedure, Comer and colleagues assessed opioid-dependent subjects that were maintained on buprenorphine (Comer et al. 2013). When the high doses of morphine and oxycodone were compared directly, participants strongly preferred oxycodone. However, due to several drawbacks of the study (including carry over effects, drug interactions with buprenorphine, and tolerance), the authors were cautious in claiming that oxycodone has greater behavioral reinforcing effects than morphine. Nevertheless, the subjective ratings of oxycodone were consistently higher.
The same methodology roadblocks to measure the reinforcing behavior effects of oxycodone are also confounds in animals drug discrimination studies. The affective state of the subject will impact the results in such studies. Opioids can act as analgesics as well as possess antidepressant and anxiolytic effects. Thus, affective states that are alleviated by opioids will accentuate both liking and wanting via negative reinforcement. Indeed, a high proportion of those with opioid use disorder and that transition from using opioids for analgesics to developing an opioid use disorder are comorbid with mental disorders such as anxiety and depression (Hser et al. 2017).
High Likability and Incentive Salience of Oxycodone Assessed in Preclinical Studies
Humans are able to verbalize their preference for oxycodone among an array of administrated opioids (Comer et al. 2008; Remillard et al. 2019). In animals, measurement of incentive salience associated to a specific drug is restricted to the observation of motivation, drug-seeking and drug-taking behaviors. There are very few drug discrimination or drug likability studies comparing the reinforcing effect and incentive salience of oxycodone compared to other opioids. In a study where mice were trained to discriminate oxycodone from vehicle in a food-reinforced, two-lever choice paradigm, fentanyl and other fentanyl analogs completely generalized to the oxycodone discriminative stimulus, suggesting that interoceptive and subjective effects of the tested fentanyl drugs overlap with those of oxycodone (Wade et al. 2015; Walentiny et al. 2019). We assume that one of the reasons for the general lack of opioid discrimination studies is due to the difficulty to observe differences of behavior associated to interoceptive and subjective effects within the same drug family. Nevertheless, preclinical rodent studies are essential in advancing our understanding the changes in the cellular and molecular mechanisms underlying substance abuse and addiction (Lynch et al. 2010). A study from Neelakantan and colleagues compared the ability of mice to discriminate equianalgesic doses of morphine and oxycodone from saline with and without the presence of pain (Neelakantan et al. 2015). Both male and female mice were able to equally discriminate both drugs from saline. However, in the presence of acid acetic-induced pain, male mice were not able to discriminate morphine from saline, but continued to discriminate saline from oxycodone. However, negative reinforcement due to the analgesic efficacy of oxycodone to relieve visceral pain likely confounded these results. To identify potential differences in compulsive drug-seeking and -taking between oxycodone, fentanyl, and buprenorphine in rats, Wade and colleagues used an intravenous self-administration paradigm in rats (Wade et al. 2015). However, there was no difference in drug escalation with extended access and all groups presented similar increased progressive ratio breakpoints. These results suggest similar enhancement in motivation to seek oxycodone, fentanyl, and heroin. The opioid discrimination studies in rodents concluded that oxycodone, heroin, fentanyl and derivates produce the same motivational drive for consumption. However, this was not assessed in different affective states and other neurochemical approaches may provide more information. For example, oxycodone induces stronger cellular or neurotransmission (i.e., incentive sensitization) changes compared to morphine that may account for the increased ‘wanting’ and compulsive-like behavior induced by oxycodone compared to other opioids (Vander Weele et al. 2014).
Oxycodone Differentially Modulates Reward Circuitry
Several brain structures and pathways compose the reward system. Within this system, the mesolimbic dopaminergic circuit is the most extensively studied. It connects a region of the central midbrain, the ventral tegmental area (VTA), to the striatum, which is divided into the dorsal and ventral components. The ventral striatum includes the NAc, which is further subdivided into core and shell, and again into medial, lateral, dorsal ventral subareas. VTA dopaminergic neurons are known to fire action potentials tonically or phasically following exposure to a salient, motivational stimuli by burst firing (Bromberg-Martin et al. 2010), which is suppressed by tonic GABA input (Lobb et al. 2010; Jalabert et al. 2011; Matsui et al. 2014). Dopamine release in the NAc triggers network communication with the prefrontal cortex, which is required for reasoning, decision-making, and planning. The prefrontal cortex incorporates these positive/pleasant feelings into memory, reinforcing the behavior by motivating the person to perform again (Lynch et al. 2010). As mentioned above, the dopamine neurotransmission within the reward system is responsible for incentive salience and is involved in the development, maintenance, and compulsive intake of abused drugs (Phillips et al. 2003; Robinson and Berridge 2003; Hyman et al. 2006; Vander Weele et al. 2014). All abused opioids increase DA transmission within the NAc, including oxycodone (Di Chiara and Imperato 1988; Nestler and Malenka 2004; Vander Weele et al. 2014). Although the loss of phasic dopamine release in the ventral striatum has also correlated with escalated drug intake (Caprioli et al. 2014; Willuhn et al. 2014).
While a number of functional magnetic resonance imaging studies have reported the effects of oxycodone on brain activity, to our knowledge, there are no human brain imaging studies that have directly compared effects of different opioids. Brain imaging studies have reported the effects of oxycodone on neuronal activity and brain connectivity when compared to placebo or non-opioids compounds (Upadhyay et al. 2010; Hansen et al. 2018), where the majority of these studies focused on regions such as the anterior cingulate cortex and amygdala, structures involved in pain and emotions (Gorka et al. 2014). Mixed results were reported on the effects of oxycodone in brain imaging studies in rodents, where either an increase (Nasseef et al. 2019), a decrease (Moore et al. 2016) or no change (Iriah et al. 2019) in blood flow in the reward systems, including the NAc, caudate putamen, and amygdala were reported after a single acute analgesic dose of oxycodone was administered. Habituation to the environment, to an injection and to handling is necessary for obtaining reproducible outcomes. Nevertheless, it is generally accepted that an acute oxycodone injection decreases whole-brain functional connectivity (communication) (Moore et al. 2016; Nasseef et al. 2019). In contrast, repeated oxycodone injections showed increased brain activity in areas affected by drug use such as the NAc, forebrain areas, amygdala, and brainstem (Iriah et al. 2019).
One of the most revealing studies that demonstrated differences in oxycodone and morphine evoked dopamine release used two different neurochemical detection techniques. Using fast-scan cyclic voltammetry and rapid (1 min) microdialysis coupled with mass spectrometry analysis in rats, Vander Weele and colleagues demonstrated differential effects on extracellular dopamine content in the NAc produced by oxycodone and morphine (Vander Weele et al. 2014). In this latter study, intravenous infusion of oxycodone led to an elevation in extracellular dopamine concentration, a more sustained release of dopamine, as well as a significant increase in the frequency and amplitude of phasic dopamine release (dopamine transients) in the NAc than an equipotent dose of morphine. Specific and significant increases in both dopamine transients, and NAc shell dopamine activity are characteristics of the primary rewarding effects of drugs of abuse (or correlated with increased abuse and dependence) (Pontieri et al. 1995; Ito et al. 2000; Frank et al. 2008). Interestingly, morphine, but not oxycodone, induced a succinct (~ 1 min) elevation in dopamine that was associated with a surge in GABA concentration (Vander Weele et al. 2014). This increase in GABA release caused by morphine, but not oxycodone, may explain the quick fall of extracellular dopamine concentration to baseline levels following administration of morphine but not oxycodone. The authors proposed two mechanisms for the quick but short-lived increase in extracellular dopamine in the NAc by morphine in comparison to the long-lasting and stable dopamine response evoked by oxycodone (Fig. 2). First, morphine-induced increase of GABA in the NAc may activate GABA-A receptors on dopamine neuronal terminals, which would consequently inhibit further dopamine release (Aono et al. 2008; Saigusa et al. 2008; Yang et al. 2018). GABA could be released from the medium spiny neurons (MSNs) (Yang et al. 2018), tyrosine-hydroxylase (TH)-expressing interneurons (Ibáñez-Sandoval et al. 2010; Tepper et al. 2010), and neuropeptide Y interneurons (English et al. 2011; Ibáñez-Sandoval et al. 2011) within the NAc. Second, increases of GABA transmission within the NAc following morphine, but not oxycodone, may be attributable to enhanced morphine-induced activation of dopamine/GABAergic neurons in the VTA that project to the NAc (van Zessen et al. 2012; Tritsch et al. 2014; Kim et al. 2015; Berrios et al. 2016; Morales and Margolis 2017). Modulation of dopamine released from dopaminergic terminals in the NAc influences motivated behavior. Again, with fast scanning cyclic voltammetry, Borgland and colleagues demonstrated that activation of GABA-B receptors on dopamine neuronal terminals could decrease pulse-evoked dopamine release and reduce probability of dopamine release (Pitman et al. 2014). While a substantial source of GABA in the NAc is from the MSNs, there is now convincing evidence that dopaminergic neurons can co-express and release GABA or glutamate neurotransmitters (Bayer and Pickel 1990). Indeed, enhanced reward seeking behavior measured by optical self-stimulation was evident when GABA release was suppressed from tyrosine hydroxylase-expressing neurons (Berrios et al. 2016). It is therefore possible, that the differences in morphine- and oxycodone-induced increases in extracellular dopamine release may be due to the activation of different VTA dopaminergic neurons that project to the NAc. The differences could also be due to the drug effects on cholinergic interneurons, which also could modulate dopamine release, via direct interaction with the dopaminergic neurons or via its modulation of GABAergic interneurons (Gould et al. 2019). The enhanced activity of dopamine neurons by oxycodone compared to morphine is further supported by a study examining the modulatory effects of oxycodone, hydrocodone and morphine on the response and excitation of D2/D3 type dopamine receptors (Emery et al. 2015). By testing the locomotor activity in mice that received quinpirole, a D2/D3 DA receptor agonist, they showed that oxycodone pretreatment produced greater locomotor sensitization to quinpirole compared to either hydrocodone or morphine. The authors suggested that this sensitization of the reward system may lead to changes in general impulsivity, which is known to be a risk factor for drug addiction. A subsequent study by the same authors provided further evidence that oxycodone and hydrocodone produce different intracellular changes in D2 receptor ß-arrestin signaling compared to morphine (Emery et al. 2016).

Action of oxycodone and morphine on potential GABAergic neurons responsible for the difference in the extracellular dopamine (DA) within the reward system. Oxycodone and morphine provoke distinct changes in DA release in the nucleus accumbens (NAc) that could potentially explain drug users’ selective preference for oxycodone over other opioids (Vander Weele et al. 2014). Intravenous infusion of oxycodone leads to greater global and phasic releases of DA in the NAc (especially in the shell), while morphine causes a short-lived DA release coupled to transient GABA release within the same structure (Vander Weele et al. 2014). Dopaminergic neurons of the mesolimbic pathway project from the VTA in the midbrain onto the GABAergic inhibitory medium spiny neurons (MSNs) in the NAc (Malenka et al. 2009; Ikemoto et al. 2010; Yager et al. 2015; Morales and Margolis 2017). These MSNs represent 95% of the striatum and express the D1 and/or D2 dopaminergic receptors (Kemp and Powell 1971; Yager et al. 2015; Morales and Margolis 2017). Besides DA, GABA is one of the principal neurotransmitters that mediate reward signaling within the NAc (Meredith et al. 1992; Shirayama et al. 2006). Terminals of dopaminergic neurons in the NAc from the VTA are presumed to express GABA receptors through which GABA released from the MSNs may induce tonic inhibition (Yang et al. 2018). MOR has been shown to be expressed on MSNs (Svingos et al. 1999; Shippenberg et al. 2008) and in an undetermined subset of GABAergic neurons within the mouse NAc (Ford et al. 2006; Margolis et al. 2012, 2014; Hinkle et al. 2019). Several lines of indirect functional evidence support the presence of MOR on GABAergic neurons in the VTA (Margolis et al. 2014). As posits the canonical model, opioids induce reward by disinhibiting VTA and NAc dopaminergic neurons through inhibition of GABAergic inputs (Johnson and North 1992; Labouèbe et al. 2007; Barrot et al. 2012; Bourdy and Barrot 2012; Margolis et al. 2012). Therefore, oxycodone may cause robust and stable DA release by decreasing the excitability of GABAergic MSNs (see label 1 on the figure), thus blocking their potential tonic inhibitory action on the DA terminals in the NAc. Oxycodone may also inhibit other NAc interneurons that innervate directly the dopaminergic terminals (label 1) and the GABAergic inputs on these dopaminergic neurons in the VTA (label 1). As our unpublished data suggest, the robust and stable increase in global and phasic releases of DA in the presence of oxycodone might involve the selective stimulation of putative opioid receptor heterodimers or potentially different signaling cascades compared to morphine. The morphine-dependent short-lived DA response could be caused by the additional surge of GABA release following morphine. One hypothesis for the transient rise of GABA is that inhibition of GABAergic neurons by morphine could be short-lived compared to oxycodone, thus leading to a brief inhibition of GABA release (bottom figure). However, this needs to be demonstrated. The surplus of GABA could also be secreted by VTA DA/GABAergic TH-expressing neurons that project to the NAc (labels 2 and 3). We speculate that these latter neurons could be activated to release GABA via disinhibition by morphine (label 2), whereas they would be inhibited in the presence of oxycodone via a potential selective agonist action on opioid receptor heterodimers-expressing GABAergic neurons (label 3)
While the studies summarized above give some indications on how exactly oxycodone modifies the dopamine system, they strongly suggest that while both opioids activate opioid receptors, there is either distinct cellular engagement and intracellular signaling, or off target modulation that results in differential behavioral outcomes. Because of how opioid agonists have unique effects on dopamine neurotransmission within the reward system, we can surmise that different opioid agonists will elicit various strengths of incentive salience. Thus, the differences in engagement of the mesolimbic dopaminergic circuits could elicit diverse effects on incentive salience or motivation and potentially analgesic mechanisms (Ma et al. 2012). These arguments favor oxycodone’s higher abuse liability, despite similar reinforcing profile between oxycodone and other opioid agonists in the drug discrimination studies. Potential molecular mechanisms including biased signaling, as well as pharmacokinetic and pharmacodynamic properties unique to oxycodone may underlie the differences in dopamine-evoked transmission between oxycodone and morphine (Nielsen et al. 2007; Lemberg et al. 2009; Lester and Traynor 2006; Peckham and Traynor 2006; Melief et al. 2010; Vander Weele et al. 2014).
Potential Molecular Mechanisms of Action of Oxycodone
The literature reviewed above describes evidence for high addiction liability associated with oxycodone and a description of some of the mechanisms that may underlie this trait. There are many cellular and signaling differences between opioid agonists that may also contribute to differences between opioids and the reader is referred to recent reviews on biased agonism (Grim et al. 2020), desensitization and intracellular signaling (Williams et al. 2013). The following discussion will support the hypothesis that oxycodone processes novel pharmacology via binding to an allosteric site on the MOR, which in turn would contribute to oxycodone’s unique pharmacology.
Pharmacokinetics, Pharmacodynamics and Intracellular Signaling Pathways
From the early hypothesis by Beckett and Casy (1954), it is apparent that all opiates (chemical entities present in opium) and semi-synthetic opiates (which includes oxycodone) must contain a hydroxyl moiety at the 3-position of the aromatic A in order to exhibit high affinity binding and analgesic potency. The high affinity binding requirement is demonstrated from the crystal structure analysis of the opioid receptors, in which the phenolic hydroxyl of the aromatic ring of the morphinan is shown to form H-bond with 2 water molecules and the H2976.52 of the mu- or H2786.52 of the delta opioid receptor (DOR) (Manglik et al. 2012; Granier et al. 2012). Oxycodone, with a methoxy group at the 3-position of the aromatic ring A, has been shown to exhibit lower affinity for the receptor than oxymorphone, a metabolite of oxycodone via cytochrome P450 metabolism (Cone et al. 1983; Lalovic et al. 2006) (Fig. 1). Oxymorphone can produce 8- to 30-fold higher G-protein activation than oxycodone (Thompson et al. 2004; Lalovic et al. 2006; Peckham and Traynor 2006). This high potency is consistent with our unpublished data that show that oxymorphone exhibited a 100-fold higher affinity for the receptor than oxycodone in competition binding studies using 3H-diprenorphone and membrane isolated from HEK293 cells overexpressing rat MOR. This pharmacology likely contributes to the higher analgesic potency of oxymorphone compared to oxycodone and more serious side effects that are associated with oxymorphone (Murphy et al. 2018). Mortality rates remain higher with oxycodone (Hirsch et al. 2014), but this is reflective of rates of use where oxycodone is much more commonly prescribed than oxymorphone. It is generally accepted that activation of MORs by oxymorphone, though not the prominent metabolic pathway (Fig. 1), contributes to the pharmacodynamic effects of oxycodone, including adverse effects such as respiratory depression, miosis, euphoria, constipation, tolerance, dependence and addiction (Cicero et al. 2005). However, only about 10% of oxycodone is metabolized to oxymorphone (Lalovic et al. 2006), and noroxycodone after further N-demethylation of oxymorphone, which is the major metabolite in rats and human, as shown in Fig. 1 (Chan et al. 2008; Lalovic et al. 2004). Ratios of oxymorphone/oxycodone in the brain as detected with mass spectrometric analysis after either subcutaneous or intracerebral administration of oxycodone revealed relatively low level of the metabolite, 1.9% and 1.5%, respectively (Yang et al. 2016). Moreover, in pharmacokinetic studies, plasma concentrations of oxymorphone are either very low or absent following the systemic or oral administration of oxycodone in human (Kaiko et al. 1996; Pöyhiä et al. 1991). Because oxycodone has been shown to be more potent than morphine in the relief of post-operative and cancer pain when administered intravenously or orally in humans (Kalso et al. 1991; Bruera et al. 1998) and in rat after systemic administration (Pöyhiä and Kalso 1992), the pharmacodynamics observed must be due to the activation of receptors by oxycodone itself or metabolites other than oxymorphone. Important to consider is how oxycodone metabolism varies with engagement of cytochrome P450 enzymes as well their expression in the brain (McMillan and Tyndale 2018). Also important is that metabolism of drugs by P450, conjugation and other drug metabolic enzymes is highly variable between individuals because of frequent polymorphisms. Individual polymorphisms often drive the ‘likability’, efficacy, and addiction potential of drugs. For example, the efficacy of demethylation of codeine to morphine is highly dependent on CYP-2D6 polymorphisms such that given some polymorphisms codeine is an ineffective opioid.
The opioid receptor type activated by oxycodone to elicit its pharmacodynamics is controversial. Ross and Smith (1997) reported that intracerebroventricular pretreatment with kappa opioid receptor (KOR) antagonist, nor-binaltorphimine (nor-BNI) blocked the antinociceptive effect of oxycodone but not morphine when administered by the same route of delivery. This data implicated KOR agonist-involvement for oxycodone action, and is of interest because the central KOR activation is dysphoric and inhibits dopamine release in reward mesolimbic structures (Ehrich et al. 2015; Liu et al. 2019). It has been indeed shown that KOR antagonism increases extracellular dopamine concentrations measured by fast-scan cyclic voltammetry in the NAc. In contrast, KOR activation by its agonist U69,593 acutely inhibited VTA dopaminergic neuron firing (Ehrich et al. 2015). Moreover, oxycodone appears to require KOR activation to mediate withdrawal-induced escalated intracranial self-administration, as suggested by a slowed escalation of responding in rats pretreated with nor-BNI before extended access to oxycodone self-administration (Nguyen et al. 2019). Many drugs of abuse increase intracranial self-stimulation reward thresholds to extended compared to short access including heroin (Kenny et al. 2006) and psychostimulants such as cocaine or methamphetamine (Jang et al. 2013; Ahmed et al. 2002). The escalation of oxycodone self-administration appears to involve the KOR-dependent negative motivational state associated with oxycodone withdrawal. One could speculate that the presumed partial agonism of oxycodone on KOR could be involved in the differential regulation of dopamine neurotransmission between oxycodone and morphine (Vander Weele et al. 2014). One study reported that the intrinsic antinociceptive effects of oxycodone were dependent on KOR activation as only KOR blockade but not MOR or DOR selective antagonists blocked oxycodone antinociception in the tail flick test (Ross and Smith 1997). The role of the peripheral KOR in oxycodone-induced analgesia has been described in the literature, especially regarding visceral pain (Ruan et al. 2017). Indeed, KOR expression is extensive in peripheral neurons and nociceptors (De Schepper et al. 2004), and both MOR and KOR have been found to be expressed in the stomach, duodenum, jejunum, ileum as well as the proximal and distal colon, where their stimulation has been suggested to modulate visceral pain (Kapitzke et al. 2005; Davis et al. 2012). However, others reported that oxycodone antinociceptive action is mostly mediated by MORs, and that oxycodone action could not be reversed with DOR or KOR antagonists (Nozaki et al. 2006; Narita et al. 2008; Lemberg et al. 2006). Our unpublished data measuring the inhibition of intracellular cAMP levels in HEK293 cells expressing either the rat or human opioid receptor support the latter, where oxycodone preferentially activates the MOR in cell-based assays. Such highly efficient coupling between the oxycodone-receptor complex with the intracellular signaling molecules could be the partial explanation for the higher analgesic potency observed with oxycodone in both human and rodents (Kalso et al. 1991; Bruera et al. 1998; Pöyhiä and Kalso 1992; Codd et al. 1995; Olson et al. 2019).
In addition to MORs, oxycodone or its metabolites appear to involve the activation of DORs based on behavioral outcomes in genetically modified animals. In MOR knockout mice, oxycodone retained residual antinociceptive action, while morphine antinociceptive activity was completely absent (Yang et al. 2016). Additionally, in non-dependent human volunteers with a history of recreational prescription opioid exposure, oxycodone positive subjective effects of drug ‘liking’ and ‘good effect’ was influenced by genetic variants in the MOR (rs6848893) as well as individuals with a genetic variant in the DOR (rs581111) (Jones et al. 2019). Oxycodone antinociceptive activity can be blocked by DOR selective antagonists, naltrindole or ICI 154,129 (Yang et al. 2019). Furthermore, the constipation effect and the condition place preference response induced by oxycodone can also be blocked by the DOR specific antagonist naltrindole (Yang et al. 2019). Because of the relative low potency of oxycodone exhibited in the in vitro cell lines expressing DORs and only residual antinociceptive response was observed in the MOR knockout mice, the involvement of metabolites or putative opioid receptor heterodimers, as suggested by Nielsen et al. (Nielsen et al. 2007), should be considered as the putative selective targets for oxycodone in vivo actions. It is also possible that oxycodone activation of MOR or MOR-receptor complexes is distinct from other MOR agonists via interactions with the orthosteric sites on the receptor. Our studies and others have demonstrated unequivocally the existence of biased agonism within the opioid receptor (Zheng et al. 2008; Conibear and Kelly 2019). MOR activation of a specific signaling molecule, such as ERK1/2, could involve agonist-specific multiple pathways, such as ß-arrestin or PKC dependence. Agonists such as etorphine or DAMGO will activate ERK1/2 via the ß-arrestin-dependent pathway, while activation of ERK1/2 by an agonist such as morphine is PKC-dependent (Johnson et al. 2006; Smith et al. 2007; Chu et al. 2010; Zheng et al. 2011) (Fig. 3). A recent in vivo study revealed that oxycodone self-administrated to rats activates PKC, ERK1/2, and the mitogen-activated protein kinase/mitogen stress-activated protein kinase (MAPK-MSK) signaling pathway as well as CREB in the dorsal striatum (Blackwood et al. 2020). It was also found that escalated doses of self-administered oxycodone increased histone H3 phospho-acetylation, a substrate of MSK1/2, which would induce chromatin decondensation for gene regulation. Although in these in vivo studies do not distinguish cellular mechanisms and pathways to second-messenger activation, preliminary results from our in vitro studies also showed that oxycodone activates PKC and ERK1/2. They also suggest that oxycodone regulation of ERK1/2 activity might involve additional PKC-independent pathways. Oxycodone-induced receptor activation leads to activation of multiple intracellular signaling cascades that may account for its distinct pharmacodynamics (Fig. 3). However, this hypothesis needs to be confirmed with future studies.

Potential biased signaling of oxycodone and morphine. Opioid receptors are both pre- and post-synaptic and are coupled to the Gi/Go proteins. In contrast to morphine, oxycodone is speculated in this review to interact with sodium (Na+) allosteric sites on the mu opioid receptor (MOR) and to act as an “efficacy switch” in the receptor signal transduction. In acute pain conditions, the activation of MOR by either morphine or oxycodone triggers the classical intracellular Gi/Go-protein signaling pathway that leads to analgesia (Law et al. 2000; Williams et al. 2013; Pena et al. 2018) and the Protein Kinase C (PKC)-mediated signaling pathway that is responsible for the receptor desensitization. The classical Go/Gi-protein signaling pathway has already been extensively described in the literature: it leads to (1) the inhibition of the adenylyl cyclase (AC)/cyclic AMP (cAMP)/ protein kinase A (PKA) or the exchange protein directly activated by cAMP (EPAC) pathways (Pena et al. 2018), (2) the stimulation of G-protein-couple inwardly-rectifying potassium channels (GIRKs) and (3) the inhibition of voltage-gated calcium channels (Ca2+ conductance) causing a decreased neurotransmitter release from the pre-synaptic nerve terminal. After receptor activation, there is a progressive reduction in signal transduction which corresponds to MOR desensitization (Williams et al. 2013). MOR desensitization by morphine and oxycodone is mainly PKC-dependent as it minimally engages GRK-ß-arrestin regulation like fentanyl (Chu et al. 2010; Zheng et al. 2011). However, unlike morphine, we hypothesize that oxycodone requires other pathways in addition to that of PKC to induce MOR desensitization. These potential additional PKC-independent pathways need to be determined, although specific signal transduction activated by oxycodone has already been demonstrated, such as the Epithelial growth factor receptor (EGFR)/ERK/Akt pathway in the context of oxidative stress (Yu et al. 2020). The mechanism by which PKC mediates morphine- and oxycodone-promoted MOR desensitization is still not clear. Strong evidences suggest that morphine induces MOR desensitization through low level of receptor phosphorylation and activation of PKCα, ɣ and ɛ to induce ERK phosphorylation and tolerance (Smith et al. 2003; Song et al. 2010; Zheng et al. 2011). After MOR activation, PKC can be stimulated by second messengers such as DAG and calcium made available thanks to PLC activation by Gßɣ subunit, non-coupled tyrosine kinases, or small G-protein (Pena et al. 2018). Many substrates of PKC have been proposed including Phosphatidylethanolamine‐binding protein 1 (PEBP1) which inhibits the MAPK/ERK pathway, scaffold proteins such as annexin 6 which also inhibits ERK activation, neurogranin and calmodulin whose stimulation by PKC leads to activation of CAMKII and TRPV1. These PKC-signaling signaling cascades of events are involved in the development of tolerance both in the spinal cord and the nucleus accumbens (NAc) (Song et al. 2010). PKC has also an important role in inhibition of receptor recycling (Bailey et al. 2006; Halls et al. 2016)
Speculative Actions of Oxycodone That May Influence Addiction Potential
Pharmacology research in recent years has identified many reasons for signaling and behavioral differences within individual drug classes such as opioid analgesics. We have addressed above the aspects of agonist bias, metabolites and selectivity for different opioid receptor types, but other possibilities should be considered. Pharmacodynamics of oxycodone or its metabolites may allow for regional and cellular selectivity of opioid receptor activation. Recent data show evidence for intracellular GPCR signaling both within vesicles and on Golgi that may influence cellular signaling and lead to drug location bias of receptor activation (Jullié et al. 2020). Accessibility to receptors in different brain regions and with different outcome functions, such as analgesia and reward, can be influenced by blood brain barrier permeability and P-Glycoprotein (drug efflux pump) interactions. In addition to homo- and heterodimers, opioid receptor signaling may be modulated by receptors existing in many different forms depending upon phosphorylation and other post-translational modified states. Furthermore, environments such as lipid rafts or association with different proteins can dramatically influence ligand interactions with the receptor. Finally, receptor regulation can include allosteric modulation. Selective allosteric ligands for MOR, DOR, and KOR have been described (Livingston and Traynor 2018; Livingston et al. 2018; Jutkiewicz et al. 2019) that bind to allosteric sites on the receptor distinct from the orthosteric site to which endogenous ligands or traditional agonists such as morphine, codeine, and fentanyl bind to produce their biological effects. Since oxycodone is a morphinan it has been assumed that this drug is solely interacting with the orthosteric site; however, given its unique pharmacology, allosteric interactions should be considered.
Positive allosteric modulators of opioid receptors represent one of the most recent potential strategies to develop therapeutically safer analgesics (Livingston and Traynor 2018) and selective allosteric ligands for MOR, DOR, and KOR have been described (Livingston and Traynor 2018; Livingston et al. 2018; Jutkiewicz et al. 2019). The allosteric modulators’ main advantage lies in their higher specificities to a receptor and lower risks of toxic side effects (Livingston and Traynor 2014; Sheik Amamuddy et al. 2020). The first selective positive allosteric modulators of MOR identified included small molecules such as BMS-986121 and BMS-986122. They were discovered in 2013 by Burford et al. (2013) using chemoinformatic analysis (i.e., a high-throughput screen (HTS) monitoring) for their ability to enhance a low concentration of the putative endogenous MOR agonist, endomorphin-1, to recruit β-arrestin to MOR (Livingston and Traynor 2018). BMS-986122 increased the potency and affinity of methadone, DAMGO, and the endogenous opioid peptides including Leu5- and Met5-enkephalin, β-endorphin as well as endomorphin-1, while it augments agonist efficacy for morphine and nalbuphine without altering the affinity (Burford et al. 2013; Livingston and Traynor 2014, 2018). BMS-986122 is specific to MOR and does not present positive allosteric activity at DOR (Burford et al. 2013). MS1 is another allosteric modulator of MOR (Bisignano et al. 2015). DOR allosteric modulators including BMS-986187 were identified using a similar strategy that involved the β-arrestin recruitment HTS assay (Burford et al. 2014, 2015; Bertekap et al. 2015). This compound is structurally distinct from the MOR positive allosteric modulator BMS-986122 and exhibits 100-fold selectivity in promoting DOR over MOR agonism (Burford et al. 2015). Most importantly, BMS-986187 has the particularity to stimulate DOR intracellular signaling pathways even in the absence of orthosteric agonist and does not bind to the orthosteric site. BMS‐986,187 increased the potency of the orthosteric DOR agonist SNC80 to decrease immobility in the forced swim test but not to elicit convulsions (Jutkiewicz et al. 2019). Other compounds that have been suggested as positive or negative allosteric modulators of opioid receptors include the natural products cannabidiol, THC (Vaysse et al. 1987; Kathmann et al. 2006), ignavine (Ohbuchi et al. 2016) and salvinorin A (Rothman et al. 2007) as well as SCH-202676 (Fawzi et al. 2001). All of these allosteric ligands could produce analgesic responses without binding to the orthosteric site of the opioid receptor.
The hallmark for allosteric agonism was first described by Pasternak and Snyder (Pasternak and Snyder 1975) in their initial observation of sodium (Na+) effect on opioid agonist binding affinity. Na+ has been reported to inhibit agonist binding by 65% for the MOR and the DOR and only 20% for the KOR (Blume 1978; Werling et al. 1986). It was further demonstrated that the coupling between the opioid receptor and adenylyl cyclase required the presence of Na+ and guanosine triphosphate (Blume et al. 1979). Interestingly, the positive MOR modulator, BMS-986122, allosterically inhibited the binding of Na+ to the MOR by disabling stabilization of the inactive state of the MOR, pushing it toward an active conformation, which explains its marked positive allosteric modulator abilities (Livingston and Traynor 2014). This mechanism is supported by recent crystal structure analysis of the human DOR, which revealed the presence of Na+ allosteric site within the polar interaction network of the 7-transmembrane bundle core stabilizing a reduced agonist affinity state and thereby inhibit signal transduction (Fenalti et al. 2014). Changing the allosteric site from an Asn131 residue to an Ala or Val residue increases β-arrestin-mediated signaling (Fenalti et al. 2014; Remesic et al. 2017). The residues involved in the Na+ allosteric site thus can act as “efficacy switches” in the receptor signal transduction (Fenalti et al. 2014).
From all of the literature reports, it is clear that oxycodone interacts with all three opioid receptor orthosteric sites with low affinity, and it possesses relatively high analgesic potency. One probable explanation for such observations is that oxycodone acts on the allosteric sites modulating the endogenous peptides’ activities or oxycodone acts as an allo-agonist. If oxycodone interacts with the allosteric Na+ sites as it was suggested for BMS-986122, it should follow that oxycodone, could either promote active conformation and thus increase agonist binding, or similar to Na+, oxycodone could reduce agonist binding, required for the receptor-effector coupling. Our unpublished observations indicate that oxycodone is acting at the Na+ site of the receptor. At the concentration of 1 µM that exhibits minimal effect in 3H-diprenorphine competition binding assays, oxycodone could potentiate the Na+ ability to reduce the agonist affinity (unpublished data). Met5-enkephalin’s ability to induce multiple affinity binding was dependent on the Na+, with 5 mM Na+ exhibiting minimal effect, 20 mM Na+ exhibiting intermediate effect, while 150 mM Na+ exhibited the maximal effect. In the presence of 1 µM oxycodone, all three concentrations of Na+ exhibited the maximal effect on the ability of Met5-enkephalin to induce multiple affinity agonist binding. Thus, we hypothesize that oxycodone might enhance the endogenous opioid efficacy for MOR by acting at the Na+ allosteric sites. Oxycodone might act as an “efficacy-switch” in the receptor signal transduction. Of course, this theory needs to be proved with additional computational modeling and further experiments that can show oxycodone potentiating the Na+ effect on endogenous opioids. Finally, as it was demonstrated for the positive allosteric DOR modulator, BMS-986187 (Jutkiewicz et al. 2019), we hypothesize that oxycodone could increase the efficacy of the orthosteric endogenous ligands at MOR to induce their analgesic effects with fewer side effects compared to other opioids like morphine. Oxycodone binding alone on the allosteric site of the receptor to engage alone MOR-dependent intracellular signaling to reduce pain is not to be excluded as it was described for BMS-986187 (Livingston and Traynor 2018; Jutkiewicz et al. 2019).
Alternative Perspectives on Novel MOR Agonists to Replace Oxycodone
Research on developing new non-addictive therapeutics against chronic pain is challenging considering the biased agonism which we discussed above. The reduced side effects or subjective negative effects compared to morphine, is a determinant factor in the choice of oxycodone for the treatment of moderate-to-severe long-term pain. Physicians will continue to favor oral oxycodone in the absence of efficient, safe, non-addictive and clinically approved alternatives to current analgesics. It is important to extend our discussion on oxycodone to other therapeutically encouraging and potentially safe strategies under development. Some of the new approaches tested involve either new MOR agonists or compounds acting on multiple receptors such as MOR/DOR, MOR-mGluR5, and MOR/Nociceptin/orphanin FQ peptide receptor (NOR)) which show in some cases markedly enhanced analgesia but with reduced side effects (Ding et al. 2018; Litman et al. 2018; Kopruszinski et al. 2020; Zhang et al. 2020).
Bifunctional Agonists and Novel MOR Agonists
An outcome of the recent appreciation of the complexity of pharmacological outcomes is the continuing need to synthesize and test new opioid drugs. One recent example is AT-121, a bifunctional new non-morphinan partial NOR/MOR agonist, that has been proposed to have the translational potential to be a safe, non-addictive analgesic and possible treatment for prescription opioid abuse (Ding et al. 2018). Medicinal chemistry, computer modeling, and structure-based drug design were used to design and develop AT-121 (Ding et al. 2018). AT-121 was shown to induce substantial acute thermal antinociceptive effects with a potency 100-fold higher than morphine in rhesus monkeys (Macaca mulatta) (Ding et al. 2018). Pretreatment with NOR receptor antagonist J-113397 combined with the MOR antagonist naltrexone confirmed the role of both NOR and MOR in these antinociceptive effects as shown by a rightward shift of the antinociceptive dose–response curve. Interestingly, AT-121 does not appear to be reinforcing as displayed by similar reinforcing strengths between AT-121 and saline in a self-administration task with a progressive ratio schedule of reinforcement. Moreover, AT-121 pretreatment effectively and selectively attenuated the reinforcing effects of oxycodone but not cocaine and remifentanil. These observations suggest not only a low abuse liability of AT-121 but also the therapeutic potential of AT-121 for opioid addiction. Unlike heroin, 10 times the analgesic doses of AT-121 do not impair respiratory function, cardiovascular activity, body temperature, motricity and sedation of monkeys. Finally, chronic administration of AT-121 does not cause physical dependence (3-day chronic exposure), opioid-induced hyperalgesia nor tolerance (30-day repeated administration). The authors of this study hypothesized that the reason why AT-121 induces analgesia without generating the undesired effects of respiratory depression and reinforcement (abuse potential) in primates was due to the activity at NOR. Indeed, NOR agonists potentiate MOR-mediated antinociception without also increasing side effects (Hu et al. 2010; Cremeans et al. 2012) and can inhibit dopamine release and neurotransmission, which was surmised to account for the lack of reward and incentive salience (Di Giannuario and Pieretti, 2000). For new drug compounds to be considered as analgesic alternatives to opioids, it is necessary that they do not exhibit reinforcing actions after repeated exposure. For example, cebranopadol (GRT6005), a full agonist of NOR and MOR (Linz et al. 2014), is currently tested in clinical trials for acute and chronic pain (Raffa et al. 2017). However, in contrast to AT-121, cebranopadol may be rewarding and possess abuse liability as it generalized to a morphine discriminative stimulus and displayed a conditioned place preference in rodents (de Guglielmo et al. 2017; Tzschentke et al. 2018). Although, concomitant activation of NOR and MOR in several scenarios appears to be detrimental for analgesia (Tian et al. 1997; Lutfy et al. 2003), the promising attributes of AT-121 speaks to the selective properties of individual ligands activating MOR receptors. Another agonist with dual mode of action, prolonged release Tapentadol® is an FDA- and European Union-approved MOR agonist and noradrenaline reuptake inhibitor prescribed for the treatment of moderate and severe chronic pain in adults (Dart et al. 2015; Deeks 2018). The use of Tapentadol® may be a reasonable treatment for chronic pain treatment as it was reported to be effective in relieving moderate-to-severe musculoskeletal pain for up to 2 years of treatment, yet no tolerance and minimal withdrawal symptoms were reported (Buynak et al. 2015; Deeks 2018). Overall, bifunctional NOR/MOR agonists or other ligands with dual mode of action should be developed and considered as alternatives to long-term opioids therapy with opioids such as oxycodone.
The development of novel MOR agonists is another alternative with encouraging therapeutic potential. For example, the novel synthetic opioid manufactured by the pharmaceutical company Trevena, Olinvyk, which is the brand name for TRV-130 or oliceridine (based on agonist bias for G-protein as opposed to arrestin signaling efficacy), has just been approved recently on August 7 2020, by the FDA for the intravenous treatment of moderate-to-severe acute pain in adults,Footnote 23.Footnote 24 It is expected to be available in the fourth quarter of 2020. The approval for Olinvyk was based on positive results from three phase III clinical trials (Bergese et al. 2019; Viscusi et al. 2019; Singla et al. 2019) conducted over 150027 patients with moderate-to-severe acute pain. Two of these phase III clinical trials (randomized, placebo and active-controlled), APOLLO-1 and APOLLO-2, demonstrated that oliceridine has a rapid analgesic efficacy statistically significant compared to placebo and is safe for the intravenous relief of moderate-to-severe acute post-operative pain in 418 patients following bunionectomy (Viscusi et al. 2019) and in 401 patients undergoing abdominoplasty (Singla et al. 2019). Oliceridine had the advantage of being as potent and efficacious as morphine and presented fewer side effects (Viscusi et al. 2016). Indeed, the number of patients experiencing nausea or vomiting after abdominoplasty post-operative treatment was lower with the two equianalgesic dose regimens of 0.35 and 0.5 mg intravenous oliceridine compared to morphine (Singla et al. 2019). The third phase III clinical trial, a large, open-label safety study called ATHENA, concluded that Olinvyk was safe and well-tolerated in a “broad, real-world patient population” with various types of painful medical conditions, including post-operative surgical patients and non-surgical patients (Bergese et al. 2019). More than just lowering the incidence of nausea, vomiting, and constipation, oliceridine seems to generate less respiratory depression than morphine, though the published data on this are controversial as to related to signaling bias (Kliewer et al. 2020) and unconvincing in the published clinical data. An improved respiratory profile would of course represent a significant advantage for the use of oliceridine over morphine. The reduced occurrence of adverse effects has been proposed to reflect oliceridine’s selective activation of the G-protein–coupled signaling pathway with potency and efficacy similar to that of morphine, but with decreased activation of the β-arrestin pathway and receptor internalization (Dahan et al. 2020). Despite these encouraging observations on oliceridine analgesic efficacy and safety, repeated administration of oliceridine or TRV-130 retained abuse liability in a rodent model (Altarifi et al. 2017). Therefore, the FDA restricted its prescription to moderate-to-severe acute pain in adults “where the pain is severe enough to require an intravenous opioid and for whom alternative treatments are inadequate”.Footnote 25 Olinvyk is indicated for high-risk patients, such as elderly, obese, renally impaired and/or comorbid, in short-term hospitals or other controlled clinical settings use28 (Bergese et al. 2019). In contrast to Olinvyk, the abuse-deterrent NKRT-181 (oxycodogel) (Kopruszinski et al. 2020), another novel MOR agonist under evaluation in human clinical trials for the intravenous treatment of severe acute pain, was not approved by the FDA. There have been indeed concerns about the benefit of the drug not exceeding the risk.Footnote 26 The NKRT-181 is composed of a morphinan structure covalently bound to a polyethylene glycol oligomer and was designed to slow rate of entry into the central nervous system to decrease abuse liability without affecting the analgesic properties (Miyazaki et al. 2017; Webster et al. 2018). Despite the slow onset of antinociception, NKTR-181 has similar efficacy to morphine (Kopruszinski et al. 2020). Oral tablets of NKTR-181 was studied in Phase III for patients with moderate-to-severe chronic low back pain (Markman et al. 2019) and in Phase II for patients with osteoarthritis knee pain.Footnote 27 The phase III clinical trial of NKTR-181, in patients with chronic low back pain, reported that NKTR-181 displayed low potential for opioid withdrawal and abuse liability almost similar to that of placebo (Markman et al. 2019). Despite the encouraging results, Nektar decided to withdraw NKTR-181 marketing application because of insufficient evidences determining the possible abuse when snorted or injected, and the potential for liver toxicity.Footnote 28,Footnote 29
Abuse-Deterrent Formulations of Oxycodone and Other Opioids
While research is focusing on exploring new alternatives to oxycodone or opioids in general, one of the strategies encouraged by the White House Executive Office and numerous federal agencies, including the FDA, is to promote the development of abuse-deterrent formulations (ADF) for oral administration.Footnote 30 As we stated previously, orally prescribed oxycodone leads to diversion for illegal abuse, and ADF technologies aim at stopping the diversion, reducing abuse and overdose of orally prescribed opioids by providing physical and chemical barriers (Adler and Mallick-Searle 2018; Litman et al. 2018). There are now ten oral opioid formulations that were granted abuse-deterrent labeling by the FDA (Litman et al. 2018). Five of them are extended release reformulation of oxycodone, including ADF extended release OxyContin® in 2013. Intranasal administration was found to be a route preferred among illicit users for the delivery of opioids, including oxycodone (Cone et al. 2013; Osgood et al. 2012). The ADF extended release OxyContin® tablet is coated with high-molecular-weight polyethylene oxide that hinders tablet crushing into powder and prevents access to the active drug. If a liquid is added to the pill, it turns into a gel making it difficult to inject or inhale. With the abuse-deterrent Oxycontin®, the intranasal oxycodone abuse potential was reported to be lower (Harris et al. 2014). The second available oxycodone extended formulation, Xtampza® ER, received ADF labeling for the intravenous, nasal, and oral routes of administration in 2016. Xtampza® ER, also called oxycodone DETERx®, are microsphere-in-capsules made from the combination of the active oxycodone base with inactive ingredients conferring resistance to crushing, chewing, heating and dissolving.Footnote 31 Approved in 2016 for the intravenous, nasal, and oral routes, but not available in the USA, Troxyca® ER capsules contains pellets of oxycodone surrounding an isolated naltrexone core.Footnote 32 When taken orally, the naltrexone remains sequestered and the patient will experience pain relief from the extended release oxycodone. If the capsule is crushed or chewed, the naltrexone will be released and will block the pharmacological effects of oxycodone (Litman et al. 2018). Both Xtampza® ER and Troxyca® ER have an advantage in patients with difficulty swallowing because they can be sprinkled on food. Targiniq® ER is another combination agonist/antagonist ADF approved by the FDA in 2014 and received the labeling for the intravenous and nasal routes. Targiniq® ER tablets contain oxycodone surrounding naloxone in a 2:1 ratio (Alexander et al. 2014) and are coated by the same PEO barrier as the reformulated OxyContin® ER. Naloxone low oral bioavailability does not interfere with oral Targiniq® ER-induced analgesia. However, naloxone will exhibit opioid antagonism with nasal or intravenous administration (Smith et al. 2012) and the polyethylene oxide barrier provides resistance to crushing and solubilization. Finally, another ADF of oxycodone RoxyBond®, the only immediate release of oxycodone available, was approved in 2017. When RoxyBond® tablet is crushed, a layer of xanthan gum and hypromellose will absorb the oxycodone, which prevents its release into the bloodstream (Maincent and Zhang 2016). Both Targiniq® ER and RoxyBond® have received the ADF labeling for the intravenous and nasal routes, but are not marketed in the USA. Hysingla® ER and Vantrela® ER are both approved ADFs of hydrocodone. Embeda®, MorphaBond® ER and Arymo® ER are morphine extended release formulations. A lot of effort has been mobilized to develop opioid ADF, as illustrated by a recent rejection by the FDA of a new extended release ADF of oxycodone, Aximris XR™, designed by the pharmaceutical company, Intellipharmaceutics,Footnote 33,Footnote 34 Nonetheless, the efficacy of these ADFs to reduce opioid abuse remains questionable, as it is unknown if it will lead to transition to heroin use. An estimate of 80% of the three-fold increase in heroin mortality since 2010 has been attributed to the OxyContin® reformulationFootnote 35 and priming for opioid misuse. Finally, the ADF does not address the problem of patients receiving long-term opioid treatment for chronic pain since they still develop dependence, opponent processes, misuse and abuse disorder.
Conclusion
The prescription drug abuse epidemic in the USA and other westernized countries has had a significant impact on morbidity and mortality. Opioid overdoses continue to rise despite many attempts to curb the epidemic, such as increased regulations, monitoring, and a much-needed emphasis on safe opioid prescription. It is important to emphasize that pain continues to be one of the leading causes of distress and morbidity due to negative affect in our population and the conundrum of effective treatment of pain, while minimizing opioid prescription abuse, is a major challenge that remains unresolved. Here, we have provided evidence of inter-opioid drug differences, comparing oxycodone to other opioid analgesics, which could contribute to diverse reporting of likability and cravings, qualities that contribute to the development of substance use disorders. Although there are individual differences in the propensity to develop a substance use disorder, the reality of the widespread abuse of prescription opioid analgesics makes it imperative to understand the extent opioid choice affects these outcomes. Here, we have highlighted potential mechanisms making it more efficacious in coupling to various signaling cascades recruited to produce their cellular effects. Clearly, drug choice and compliance are an important factor in pain treatment, but prescribing choice must take into account the high rating of abuse risk of immediate release oxycodone compared to other opioid prescription medications (Butler et al. 2011). Nevertheless, no opioid analgesic is without risk of misuse, abuse, and addiction. It is with optimism that advances in our understanding of agonist–receptor interactions and cellular signaling will help identify new improved analgesic drugs with minimal life-threatening consequences.
Notes
International Narcotics Control Board (2019). Estimated World Requirements for 2020—Statistics for 2018. https://www.incb.org/incb/en/narcotic-drugs/Technical_Reports/narcotic_drugs_reports.html. Accessed 04 April 2020.
Centers for Disease Control and Prevention (2019) CDC Guideline for Prescribing Opioids for Chronic Pain. https://www.cdc.gov/drugoverdose/prescribing/guideline.html. Accessed 04 April 2020.
FDA (2020) FDA Briefing Document Joint Meeting of Anesthetic and Analgesic Drug Products Advisory Committee and Drug Safety and Risk Management Advisory Committee January 15, 2020. https://www.fda.gov/advisory-committees/advisory-committee-calendar/january-15-2020-joint-meeting-anesthetic-and-analgesic-drug-products-advisory-committee-and-drug and https://www.fda.gov/media/134150/download. Accessed 04 April 2020.
Centers for Disease Control and Prevention (CDC) (2020) Prescription opioid overdose data. cdc.gov/drugoverdose/data/overdose.html. Accessed 04 April 2020.
FDA (2020) FDA Briefing Document Joint Meeting of Anesthetic and Analgesic Drug Products Advisory Committee and Drug Safety and Risk Management Advisory Committee January 15, 2020. https://www.fda.gov/advisory-committees/advisory-committee-calendar/january-15-2020-joint-meeting-anesthetic-and-analgesic-drug-products-advisory-committee-and-drug and https://www.fda.gov/media/134150/download. Accessed 04 April 2020.
DEA Diversion Control Division (2020) Oxycodone. https://www.deadiversion.usdoj.gov/drug_chem_info/oxycodone/oxycodone.pdf?amp=1. Accessed 06 June 2020.
FDA (2018) FDA analysis of long-term trends in prescription opioid analgesic products: quantity, sales, and price trends. https://www.fda.gov/media/111695/download. Accessed 04 April 2020.
International Narcotics Control Board (2019). Estimated World Requirements for 2020—Statistics for 2018. https://www.incb.org/incb/en/narcotic-drugs/Technical_Reports/narcotic_drugs_reports.html. Accessed 04 April 2020.
Rappaport B (2008) Overview of the November 13, 2008 ALSDAC Meeting to Discuss NDA 22-324 for Remoxy (Oxycodone Hydrochloride Controlled Release) Capsules. A. Division of Anesthesia, and Rheumatology Products, Office of Drug Evaluation II, editor. Accessed 25 June 2020.
Underhill, District Judge (2013) Purdue Pharma v. Kentucky. FindLaw. https://caselaw.findlaw.com/us-2nd-circuit/1619840.html. Accessed 14 April 2020.
Underhill, District Judge (2013) Purdue Pharma v. Kentucky. FindLaw. https://caselaw.findlaw.com/us-2nd-circuit/1619840.html. Accessed 14 April 2020.
U.S. Government Accountability Office (GAO) (2003) Prescription Drugs: OxyContin Abuse and Diversion and Efforts to Address the Problem. Washington, DC: General Accounting Office. https://www.gao.gov/htext/d04110.html. Accessed 26 June 2020.
FDA (2015) Oxycontin prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022272s027lbl.pdf. Accessed 14 April 2020.
Stolberg SG, Gerth J (2000). High-tech stealth being used to sway doctor prescriptions. The New York Times. http://query.nytimes.com/gst/fullpage.html?res=9502EEDF153BF935A25752C1A9669C8B63&sec=&spon=&pagewanted=1. Accessed 14 April 2020.
U.S. Department of Justice (2007) Justice Department Recovers $2 Billion for Fraud Against the Government in Fy 2007; More Than $20 Billion Since 1986. https://www.justice.gov/archive/opa/pr/2007/November/07_civ_873.html. Accessed 14 April 2020.
Meier (2007) U.S. maker of OxyContin painkiller to pay $600 million in guilty plea. The New York Times. http://www.nytimes.com/2007/05/11/business/worldbusiness/11iht-oxy.1.5665287.html. Accessed June 24 2020.
Planalp C and Lahr M (2018) The opioid epidemic: national trends in opioid-related overdose eaths from 2000 to 2016. shadac.https://shadac.org/sites/default/files/publications/NATIONAL_opioid_brief_2000-2016.pdf. Accessed 24 June 2020.
Jones JP (2007) United States of America v. The Purdue Frederick Company, INC., et al. United States District Court for the Western District of Virginia Abingdon Division. http://www.vawd.uscourts.gov/OPINIONS/JONES/107CR00029.PDF. Accessed 24 June2020.
Moritz JC (2018) 6 states sue maker of OxyContin as they battle expenses, human costs of opioid crisis. USA TODAY. https://www.usatoday.com/story/news/nation-now/2018/05/15/six-attorney-generals-opioid-lawsuits/612721002/. Accessed 24 June 2020.
Alpert A, Powell D, Liccardo Pacula R (2017) Supply-Side Drug Policy in the Presence of Substitutes: Evidence from the Introduction of Abuse-Deterrent Opioids. National Bureau of Economic Research. http://www.nber.org/papers/w23031. Accessed 24 June 2020.
Standford School of Medicine. Palliative care. https://palliative.stanford.edu/opioid-conversion/equivalency-table/. Accessed 24 June 2020.
SAMHSA (2006). Treatment Episodes Data Set (TEDS): Highlights—2005. DASIS Series: S-36, DHHS Publication No. SMA 07-4229. Department of Health and Human Services (DHHS): Rockville, MD. Office of Applied Studies. https://wwwdasis.samhsa.gov/dasis2/teds_pubs/2006_teds_highlights_rpt.pdf. Accessed 24 June 2020.
Olinvyk™ is now approved. Trevena. https://olinvyk.com/. Accessed 14 September 2020.
FDA (2020) FDA Approves New Opioid for Intravenous Use in Hospitals, Other Controlled Clinical Settings. https://www.fda.gov/news-events/press-announcements/fda-approves-new-opioid-intravenous-use-hospitals-other-controlled-clinical-settings. Accessed 14 September 2020.
FDA (2020) FDA Approves New Opioid for Intravenous Use in Hospitals, Other Controlled Clinical Settings. https://www.fda.gov/news-events/press-announcements/fda-approves-new-opioid-intravenous-use-hospitals-other-controlled-clinical-settings. Accessed 14 September 2020.
Nektar Therapeutics (2020) Nektar Issues Statement Regarding FDA Advisory Committee Vote for Oxycodegol. Nektar. https://ir.nektar.com/news-releases/news-release-details/nektar-issues-statement-regarding-fda-advisory-committee-vote. Accessed 24 June 2020.
Nektar Therapeutics (2013) Nektar Announces Preliminary Topline Results from Phase 2 Efficacy Study for NKTR-181 in Chronic Pain Patients with Osteoarthritis of the Knee. Nektar. https://ir.nektar.com/news-releases/news-release-details/nektar-announces-preliminary-topline-results-phase-2-efficacy#. Accessed 24 June 2020.
FirstWord Pharma. FDA panel rejects Nektar's opioid analgesic NKTR-181 citing safety concerns, lack of data. https://m.firstwordpharma.com/fda-panel-rejects-nektars-opioid-analgesic-nktr-181-citing-safety-concerns-lack-data. Accessed 24 June 2020.
FDA (2020) FDA Briefing Document, Joint Meeting of the Anesthetic and Analgesic Drug Products Advisory Committee and the Drug Safety and Risk Management Advisory Committee, January 14, 2020. https://www.fda.gov/media/134082/download. Accessed 24 June 2020.
The White House (2017) The president’s commission on combatting drug addiction and the opioid crisis. Office of National Drug Control Policy USA: National Drug Control Strategy. https://www.whitehouse.gov/sites/whitehouse.gov/files/images/Final_Report_Draft_11-15-2017.pdf. Accessed 24 June 2020.
Collegium Pharmaceutical Inc (2020) Xtampza ER. Collegium Pharmaceutical. http://www.collegiumpharma.com/technology/overview. Accessed 24 June 2020.
Pfizer Inc (2017) TROXYCA ER- oxycodone hydrochloride and naltrexone hydrochloride capsule, extended release. Physician prescribing information. Pfizer. http://labeling.pfizer.com/ShowLabeling.aspx?id=4047. Accessed 24 June 2020.
FDA (2020) FDA Briefing Document Joint Meeting of Anesthetic and Analgesic Drug Products Advisory Committee and Drug Safety and Risk Management Advisory Committee January 15, 2020. https://www.fda.gov/advisory-committees/advisory-committee-calendar/january-15-2020-joint-meeting-anesthetic-and-analgesic-drug-products-advisory-committee-and-drug and https://www.fda.gov/media/134150/download. Accessed 04 April 2020.
Intellipharmaceutics (2020) https://www.intellipharmaceutics.com/news-media/press-releases/detail/217/intellipharmaceutics-provides-update-on-fda-advisory. Accessed 24 June 2020.
Alpert A, Powell D, Pacula RL (2017) Supply-side drug policy in the presence of substitutes: Evidence from the introduction of abuse-deterrent opioid. The national bureau of economic research. http://www.nber.org/papers/w23031. Accessed 24 June 2020.
References
Abel GA, Penson RT, Joffe S, Schapira L, Chabner BA, Lynch TJ (2006) Direct-to-consumer advertising in oncology. Oncologist 11(2):217–226. https://doi.org/10.1634/theoncologist.11-2-217
Adler JA, Mallick-Searle T (2018) An overview of abuse-deterrent opioids and recommendations for practical patient care. J Multidiscip Healthc 11:323–332. https://doi.org/10.2147/JMDH.S166915
Ahmed SH, Kenny PJ, Koob GF, Markou A (2002) Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat Neurosci 5(7):625–626. https://doi.org/10.1038/nn872
Alexander L, Mannion RO, Weingarten B, Fanelli RJ, Stiles GL (2014) Development and impact of prescription opioid abuse deterrent formulation technologies. Drug Alcohol Depend 138:1–6. https://doi.org/10.1016/j.drugalcdep.2014.02.006
Altarifi AA, David B, Muchhala KH, Blough BE, Akbarali H, Negus SS (2017) Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J Psychopharmacol 31(6):730–739. https://doi.org/10.1177/0269881116689257
Anselme P, Robinson MJ (2016) “Wanting,” “liking,” and their relation to consciousness. J Exp Psychol Anim Learn Cogn 42(2):123–140. https://doi.org/10.1037/xan0000090
Aono Y, Saigusa T, Mizoguchi N, Iwakami T, Takada K, Gionhaku N, Oi Y, Ueda K, Koshikawa N, Cools AR (2008) Role of GABAA receptors in the endomorphin-1-, but not endomorphin-2-, induced dopamine efflux in the nucleus accumbens of freely moving rats. Eur J Pharmacol 580(1–2):87–94. https://doi.org/10.1016/j.ejphar.2007.10.020
Atluri S, Sudarshan G, Manchikanti L (2014) Assessment of the trends in medical use and misuse of opioid analgesics from 2004 to 2011. Pain Physician 17(2):E119-128
Backlund M, Lindgren L, Kajimoto Y, Rosenberg PH (1997) Comparison of epidural morphine and oxycodone for pain after abdominal surgery. J Clin Anesth 9(1):30–35. https://doi.org/10.1016/S0952-8180(96)00212-7
Bailey CP, Smith FL, Kelly E, Dewey WL, Henderson G (2006) How important is protein kinase C in mu-opioid receptor desensitization and morphine tolerance? Trends Pharmacol Sci 27(11):558–565. https://doi.org/10.1016/j.tips.2006.09.006
Ballantyne JC (2015) Opioid therapy in chronic pain. Phys Med Rehabil Clin N Am 26(2):201–218. https://doi.org/10.1016/j.pmr.2014.12.001
Barrot M, Sesack SR, Georges F, Pistis M, Hong S, Jhou TC (2012) Braking dopamine systems: a new GABA master structure for mesolimbic and nigrostriatal functions. J Neurosci 32(41):14094–14101. https://doi.org/10.1523/JNEUROSCI.3370-12.2012
Bayer VE, Pickel VM (1990) Ultrastructural localization of tyrosine hydroxylase in the rat ventral tegmental area: relationship between immunolabeling density and neuronal associations. J Neurosci 10(9):2996–3013. https://doi.org/10.1523/JNEUROSCI.10-09-02996.1990
Beckett AH, Casy AF (1954) Stereochemistry of certain analgesics. Nature 173(4417):1231–1232. https://doi.org/10.1038/1731231a0
Bercovitch M, Adunsky A (2006) High dose controlled-release oxycodone in hospice care. J Pain Palliat Care Pharmacother 20(4):33–39
Bergese SD, Brzezinski M, Hammer GB, Beard TL, Pan PH, Mace SE, Berkowitz RD, Cochrane K, Wase L, Minkowitz HS, Habib AS (2019) ATHENA: a phase 3, open-label study of the safety and effectiveness of oliceridine (TRV130), a G-protein selective agonist at the micro-opioid receptor, in patients with moderate to severe acute pain requiring parenteral opioid therapy. J Pain Res 12:3113–3126. https://doi.org/10.2147/JPR.S217563
Berridge KC (2000) Measuring hedonic impact in animals and infants: microstructure of affective taste reactivity patterns. Neurosci Biobehav Rev 24(2):173–198. https://doi.org/10.1016/s0149-7634(99)00072-x
Berridge KC (2018) Evolving concepts of emotion and motivation. Front Psychol 9:1647. https://doi.org/10.3389/fpsyg.2018.01647
Berridge KC, Kringelbach ML (2015) Pleasure systems in the brain. Neuron 86(3):646–664. https://doi.org/10.1016/j.neuron.2015.02.018
Berridge KC, Robinson TE (2016) Liking, wanting, and the incentive-sensitization theory of addiction. Am Psychol 71(8):670–679. https://doi.org/10.1037/amp0000059
Berridge KC, Valenstein ES (1991) What psychological process mediates feeding evoked by electrical stimulation of the lateral hypothalamus? Behav Neurosci 105(1):3–14
Berridge KC, Venier IL, Robinson TE (1989) Taste reactivity analysis of 6-hydroxydopamine-induced aphagia: implications for arousal and anhedonia hypotheses of dopamine function. Behav Neurosci 103(1):36–45. https://doi.org/10.1037//0735-7044.103.1.36
Berridge KC, Robinson TE, Aldridge JW (2009) Dissecting components of reward: “liking”, “wanting”, and learning. Curr Opin Pharmacol 9(1):65–73. https://doi.org/10.1016/j.coph.2008.12.014
Berrios J, Stamatakis AM, Kantak PA, McElligott ZA, Judson MC, Aita M, Rougie M, Stuber GD, Philpot BD (2016) Loss of UBE3A from TH-expressing neurons suppresses GABA co-release and enhances VTA-NAc optical self-stimulation. Nat Commun 7:10702. https://doi.org/10.1038/ncomms10702
Bertekap RL, Burford NT, Li Z, Alt A (2015) High-throughput screening for allosteric modulators of GPCRs. Methods Mol Biol 1335:223–240. https://doi.org/10.1007/978-1-4939-2914-6_15
Bisignano P, Burford NT, Shang Y, Marlow B, Livingston KE, Fenton AM, Rockwell K, Budenholzer L, Traynor JR, Gerritz SW, Alt A, Filizola M (2015) Ligand-based discovery of a new scaffold for allosteric modulation of the mu-opioid receptor. J Chem Inf Model 55(9):1836–43. https://doi.org/10.1021/acs.jcim.5b00388
Blackwood CA, McCoy MT, Ladenheim B, Cadet JL (2020) Oxycodone self-administration activates the mitogen-activated protein kinase/mitogen- and stress-activated protein kinase (MAPK-MSK) signaling pathway in the rat dorsal striatum. bioRxiv. https://doi.org/10.1101/2020.08.31.276253
Blume AJ (1978) Interaction of ligands with the opiate receptors of brain membranes: regulation by ions and nucleotides. Proc Natl Acad Sci USA 75(4):1713–1717. https://doi.org/10.1073/pnas.75.4.1713
Blume AJ, Lichtshtein D, Boone G (1979) Coupling of opiate receptors to adenylate cyclase: requirement for Na+ and GTP. Proc Natl Acad Sci USA 76(11):5626–5630. https://doi.org/10.1073/pnas.76.11.5626
Bostrom E, Hammarlund-Udenaes M, Simonsson US (2008) Blood–brain barrier transport helps to explain discrepancies in in vivo potency between oxycodone and morphine. Anesthesiology 108(3):495–505. https://doi.org/10.1097/ALN.0b013e318164cf9e
Bourdy R, Barrot M (2012) A new control center for dopaminergic systems: pulling the VTA by the tail. Trends Neurosci 35(11):681–690. https://doi.org/10.1016/j.tins.2012.06.007
Brauer LH, De Wit H (1997) High dose pimozide does not block amphetamine-induced euphoria in normal volunteers. Pharmacol Biochem Behav 56(2):265–272. https://doi.org/10.1016/s0091-3057(96)00240-7
Bromberg-Martin ES, Matsumoto M, Hikosaka O (2010) Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 68(5):815–834. https://doi.org/10.1016/j.neuron.2010.11.022
Bruera E, Belzile M, Pituskin E, Fainsinger R, Darke A, Harsanyi Z, Babul N, Ford I (1998) Randomized, double-blind, cross-over trial comparing safety and efficacy of oral controlled-release oxycodone with controlled-release morphine in patients with cancer pain. J Clin Oncol 16(10):3222–3229. https://doi.org/10.1200/JCO.1998.16.10.3222
Burford NT, Clark MJ, Wehrman TS, Gerritz SW, Banks M, O’Connell J, Traynor JR, Alt A (2013) Discovery of positive allosteric modulators and silent allosteric modulators of the mu-opioid receptor. Proc Natl Acad Sci USA 110(26):10830–5. https://doi.org/10.1073/pnas.1300393110
Burford NT, Wehrman T, Bassoni D, O’Connell J, Banks M, Zhang L, Alt A (2014) Identification of selective agonists and positive allosteric modulators for micro- and delta-opioid receptors from a single high-throughput screen. J Biomol Screen 19(9):1255–1265. https://doi.org/10.1177/1087057114542975
Burford NT, Livingston KE, Canals M, Ryan MR, Budenholzer LM, Han Y, Shang Y, Herbst JJ, O’Connell J, Banks M, Zhang L, Filizola M, Bassoni DL, Wehrman TS, Christopoulos A, Traynor JR, Gerritz SW, Alt A (2015) Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the delta-opioid receptor. J Med Chem 58(10):4220–4229. https://doi.org/10.1021/acs.jmedchem.5b00007
Butler SF, Fernandez KC, Chang A, Benoit C, Morey LC, Black R, Katz N (2010) Measuring attractiveness for abuse of prescription opioids. Pain Med 11(1):67–80. https://doi.org/10.1111/j.1526-4637.2009.00736.x
Butler SF, Black RA, Cassidy TA, Dailey TM, Budman SH (2011) Abuse risks and routes of administration of different prescription opioid compounds and formulations. Harm Reduct J 8:29. https://doi.org/10.1186/1477-7517-8-29
Buynak R, Rappaport SA, Rod K, Arsenault P, Heisig F, Rauschkolb C, Etropolski M (2015) Long-term safety and efficacy of tapentadol extended release following up to 2 years of treatment in patients with moderate to severe, chronic pain: results of an open-label extension trial. Clin Ther 37(11):2420–2438. https://doi.org/10.1016/j.clinthera.2015.08.014
Cahill CM (2020) Opioid dose regimen shapes mesolimbic adaptations. Neuropsychopharmacology. https://doi.org/10.1038/s41386-020-0679-y
Caldwell JR, Hale ME, Boyd RE, Hague JM, Iwan T, Shi M, Lacouture PG (1999) Treatment of osteoarthritis pain with controlled release oxycodone or fixed combination oxycodone plus acetaminophen added to nonsteroidal antiinflammatory drugs: a double blind, randomized, multicenter, placebo controlled trial. J Rheumatol 26(4):862–869
Caprioli D, Calu D, Shaham Y (2014) Loss of phasic dopamine: a new addiction marker? Nat Neurosci 17(5):644–646. https://doi.org/10.1038/nn.3699
Caraceni AT, Brunelli C, Rocco P, Minghetti P (2013) Trends in opioid analgesics sales to community pharmacies and hospitals in Italy (2000–2010). Minerva Anestesiol 79(8):906–914
Carlson RG, Nahhas RW, Martins SS, Daniulaityte R (2016) Predictors of transition to heroin use among initially non-opioid dependent illicit pharmaceutical opioid users: A natural history study. Drug Alcohol Depend 160:127–134. https://doi.org/10.1016/j.drugalcdep.2015.12.026
Castro D, Berridge KC (2012) Mu opioid, delta or kappa opioid receptor activation in nucleus accumbens hotspot enhances 'liking' reaction to hedonic impact of tastes. Society for Neuroscience Abstracts
Castro DC, Berridge KC (2014) Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness “liking” and “wanting.” J Neurosci 34(12):4239–4250. https://doi.org/10.1523/JNEUROSCI.4458-13.2014
Castro DC, Berridge KC (2017) Opioid and orexin hedonic hotspots in rat orbitofrontal cortex and insula. Proc Natl Acad Sci USA 114(43):E9125–E9134. https://doi.org/10.1073/pnas.1705753114
Chan S, Edwards SR, Wyse BD, Smith MT (2008) Sex differences in the pharmacokinetics, oxidative metabolism and oral bioavailability of oxycodone in the Sprague-Dawley rat. Clin Exp Pharmacol Physiol 35(3):295–302. https://doi.org/10.1111/j.1440-1681.2007.04821.x
Childress AR, Ehrman RN, Wang Z, Li Y, Sciortino N, Hakun J, Jens W, Suh J, Listerud J, Marquez K, Franklin T, Langleben D, Detre J, O’Brien CP (2008) Prelude to passion: limbic activation by “unseen” drug and sexual cues. PLoS ONE 3(1):e1506. https://doi.org/10.1371/journal.pone.0001506
Chu J, Zheng H, Zhang Y, Loh HH, Law PY (2010) Agonist-dependent mu-opioid receptor signaling can lead to heterologous desensitization. Cell Signal 22(4):684–696. https://doi.org/10.1016/j.cellsig.2009.12.003
Cicero TJ, Ellis MS (2015) Abuse-deterrent formulations and the prescription opioid abuse epidemic in the United States: lessons learned from OxyContin. JAMA Psychiatry 72(5):424–430. https://doi.org/10.1001/jamapsychiatry.2014.3043
Cicero TJ, Inciardi JA, Munoz A (2005) Trends in abuse of Oxycontin and other opioid analgesics in the United States: 2002–2004. J Pain 6(10):662–672. https://doi.org/10.1016/j.jpain.2005.05.004
Cicero TJ, Ellis MS, Paradis A, Ortbal Z (2010) Determinants of fentanyl and other potent micro opioid agonist misuse in opioid-dependent individuals. Pharmacoepidemiol Drug Saf 19(10):1057–1063. https://doi.org/10.1002/pds.1989
Cicero TJ, Ellis MS, Surratt HL (2012) Effect of abuse-deterrent formulation of OxyContin. N Engl J Med 367(2):187–189. https://doi.org/10.1056/NEJMc1204141
Cicero TJ, Ellis MS, Surratt HL, Kurtz SP (2013) Factors influencing the selection of hydrocodone and oxycodone as primary opioids in substance abusers seeking treatment in the United States. Pain 154(12):2639–2648. https://doi.org/10.1016/j.pain.2013.07.025
Codd EE, Shank RP, Schupsky JJ, Raffa RB (1995) Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther 274(3):1263–1270
Comer SD, Walker EA, Collins ED (2005) Buprenorphine/naloxone reduces the reinforcing and subjective effects of heroin in heroin-dependent volunteers. Psychopharmacology (Berl) 181(4):664–675. https://doi.org/10.1007/s00213-005-0023-6
Comer SD, Sullivan MA, Whittington RA, Vosburg SK, Kowalczyk WJ (2008) Abuse liability of prescription opioids compared to heroin in morphine-maintained heroin abusers. Neuropsychopharmacology 33(5):1179–1191. https://doi.org/10.1038/sj.npp.1301479
Comer SD, Metz VE, Cooper ZD, Kowalczyk WJ, Jones JD, Sullivan MA, Manubay JM, Vosburg SK, Smith ME, Peyser D, Saccone PA (2013) Comparison of a drug versus money and drug versus drug self-administration choice procedure with oxycodone and morphine in opioid addicts. Behav Pharmacol 24(5–6):504–516. https://doi.org/10.1097/FBP.0b013e328363d1c4
Cone EJ, Darwin WD, Buchwald WF, Gorodetzky CW (1983) Oxymorphone metabolism and urinary excretion in human, rat, guinea pig, rabbit, and dog. Drug Metab Dispos 11(5):446–450
Cone EJ, Giordano J, Weingarten B (2013) An iterative model for in vitro laboratory assessment of tamper deterrent formulations. Drug Alcohol Depend 131(1–2):100–105. https://doi.org/10.1016/j.drugalcdep.2012.12.006
Conibear AE, Kelly E (2019) A biased view of mu-opioid receptors? Mol Pharmacol 96(5):542–549. https://doi.org/10.1124/mol.119.115956
Connors AL (2009) Big bad pharma: an ethical analysis of physician-directed and consumer-directed marketing tactics. Albany Law Rev 73(1):243–282
Cremeans CM, Gruley E, Kyle DJ, Ko MC (2012) Roles of mu-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J Pharmacol Exp Ther 343(1):72–81. https://doi.org/10.1124/jpet.112.194308
Dahan A, van Dam CJ, Niesters M, van Velzen M, Fossler MJ, Demitrack MA, Olofsen E (2020) Benefit and risk evaluation of biased mu-receptor agonist oliceridine versus morphine. Anesthesiology 133(3):559–568. https://doi.org/10.1097/ALN.0000000000003441
Dart RC, Surratt HL, Cicero TJ, Parrino MW, Severtson SG, Bucher-Bartelson B, Green JL (2015) Trends in opioid analgesic abuse and mortality in the United States. N Engl J Med 372(3):241–248. https://doi.org/10.1056/NEJMsa1406143
Davis MP (2012) Drug management of visceral pain: concepts from basic research. Pain Res Treat 2012:265605. https://doi.org/10.1155/2012/265605
de Wit H, Phillips TJ (2012) Do initial responses to drugs predict future use or abuse? Neurosci Biobehav Rev 36:1565–1576. https://doi.org/10.1016/j.neubiorev.2012.04.005
de Guglielmo G, Matzeu A, Kononoff J, Mattioni J, Martin-Fardon R, George O (2017) Cebranopadol blocks the escalation of cocaine intake and conditioned reinstatement of cocaine seeking in rats. J Pharmacol Exp Ther 362(3):378–384. https://doi.org/10.1124/jpet.117.241042
De Schepper HU, Cremonini F, Park MI, Camilleri M (2004) Opioids and the gut: pharmacology and current clinical experience. Neurogastroenterol Motil 16(4):383–394. https://doi.org/10.1111/j.1365-2982.2004.00513.x
Deeks ED (2018) Tapentadol prolonged release: a review in pain management. Drugs 78(17):1805–1816. https://doi.org/10.1007/s40265-018-1007-2
Defalque RJ, Wright AJ (2003) Scophedal (SEE) was it a fad or a miracle drug? Bull Anesth Hist 21(4):12–14. https://doi.org/10.1016/s1522-8649(03)50051-8
Di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85(14):5274–5278. https://doi.org/10.1073/pnas.85.14.5274
Di Giannuario A, Pieretti S (2000) Nociceptin differentially affects morphine-induced dopamine release from the nucleus accumbens and nucleus caudate in rats. Peptides 21(7):1125–1130. https://doi.org/10.1016/s0196-9781(00)00250-3
Ding H, Kiguchi N, Yasuda D, Daga PR, Polgar WE, Lu JJ, Czoty PW, Kishioka S, Zaveri NT, Ko MC (2018) A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aar3483
Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, Zweifel LS, Kieffer BL, Phillips PE, Chavkin C (2015) Kappa opioid receptor-induced aversion requires p38 MAPK activation in VTA dopamine neurons. J Neurosci 35(37):12917–12931. https://doi.org/10.1523/JNEUROSCI.2444-15.2015
Emery MA, Bates ML, Wellman PJ, Eitan S (2015) Differential effects of oxycodone, hydrocodone, and morphine on the responses of D2/D3 dopamine receptors. Behav Brain Res 284:37–41. https://doi.org/10.1016/j.bbr.2015.01.023
Emery MA, Bates ML, Wellman PJ, Eitan S (2016) Differential effects of oxycodone, hydrocodone, and morphine on activation levels of signaling molecules. Pain Med 17(5):908–914. https://doi.org/10.1111/pme.12918
English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsaki G, Deisseroth K, Tepper JM, Koos T (2011) GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci 15(1):123–130. https://doi.org/10.1038/nn.2984
Evans CJ, Cahill CM (2016) Neurobiology of opioid dependence in creating addiction vulnerability. F1000Res. https://doi.org/10.12688/f1000research.8369.1
Fain KM, Alexander GC (2014) Mind the gap: understanding the effects of pharmaceutical direct-to-consumer advertising. Med Care 52(4):291–293. https://doi.org/10.1097/MLR.0000000000000126
Fawzi AB, Macdonald D, Benbow LL, Smith-Torhan A, Zhang H, Weig BC, Ho G, Tulshian D, Linder ME, Graziano MP (2001) SCH-202676: an allosteric modulator of both agonist and antagonist binding to G protein-coupled receptors. Mol Pharmacol 59(1):30–7. https://doi.org/10.1124/mol.59.1.30
Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC (2014) Molecular control of delta-opioid receptor signalling. Nature 506(7487):191–196. https://doi.org/10.1038/nature12944
Ford CP, Mark GP, Williams JT (2006) Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci 26(10):2788–2797. https://doi.org/10.1523/JNEUROSCI.4331-05.2006
Frank ST, Krumm B, Spanagel R (2008) Cocaine-induced dopamine overflow within the nucleus accumbens measured by in vivo microdialysis: a meta-analysis. Synapse 62(4):243–252. https://doi.org/10.1002/syn.20489
Fraser HF, Van Horn GD, Martin WR, Wolbach AB, Isbell H (1961) Methods for evaluating addiction liability. (A) “Attitude” of opiate addicts toward opiate-like drugs. (B) a short-term “direct” addiction test. J Pharmacol Exp Ther 133:371–387
Gavin PD, Tremper L, Smith A, Williams G, Brooker C (2017) Transdermal oxycodone patch for the treatment of postherpetic neuralgia: a randomized, double-blind, controlled trial. Pain Manag 7(4):255–267. https://doi.org/10.2217/pmt-2016-0067
Gorka SM, Fitzgerald DA, de Wit H, Angstadt M, Phan KL (2014) Opioid modulation of resting-state anterior cingulate cortex functional connectivity. J Psychopharmacol 28(12):1115–1124. https://doi.org/10.1177/0269881114548436
Gould RW, Gunter BW, Bubser M, Matthews RT, Teal LB, Ragland MG, Bridges TM, Garrison AT, Winder DG, Lindsley CW, Jones CK (2019) Acute negative allosteric modulation of M5 muscarinic acetylcholine receptors inhibits oxycodone self-Administration and cue-induced reactivity with no effect on antinociception. ACS Chem Neurosci 10(8):3740–3750. https://doi.org/10.1021/acschemneuro.9b00274
Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK (2012) Structure of the delta-opioid receptor bound to naltrindole. Nature 485(7398):400–404. https://doi.org/10.1038/nature11111
Grau LE, Dasgupta N, Harvey AP, Irwin K, Givens A, Kinzly ML, Heimer R (2007) Illicit use of opioids: is OxyContin a “gateway drug”? Am J Addict 16(3):166–173. https://doi.org/10.1080/10550490701375293
Grim TW, Acevedo-Canabal A, Bohn LM (2020) Toward directing opioid receptor signaling to refine opioid therapeutics. Biol Psychiatry 87(1):15–21. https://doi.org/10.1016/j.biopsych.2019.10.020
Guo KK, Deng CQ, Lu GJ, Zhao GL (2018) Comparison of analgesic effect of oxycodone and morphine on patients with moderate and advanced cancer pain: a meta-analysis. BMC Anesthesiol 18(1):132. https://doi.org/10.1186/s12871-018-0583-8
Hagen NA, Babul N (1997) Comparative clinical efficacy and safety of a novel controlled-release oxycodone formulation and controlled-release hydromorphone in the treatment of cancer pain. Cancer 79(7):1428–1437
Halls ML, Yeatman HR, Nowell CJ, Thompson GL, Gondin AB, Civciristov S, Bunnett NW, Lambert NA, Poole DP, Canals M (2016) Plasma membrane localization of the mu-opioid receptor controls spatiotemporal signaling. Sci Signal 9(414):16. https://doi.org/10.1126/scisignal.aac9177
Hamunen K, Paakkari P, Kalso E (2009) Trends in opioid consumption in the Nordic countries 2002–2006. Eur J Pain 13(9):954–962. https://doi.org/10.1016/j.ejpain.2008.11.006
Hanks GW, Conno F, Cherny N, Hanna M, Kalso E, McQuay HJ, Mercadante S, Meynadier J, Poulain P, Ripamonti C, Radbruch L, Casas JR, Sawe J, Twycross RG, Ventafridda V (2001) Morphine and alternative opioids in cancer pain: the EAPC recommendations. Br J Cancer 84(5):587–593. https://doi.org/10.1054/bjoc.2001.1680
Hansen TM, Lelic D, Olesen AE, Drewes AM, Frokjaer JB (2018) Differential effects of oxycodone and venlafaxine on resting state functional connectivity-A randomized placebo-controlled magnetic resonance imaging study. CNS Neurosci Ther 24(9):820–827. https://doi.org/10.1111/cns.12827
Harris SC, Perrino PJ, Smith I, Shram MJ, Colucci SV, Bartlett C, Sellers EM (2014) Abuse potential, pharmacokinetics, pharmacodynamics, and safety of intranasally administered crushed oxycodone HCl abuse-deterrent controlled-release tablets in recreational opioid users. J Clin Pharmacol 54(4):468–477. https://doi.org/10.1002/jcph.235
Heiskanen T, Kalso E (1997) Controlled-release oxycodone and morphine in cancer related pain. Pain 73(1):37–45. https://doi.org/10.1016/s0304-3959(97)00072-9
Hinkle C, Dedkov E, Buono B, Ferraro T (2019) Colocalization of MOR1 and GAD67 in mouse nucleus accumbens. bioRxiv. https://doi.org/10.1101/574434
Hirsch A, Proescholdbell SK, Bronson W, Dasgupta N (2014) Prescription histories and dose strengths associated with overdose deaths. Pain Med 15(7):1187–1195. https://doi.org/10.1111/pme.12391
Hser YI, Mooney LJ, Saxon AJ, Miotto K, Bell DS, Huang D (2017) Chronic pain among patients with opioid use disorder: results from electronic health records data. J Subst Abuse Treat 77:26–30. https://doi.org/10.1016/j.jsat.2017.03.006
Hu E, Calo G, Guerrini R, Ko MC (2010) Long-lasting antinociceptive spinal effects in primates of the novel nociceptin/orphanin FQ receptor agonist UFP-112. Pain 148(1):107–113
Hyman SE, Malenka RC, Nestler EJ (2006) Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29:565–598. https://doi.org/10.1146/annurev.neuro.29.051605.113009
Ibanez-Sandoval O, Tecuapetla F, Unal B, Shah F, Koos T, Tepper JM (2010) Electrophysiological and morphological characteristics and synaptic connectivity of tyrosine hydroxylase-expressing neurons in adult mouse striatum. J Neurosci 30(20):6999–7016. https://doi.org/10.1523/JNEUROSCI.5996-09.2010
Ibanez-Sandoval O, Tecuapetla F, Unal B, Shah F, Koos T, Tepper JM (2011) A novel functionally distinct subtype of striatal neuropeptide Y interneuron. J Neurosci 31(46):16757–16769. https://doi.org/10.1523/JNEUROSCI.2628-11.2011
Ikemoto S (2010) Brain reward circuitry beyond the mesolimbic dopamine system: a neurobiological theory. Neurosci Biobehav Rev 35(2):129–50. https://doi.org/10.1016/j.neubiorev.2010.02.001
Iriah SC, Trivedi M, Kenkel W, Grant SE, Moore K, Yee JR, Madularu D, Kulkarni P, Ferris CF (2019) Oxycodone exposure: a magnetic resonance imaging study in response to acute and chronic oxycodone treatment in rats. Neuroscience 398:88–101. https://doi.org/10.1016/j.neuroscience.2018.11.042
Ito R, Dalley JW, Howes SR, Robbins TW, Everitt BJ (2000) Dissociation in conditioned dopamine release in the nucleus accumbens core and shell in response to cocaine cues and during cocaine-seeking behavior in rats. J Neurosci 20(19):7489–7495. https://doi.org/10.1523/JNEUROSCI.20-19-07489.2000
Jalabert M, Bourdy R, Courtin J, Veinante P, Manzoni OJ, Barrot M, Georges F (2011) Neuronal circuits underlying acute morphine action on dopamine neurons. Proc Natl Acad Sci USA 108(39):16446–16450. https://doi.org/10.1073/pnas.1105418108
Jang CG, Whitfield T, Schulteis G, Koob GF, Wee S (2013) A dysphoric-like state during early withdrawal from extended access to methamphetamine self-administration in rats. Psychopharmacology (Berl) 225(3):753–763. https://doi.org/10.1007/s00213-012-2864-0
Johnson SW, North RA (1992) Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci 12(2):483–488. https://doi.org/10.1523/JNEUROSCI.12-02-00483.1992
Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, Mundell SJ, Bailey CP, Kelly E, Henderson G (2006) Agonist-selective mechanisms of mu-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol 70(2):676–685. https://doi.org/10.1124/mol.106.022376
Jones JD, Mumtaz M, Manubay JM, Mogali S, Sherwin E, Martinez S, Comer SD (2019) Assessing the contribution of opioid- and dopamine-related genetic polymorphisms to the abuse liability of oxycodone. Pharmacol Biochem Behav 186:172778. https://doi.org/10.1016/j.pbb.2019.172778
Jullié D, Gondin AB, von Zastrow M, Canals M (2020) Opioid pharmacology under the microscope. Mol Pharmacol 98(4):425–432. https://doi.org/10.1124/mol.119.119321
Jutkiewicz EM, Palleiko BA, Dripps IJ, Rice KC (2019) The opioid receptor positive allosteric modulator BMS-986187 enhances some, but not all, delta-opioid receptor mediated behaviors. FASEB J 33(S1):6665. https://doi.org/10.1096/fasebj.2019.33.1_supplement.666.5
Kaiko RF, Benziger DP, Fitzmartin RD, Burke BE, Reder RF, Goldenheim PD (1996) Pharmacokinetic-pharmacodynamic relationships of controlled-release oxycodone. Clin Pharmacol Ther 59(1):52–61. https://doi.org/10.1016/S0009-9236(96)90024-7
Kalso E (2005) Oxycodone. J Pain Symptom Manage 29:S47-56. https://doi.org/10.1016/j.jpainsymman.2005.01.010
Kalso E, Vainio A (1990) Morphine and oxycodone hydrochloride in the management of cancer pain. Clin Pharmacol Ther 47(5):639–646. https://doi.org/10.1038/clpt.1990.85
Kalso E, Poyhia R, Onnela P, Linko K, Tigerstedt I, Tammisto T (1991) Intravenous morphine and oxycodone for pain after abdominal surgery. Acta Anaesthesiol Scand 35(7):642–646. https://doi.org/10.1111/j.1399-6576.1991.tb03364.x
Kapitzke D, Vetter I, Cabot PJ (2005) Endogenous opioid analgesia in peripheral tissues and the clinical implications for pain control. Ther Clin Risk Manag 1(4):279–297
Kathmann M, Flau K, Redmer A, Trankle C, Schlicker E (2006) Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn Schmiedebergs Arch Pharmacol 372(5):354–361. https://doi.org/10.1007/s00210-006-0033-x
Katz N, Fernandez K, Chang A, Benoit C, Butler SF (2008) Internet-based survey of nonmedical prescription opioid use in the United States. Clin J Pain 24(6):528–535. https://doi.org/10.1097/AJP.0b013e318167a087
Kemp JM, Powell TP (1971) The structure of the caudate nucleus of the cat: light and electron microscopy. Philos Trans R Soc Lond B Biol Sci 262(845):383–401. https://doi.org/10.1098/rstb.1971.0102
Kenny PJ, Chen SA, Kitamura O, Markou A, Koob GF (2006) Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J Neurosci 26(22):5894–5900. https://doi.org/10.1523/JNEUROSCI.0740-06.2006
Kim JI, Ganesan S, Luo SX, Wu YW, Park E, Huang EJ, Chen L, Ding JB (2015) Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 350(6256):102–106. https://doi.org/10.1126/science.aac4690
Kim MK, Ahn SE, Shin E, Park SW, Choi JH, Kang HY (2018) Comparison of analgesic efficacy of oxycodone and fentanyl after total hip replacement surgery: a randomized controlled trial. Medicine (Baltimore) 97(49):e13385. https://doi.org/10.1097/MD.0000000000013385
King SJ, Reid C, Forbes K, Hanks G (2011) A systematic review of oxycodone in the management of cancer pain. Palliat Med 25(5):454–470. https://doi.org/10.1177/0269216311401948
Kliewer A, Gillis A, Hill R, Schmiedel F, Bailey C, Kelly E, Henderson G, Christie MJ, Schulz S (2020) Morphine-induced respiratory depression is independent of beta-arrestin2 signalling. Br J Pharmacol 177(13):2923–2931. https://doi.org/10.1111/bph.15004
Koob GF, Le Moal M (1997) Drug abuse: hedonic homeostatic dysregulation. Science 278(5335):52–58. https://doi.org/10.1126/science.278.5335.52
Kopruszinski CM, Swiokla J, Lee YS, Navratilova E, VanderVeen L, Yang M, Liu Y, Miyazaki T, Schmidt WK, Zalevsky J, Porreca F (2020) Preclinical assessment of the analgesic pharmacology of NKTR-181 in rodents. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-020-00816-3
Kringelbach ML (2010) The hedonic brain: A functional neuroanatomy of human pleasure. In: Kringelbach ML, Berridge KC (eds) Pleasures of the brain. Oxford University Press, Oxford, pp 202–221
Kringelbach ML, O’Doherty J, Rolls ET, Andrews C (2003) Activation of the human orbitofrontal cortex to a liquid food stimulus is correlated with its subjective pleasantness. Cereb Cortex 13(10):1064–1071. https://doi.org/10.1093/cercor/13.10.1064
Kuypers KPC, Desousafernandesperna EB, Dolder PC, Toennes SW, Theunissen EL, Mason NL, Hutten N, Ramaekers JG (2018) Drug liking and wanting, not impulsive action or reflection is increased by 4-fluoroamphetamine. Psychopharmacology (Berl) 235(8):2349–2356. https://doi.org/10.1007/s00213-018-4931-7
Labouèbe G, Lomazzi M, Cruz HG, Creton C, Lujan R, Li M, Yanagawa Y, Obata K, Watanabe M, Wickman K, Boyer SB, Slesinger PA, Luscher C (2007) RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci 10(12):1559–1568. https://doi.org/10.1038/nn2006
Lalovic B, Phillips B, Risler LL, Howald W, Shen DD (2004) Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metab Dispos 32(4):447–454. https://doi.org/10.1124/dmd.32.4.447
Lalovic B, Kharasch E, Hoffer C, Risler L, Liu-Chen LY, Shen DD (2006) Pharmacokinetics and pharmacodynamics of oral oxycodone in healthy human subjects: role of circulating active metabolites. Clin Pharmacol Ther 79(5):461–479. https://doi.org/10.1016/j.clpt.2006.01.009
Lauretti GR, Oliveira GM, Pereira NL (2003) Comparison of sustained-release morphine with sustained-release oxycodone in advanced cancer patients. Br J Cancer 89(11):2027–2030. https://doi.org/10.1038/sj.bjc.6601365
Law PY, Hom DS, Loh HH (1983) Opiate receptor down-regulation and desensitization in neuroblastoma X glioma NG108-15 hybrid cells are two separate cellular adaptation processes. Mol Pharmacol 24(3):413–424
Law PY, Wong YH, Loh HH (2000) Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol 40:389–430. https://doi.org/10.1146/annurev.pharmtox.40.1.389
Lee K, Vuong HE, Nusbaum DJ, Hsiao EY, Evans CJ, Taylor AMW (2018) The gut microbiota mediates reward and sensory responses associated with regimen-selective morphine dependence. Neuropsychopharmacology 43(13):2606–2614. https://doi.org/10.1038/s41386-018-0211-9
Lefevre EM, Pisansky MT, Toddes C, Baruffaldi F, Pravetoni M, Tian L, Kono TJY, Rothwell PE (2020) Interruption of continuous opioid exposure exacerbates drug-evoked adaptations in the mesolimbic dopamine system. Neuropsychopharmacology. https://doi.org/10.1038/s41386-020-0643-x
Lemberg KK, Kontinen VK, Siiskonen AO, Viljakka KM, Yli-Kauhaluoma JT, Korpi ER, Kalso EA (2006) Antinociception by spinal and systemic oxycodone: why does the route make a difference? In vitro and in vivo studies in rats. Anesthesiology 105(4):801–812. https://doi.org/10.1097/00000542-200610000-00027
Lemberg KK, Siiskonen AO, Kontinen VK, Yli-Kauhaluoma JT, Kalso EA (2008) Pharmacological characterization of noroxymorphone as a new opioid for spinal analgesia. Anesth Analg 106(2):463–470. https://doi.org/10.1213/ane.0b013e3181605a15
Lemberg KK, Heiskanen TE, Kontinen VK, Kalso EA (2009) Pharmacology of oxycodone: does it explain why oxycodone has become a bestselling strong opioid? Scand J Pain 1:S18–S23. https://doi.org/10.1016/S1877-8860(09)70005-9
Lenz H, Sandvik L, Qvigstad E, Bjerkelund CE, Raeder J (2009) A comparison of intravenous oxycodone and intravenous morphine in patient-controlled postoperative analgesia after laparoscopic hysterectomy. Anesth Analg 109(4):1279–1283. https://doi.org/10.1213/ane.0b013e3181b0f0bb
Lester PA, Traynor JR (2006) Comparison of the in vitro efficacy of mu, delta, kappa and ORL1 receptor agonists and non-selective opioid agonists in dog brain membranes. Brain Res 1073–1074:290–296. https://doi.org/10.1016/j.brainres.2005.12.066
Leyton M (2010) The neurobiology of desire: dopamine and the regulation of mood and motivational states in humans. In: Kringelbach ML, Berridge KC (eds) Pleasures of the brain. Oxford University Press, Oxford, pp 222–243
Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M, Schroder W, Kogel BY, Beier H, Englberger W, Schunk S, De Vry J, Jahnel U, Frosch S (2014) Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther 349(3):535–548. https://doi.org/10.1124/jpet.114.213694
Litman RS, Pagan OH, Cicero TJ (2018) Abuse-deterrent opioid formulations. Anesthesiology 128(5):1015–1026. https://doi.org/10.1097/ALN.0000000000002031
Liu SS, Pickens S, Burma NE, Ibarra-Lecue I, Yang H, Xue L, Cook C, Hakimian JK, Severino AL, Lueptow L, Komarek K, Taylor AMW, Olmstead MC, Carroll FI, Bass CE, Andrews AM, Walwyn W, Trang T, Evans CJ, Leslie FM, Cahill CM (2019) Kappa opioid receptors drive a tonic aversive component of chronic pain. J Neurosci 39(21):4162–4178. https://doi.org/10.1523/JNEUROSCI.0274-19.2019
Livingston KE, Traynor JR (2014) Disruption of the Na+ ion binding site as a mechanism for positive allosteric modulation of the mu-opioid receptor. Proc Natl Acad Sci USA 111(51):18369–18374. https://doi.org/10.1073/pnas.1415013111
Livingston KE, Traynor JR (2018) Allostery at opioid receptors: modulation with small molecule ligands. Br J Pharmacol 175(14):2846–2856. https://doi.org/10.1111/bph.13823
Livingston KE, Stanczyk MA, Burford NT, Alt A, Canals M, Traynor JR (2018) Pharmacologic evidence for a putative conserved allosteric site on opioid receptors. Mol Pharmacol 93(2):157–167. https://doi.org/10.1124/mol.117.109561
Lobb CJ, Wilson CJ, Paladini CA (2010) A dynamic role for GABA receptors on the firing pattern of midbrain dopaminergic neurons. J Neurophysiol 104(1):403–413. https://doi.org/10.1152/jn.00204.2010
Lutfy K, Eitan S, Bryant CD, Yang YC, Saliminejad N, Walwyn W, Kieffer BL, Takeshima H, Carroll FI, Maidment NT, Evans CJ (2003) Buprenorphine-induced antinociception is mediated by mu-opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J Neurosci 23(32):10331–10337. https://doi.org/10.1523/jneurosci.23-32-10331.2003
Lynch WJ, Nicholson KL, Dance ME, Morgan RW, Foley PL (2010) Animal models of substance abuse and addiction: implications for science, animal welfare, and society. Comp Med 60(3):177–188
Ma YY, Cepeda C, Chatta P, Franklin L, Evans CJ, Levine MS (2012) Regional and cell-type-specific effects of DAMGO on striatal D1 and D2 dopamine receptor-expressing medium-sized spiny neurons. ASN Neuro. https://doi.org/10.1042/AN20110063
MacKillop J, de Wit H (2013) The Wiley-Blackwell handbook of addiction psychopharmacology. Wiley-Blackwell, Hoboken
Maddocks I, Somogyi A, Abbott F, Hayball P, Parker D (1996) Attenuation of morphine-induced delirium in palliative care by substitution with infusion of oxycodone. J Pain Symptom Manage 12(3):182–189. https://doi.org/10.1016/0885-3924(96)00050-4
Maincent J, Zhang F (2016) Recent advances in abuse-deterrent technologies for the delivery of opioids. Int J Pharm 510(1):57–72. https://doi.org/10.1016/j.ijpharm.2016.06.012
Malenka RC, Nestler EJ, Hyman SE (2009) Molecular neuropharmacology: a foundation for clinical neuroscience (2nd edn). McGraw-Hill Medical, New York, pp 147–148
Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S (2012) Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 485(7398):321–326. https://doi.org/10.1038/nature10954
Margolis EB, Toy B, Himmels P, Morales M, Fields HL (2012) Identification of rat ventral tegmental area GABAergic neurons. PLoS ONE 7(7):e42365. https://doi.org/10.1371/journal.pone.0042365
Margolis EB, Hjelmstad GO, Fujita W, Fields HL (2014) Direct bidirectional mu-opioid control of midbrain dopamine neurons. J Neurosci 34(44):14707–14716. https://doi.org/10.1523/JNEUROSCI.2144-14.2014
Markman J, Gudin J, Rauck R, Argoff C, Rowbotham M, Agaiby E, Gimbel J, Katz N, Doberstein SK, Tagliaferri M, Lu L, Siddhanti S, Hale M (2019) SUMMIT-07: a randomized trial of NKTR-181, a new molecular entity, full mu-opioid receptor agonist for chronic low-back pain. Pain 160(6):1374–1382. https://doi.org/10.1097/j.pain.0000000000001517
Mars SG, Bourgois P, Karandinos G, Montero F, Ciccarone D (2014) "Every 'never' I ever said came true": transitions from opioid pills to heroin injecting. Int J Drug Policy 25(2):257–266. doi: https://doi.org/10.1016/j.drugpo.2013.10.004
Matsui A, Jarvie BC, Robinson BG, Hentges ST, Williams JT (2014) Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron 82(6):1346–1356. https://doi.org/10.1016/j.neuron.2014.04.030
McMillan DM, Tyndale RF (2018) CYP-mediated drug metabolism in the brain impacts drug response. Pharmacol Ther 184:189–200. https://doi.org/10.1016/j.pharmthera.2017.10.008
Melief EJ, Miyatake M, Bruchas MR, Chavkin C (2010) Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci USA 107(25):11608–11613. https://doi.org/10.1073/pnas.1000751107
Meredith GE, Agolia R, Arts MP, Groenewegen HJ, Zahm DS (1992) Morphological differences between projection neurons of the core and shell in the nucleus accumbens of the rat. Neuroscience 50(1):149–162. https://doi.org/10.1016/0306-4522(92)90389-j
Minami K, Hasegawa M, Ito H, Nakamura A, Tomii T, Matsumoto M, Orita S, Matsushima S, Miyoshi T, Masuno K, Torii M, Koike K, Shimada S, Kanemasa T, Kihara T, Narita M, Suzuki T, Kato A (2009) Morphine, oxycodone, and fentanyl exhibit different analgesic profiles in mouse pain models. J Pharmacol Sci 111(1):60–72. https://doi.org/10.1254/jphs.09139fp
Miyazaki T, Choi IY, Rubas W, Anand NK, Ali C, Evans J, Gursahani H, Hennessy M, Kim G, McWeeney D, Pfeiffer J, Quach P, Gauvin D, Riley TA, Riggs JA, Gogas K, Zalevsky J, Doberstein SK (2017) NKTR-181: a novel mu-opioid analgesic with inherently low abuse potential. J Pharmacol Exp Ther 363(1):104–113. https://doi.org/10.1124/jpet.117.243030
Moore K, Madularu D, Iriah S, Yee JR, Kulkarni P, Darcq E, Kieffer BL, Ferris CF (2016) BOLD imaging in awake wild-Type and mu-Opioid receptor knock-out mice reveals on-target activation maps in response to oxycodone. Front Neurosci 10:471. https://doi.org/10.3389/fnins.2016.00471
Moradi M, Esmaeili S, Shoar S, Safari S (2012) Use of oxycodone in pain management. Anesth Pain Med 1(4):262–264. https://doi.org/10.5812/aapm.4529
Morales M, Margolis EB (2017) Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci 18(2):73–85
Morean ME, de Wit H, King AC, Sofuoglu M, Rueger SY, O’Malley SS (2013) The drug effects questionnaire: psychometric support across three drug types. Psychopharmacology (Berl) 227(1):177–192. https://doi.org/10.1007/s00213-012-2954-z
Mucci-LoRusso P, Berman BS, Silberstein PT, Citron ML, Bressler L, Weinstein SM, Kaiko RF, Buckley BJ, Reder RF (1998) Controlled-release oxycodone compared with controlled-release morphine in the treatment of cancer pain: a randomized, double-blind, parallel-group study. Eur J Pain 2(3):239–249. https://doi.org/10.1016/s1090-3801(98)90020-9
Murphy DL, Lebin JA, Severtson SG, Olsen HA, Dasgupta N, Dart RC (2018) Comparative rates of mortality and serious adverse effects among commonly prescribed opioid analgesics. Drug Saf 41(8):787–795. https://doi.org/10.1007/s40264-018-0660-4
Narita M, Nakamura A, Ozaki M, Imai S, Miyoshi K, Suzuki M, Suzuki T (2008) Comparative pharmacological profiles of morphine and oxycodone under a neuropathic pain-like state in mice: evidence for less sensitivity to morphine. Neuropsychopharmacology 33(5):1097–1112. https://doi.org/10.1038/sj.npp.1301471
Nasseef MT, Singh JP, Ehrlich AT, McNicholas M, Park DW, Ma W, Kulkarni P, Kieffer BL, Darcq E (2019) Oxycodone-mediated activation of the mu opioid receptor reduces whole brain functional connectivity in mice. ACS Pharmacol Transl Sci 2(4):264–274. https://doi.org/10.1021/acsptsci.9b00021
Neelakantan H, Ward SJ, Walker EA (2015) Discriminative stimulus effects of morphine and oxycodone in the absence and presence of acetic acid in male and female C57Bl/6 mice. Exp Clin Psychopharmacol 23(4):217–227. https://doi.org/10.1037/pha0000028
Nesse RM, Berridge KC (1997) Psychoactive drug use in evolutionary perspective. Science 278(5335):63–66. https://doi.org/10.1126/science.278.5335.63
Nestler EJ, Malenka RC (2004) The addicted brain. Sci Am 290(3):78–85. https://doi.org/10.1038/scientificamerican0304-78
Nguyen JD, Grant Y, Taffe MA (2019) Paradoxical changes in brain reward status during opioid self-administration in a novel test of the negative reinforcement hypothesis. bioRxiv. https://doi.org/10.1101/460048
Nielsen CK, Ross FB, Lotfipour S, Saini KS, Edwards SR, Smith MT (2007) Oxycodone and morphine have distinctly different pharmacological profiles: radioligand binding and behavioural studies in two rat models of neuropathic pain. Pain 132(3):289–300. https://doi.org/10.1016/j.pain.2007.03.022
Nozaki C, Saitoh A, Kamei J (2006) Characterization of the antinociceptive effects of oxycodone in diabetic mice. Eur J Pharmacol 535(1–3):145–151. https://doi.org/10.1016/j.ejphar.2006.02.002
Ohbuchi K, Miyagi C, Suzuki Y, Mizuhara Y, Mizuno K, Omiya Y, Yamamoto M, Warabi E, Sudo Y, Yokoyama A, Miyano K, Hirokawa T, Uezono Y (2016) Ignavine: a novel allosteric modulator of the mu opioid receptor. Sci Rep 6:31748. https://doi.org/10.1038/srep31748
Olson KM, Duron DI, Womer D, Fell R, Streicher JM (2019) Comprehensive molecular pharmacology screening reveals potential new receptor interactions for clinically relevant opioids. PLoS ONE 14(6):e0217371. https://doi.org/10.1371/journal.pone.0217371
Orlowski JP, Wateska L (1992) The effects of pharmaceutical firm enticements on physician prescribing patterns. There’s no such thing as a free lunch. Chest 102(1):270–273. https://doi.org/10.1378/chest.102.1.270
Osgood ED, Eaton TA, Trudeau JJ, Katz NP (2012) A brief survey to characterize oxycodone abuse patterns in adolescents enrolled in two substance abuse recovery high schools. Am J Drug Alcohol Abuse 38(2):166–170. https://doi.org/10.3109/00952990.2011.643994
Ostlund SB, LeBlanc KH, Kosheleff AR, Wassum KM, Maidment NT (2014) Phasic mesolimbic dopamine signaling encodes the facilitation of incentive motivation produced by repeated cocaine exposure. Neuropsychopharmacology 39(10):2441–2449. https://doi.org/10.1038/npp.2014.96
Pappagallo M, Campbell JN (1994) Chronic opioid therapy as alternative treatment for post-herpetic neuralgia. Ann Neurol 35:S54-56. https://doi.org/10.1002/ana.410350716
Pasternak GW, Snyder SH (1975) Identification of novel high affinity opiate receptor binding in rat brain. Nature 253(5492):563–565. https://doi.org/10.1038/253563a0
Paulozzi LJ, Budnitz DS, Xi Y (2006) Increasing deaths from opioid analgesics in the United States. Pharmacoepidemiol Drug Saf 15(9):618–627
Pecina S, Berridge KC (2000) Opioid site in nucleus accumbens shell mediates eating and hedonic “liking” for food: map based on microinjection Fos plumes. Brain Res 863(1–2):71–86. https://doi.org/10.1016/s0006-8993(00)02102-8
Pecina S, Berridge KC (2005) Hedonic hot spot in nucleus accumbens shell: where do mu-opioids cause increased hedonic impact of sweetness? J Neurosci 25(50):11777–11786. https://doi.org/10.1523/JNEUROSCI.2329-05.2005
Peckham EM, Traynor JR (2006) Comparison of the antinociceptive response to morphine and morphine-like compounds in male and female Sprague-Dawley rats. J Pharmacol Exp Ther 316(3):1195–1201. https://doi.org/10.1124/jpet.105.094276
Pena DA, Duarte ML, Pramio DT, Devi LA, Schechtman D (2018) Exploring morphine-triggered PKC-targets and their interaction with signaling pathways leading to pain via TrkA. Proteomes. https://doi.org/10.3390/proteomes6040039
Phillips PE, Robinson DL, Stuber GD, Carelli RM, Wightman RM (2003) Real-time measurements of phasic changes in extracellular dopamine concentration in freely moving rats by fast-scan cyclic voltammetry. Methods Mol Med 79:443–464. https://doi.org/10.1385/1-59259-358-5:443
Pitman KA, Puil E, Borgland SL (2014) GABA(B) modulation of dopamine release in the nucleus accumbens core. Eur J Neurosci 40(10):3472–34780. https://doi.org/10.1111/ejn.12733
Plummer JL, Cmielewski PL, Reynolds GD, Gourlay GK, Cherry DA (1990) Influence of polarity on dose-response relationships of intrathecal opioids in rats. Pain 40(3):339–347. https://doi.org/10.1016/0304-3959(90)91131-2
Pollini RA, Banta-Green CJ, Cuevas-Mota J, Metzner M, Teshale E, Garfein RS (2011) Problematic use of prescription-type opioids prior to heroin use among young heroin injectors. Subst Abuse Rehabil 2(1):173–180. https://doi.org/10.2147/SAR.S24800
Pontieri FE, Tanda G, Di Chiara G (1995) Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc Natl Acad Sci USA 92(26):12304–12308. https://doi.org/10.1073/pnas.92.26.12304
Pool E, Sennwald V, Delplanque S, Brosch T, Sander D (2016) Measuring wanting and liking from animals to humans: a systematic review. Neurosci Biobehav Rev 63:124–142. https://doi.org/10.1016/j.neubiorev.2016.01.006
Poyhia R, Kalso EA (1992) Antinociceptive effects and central nervous system depression caused by oxycodone and morphine in rats. Pharmacol Toxicol 70(2):125–130. https://doi.org/10.1111/j.1600-0773.1992.tb00441.x
Poyhia R, Olkkola KT, Seppala T, Kalso E (1991) The pharmacokinetics of oxycodone after intravenous injection in adults. Br J Clin Pharmacol 32(4):516–518. https://doi.org/10.1111/j.1365-2125.1991.tb03942.x
Raff M, Belbachir A, El-Tallawy S, Ho KY, Nagtalon E, Salti A, Seo JH, Tantri AR, Wang H, Wang T, Buemio KC, Gutierrez C, Hadjiat Y (2019) Intravenous oxycodone versus other intravenous strong opioids for acute postoperative pain control: a systematic review of randomized controlled trials. Pain Ther 8(1):19–39. https://doi.org/10.1007/s40122-019-0122-4
Raffa RB, Burdge G, Gambrah J, Kinecki HE, Lin F, Lu B, Nguyen JT, Phan V, Ruan A, Sesay MA, Watkins TN (2017) Cebranopadol: novel dual opioid/NOP receptor agonist analgesic. J Clin Pharm Ther 42(1):8–17. https://doi.org/10.1111/jcpt.12461
Rappaport B (2008) Overview of the November 13, 2008 ALSDAC Meeting to Discuss NDA 22-324 for Remoxy (Oxycodone Hydrochloride Controlled-Release) Capsules. A. Division of Anesthesia, and Rheumatology Products, Office of Drug Evaluation II, editor.
Remesic M, Hruby VJ, Porreca F, Lee YS (2017) Recent advances in the realm of allosteric modulators for opioid receptors for future therapeutics. ACS Chem Neurosci 8(6):1147–1158. https://doi.org/10.1021/acschemneuro.7b00090
Remillard D, Kaye AD, McAnally H (2019) Oxycodone’s unparalleled addictive potential: is it time for a moratorium? Curr Pain Headache Rep 23(2):15. https://doi.org/10.1007/s11916-019-0751-7
Riley J, Eisenberg E, Muller-Schwefe G, Drewes AM, Arendt-Nielsen L (2008) Oxycodone: a review of its use in the management of pain. Curr Med Res Opin 24(1):175–192. https://doi.org/10.1185/030079908x253708
Robinson TE, Berridge KC (1993) The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev 18(3):247–291. https://doi.org/10.1016/0165-0173(93)90013-p
Robinson TE, Berridge KC (2003) Addiction. Annu Rev Psychol 54:25–53
Rosenblum A, Parrino M, Schnoll SH, Fong C, Maxwell C, Cleland CM, Magura S, Haddox JD (2007) Prescription opioid abuse among enrollees into methadone maintenance treatment. Drug Alcohol Depend 90(1):64–71. https://doi.org/10.1016/j.drugalcdep.2007.02.012
Ross FB, Smith MT (1997) The intrinsic antinociceptive effects of oxycodone appear to be kappa-opioid receptor mediated. Pain 73(2):151–157. https://doi.org/10.1016/s0304-3959(97)00093-6
Rothman RB, Murphy DL, Xu H, Godin JA, Dersch CM, Partilla JS, Tidgewell K, Schmidt M, Prisinzano TE (2007) Salvinorin A: allosteric interactions at the mu-opioid receptor. J Pharmacol Exp Ther 320(2):801–810. https://doi.org/10.1124/jpet.106.113167
Ruan X, Mancuso KF, Kaye AD (2017) Revisiting Oxycodone Analgesia: A Review And Hypothesis. Anesthesiol Clin 35(2):e163–e174. https://doi.org/10.1016/j.anclin.2017.01.022
Saigusa T, Aono Y, Mizoguchi N, Iwakami T, Takada K, Oi Y, Ueda K, Koshikawa N, Cools AR (2008) Role of GABA B receptors in the endomorphin-1-, but not endomorphin-2-, induced dopamine efflux in the nucleus accumbens of freely moving rats. Eur J Pharmacol 581(3):276–282. https://doi.org/10.1016/j.ejphar.2007.12.008
Setnik B, Roland CL, Goli V, Pixton GC, Levy-Cooperman N, Smith I, Webster L (2015) Self-reports of prescription opioid abuse and diversion among recreational opioid users in a Canadian and a United States city. J Opioid Manag 11(6):463–473. https://doi.org/10.5055/jom.2015.0299
Severino AL, Shadfar A, Hakimian JK, Crane O, Singh G, Heinzerling K, Walwyn WM (2018) Pain therapy guided by purpose and perspective in light of the opioid epidemic. Front Psychiatry 9:119. https://doi.org/10.3389/fpsyt.2018.00119
Sheik Amamuddy O, Veldman W, Manyumwa C, Khairallah A, Agajanian S, Oluyemi O, Verkhivker G, Tastan Bishop O (2020) Integrated computational approaches and tools for allosteric drug discovery. Int J Mol Sci. https://doi.org/10.3390/ijms21030847
Shimura T, Imaoka H, Yamamoto T (2006) Neurochemical modulation of ingestive behavior in the ventral pallidum. Eur J Neurosci 23(6):1596–1604. https://doi.org/10.1111/j.1460-9568.2006.04689.x
Shippenberg TS, LeFevour A, Chefer VI (2008) Targeting endogenous mu- and delta-opioid receptor systems for the treatment of drug addiction. CNS Neurol Disord Drug Targets 7(5):442–453. https://doi.org/10.2174/187152708786927813
Shirayama Y, Chaki S (2006) Neurochemistry of the nucleus accumbens and its relevance to depression and antidepressant action in rodents. Curr Neuropharmacol 4(4):277–291. https://doi.org/10.2174/157015906778520773
Siegal HA, Carlson RG, Kenne DR, Swora MG (2003) Probable relationship between opioid abuse and heroin use. Am Fam Physician 67(5):942–945
Singla NK, Skobieranda F, Soergel DG, Salamea M, Burt DA, Demitrack MA, Viscusi ER (2019) APOLLO-2: A Randomized, Placebo and Active-Controlled Phase III Study Investigating Oliceridine (TRV130), a G Protein-Biased Ligand at the mu-Opioid Receptor, for Management of Moderate to Severe Acute Pain Following Abdominoplasty. Pain Pract 19(7):715–731. https://doi.org/10.1111/papr.12801
Small DM, Zatorre RJ, Dagher A, Evans AC, Jones-Gotman M (2001) Changes in brain activity related to eating chocolate: from pleasure to aversion. Brain 124:1720–1733. https://doi.org/10.1093/brain/124.9.1720
Smith KS, Berridge KC (2005) The ventral pallidum and hedonic reward: neurochemical maps of sucrose “liking” and food intake. J Neurosci 25(38):8637–8649. https://doi.org/10.1523/JNEUROSCI.1902-05.2005
Smith KS, Berridge KC (2007) Opioid limbic circuit for reward: interaction between hedonic hotspots of nucleus accumbens and ventral pallidum. J Neurosci 27(7):1594–1605. https://doi.org/10.1523/JNEUROSCI.4205-06.2007
Smith FL, Javed RR, Elzey MJ, Dewey WL (2003) The expression of a high level of morphine antinociceptive tolerance in mice involves both PKC and PKA. Brain Res 985(1):78–88. https://doi.org/10.1016/s0006-8993(03)03170-6
Smith FL, Gabra BH, Smith PA, Redwood MC, Dewey WL (2007) Determination of the role of conventional, novel and atypical PKC isoforms in the expression of morphine tolerance in mice. Pain 127(1–2):129–139. https://doi.org/10.1016/j.pain.2006.08.009
Smith KS, Berridge KC, Aldridge JW (2011) Disentangling pleasure from incentive salience and learning signals in brain reward circuitry. Proc Natl Acad Sci USA 108(27):E255-264. https://doi.org/10.1073/pnas.1101920108
Smith K, Hopp M, Mundin G, Bond S, Bailey P, Woodward J, Bell D (2012) Low absolute bioavailability of oral naloxone in healthy subjects. Int J Clin Pharmacol Ther 50(5):360–367. https://doi.org/10.5414/cp201646
Song Z, Zou W, Liu C, Guo Q (2010) Gene knockdown with lentiviral vector-mediated intrathecal RNA interference of protein kinase C gamma reverses chronic morphine tolerance in rats. J Gene Med 12(11):873–880. https://doi.org/10.1002/jgm.1514
Steiner JE, Glaser D, Hawilo ME, Berridge KC (2001) Comparative expression of hedonic impact: affective reactions to taste by human infants and other primates. Neurosci Biobehav Rev 25(1):53–74. https://doi.org/10.1016/s0149-7634(00)00051-8
Svingos AL, Clarke CL, Pickel VM (1999) Localization of the delta-opioid receptor and dopamine transporter in the nucleus accumbens shell: implications for opiate and psychostimulant cross-sensitization. Synapse 34(1):1–10
Taylor R Jr, Raffa RB, Pergolizzi JV Jr (2012) Controlled release formulation of oxycodone in patients with moderate to severe chronic osteoarthritis: a critical review of the literature. J Pain Res 5:77–87. https://doi.org/10.2147/JPR.S21965
Tepper JM, Wilson CJ, Koos T (2008) Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res Rev 58(2):272–281. https://doi.org/10.1016/j.brainresrev.2007.10.008
Tepper JM, Tecuapetla F, Koos T, Ibanez-Sandoval O (2010) Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat 4:150. https://doi.org/10.3389/fnana.2010.00150
Thompson CM, Wojno H, Greiner E, May EL, Rice KC, Selley DE (2004) Activation of G-proteins by morphine and codeine congeners: insights to the relevance of O- and N-demethylated metabolites at mu- and delta-opioid receptors. J Pharmacol Exp Ther 308(2):547–554. https://doi.org/10.1124/jpet.103.058602
Tian JH, Xu W, Fang Y, Mogil JS, Grisel JE, Grandy DK, Han JS (1997) Bidirectional modulatory effect of orphanin FQ on morphine-induced analgesia: antagonism in brain and potentiation in spinal cord of the rat. Br J Pharmacol 120(4):676–680. https://doi.org/10.1038/sj.bjp.0700942
Tritsch NX, Oh WJ, Gu C, Sabatini BL (2014) Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. Elife 3:e01936. https://doi.org/10.7554/eLife.01936
Tzschentke TM, Rutten K (2018) Mu-opioid peptide (MOP) and nociceptin/orphanin FQ peptide (NOP) receptor activation both contribute to the discriminative stimulus properties of cebranopadol in the rat. Neuropharmacology 129:100–108. https://doi.org/10.1016/j.neuropharm.2017.11.026
Upadhyay J, Maleki N, Potter J, Elman I, Rudrauf D, Knudsen J, Wallin D, Pendse G, McDonald L, Griffin M, Anderson J, Nutile L, Renshaw P, Weiss R, Becerra L, Borsook D (2010) Alterations in brain structure and functional connectivity in prescription opioid-dependent patients. Brain 133(Pt7):2098–2114. https://doi.org/10.1093/brain/awq138
Van Zee A (2009) The promotion and marketing of oxycontin: commercial triumph, public health tragedy. Am J Public Health 99(2):221–227. https://doi.org/10.2105/AJPH.2007.131714
van Zessen R, Phillips JL, Budygin EA, Stuber GD (2012) Activation of VTA GABA neurons disrupts reward consumption. Neuron 73(6):1184–1194. https://doi.org/10.1016/j.neuron.2012.02.016
Vander Weele CM, Porter-Stransky KA, Mabrouk OS, Lovic V, Singer BF, Kennedy RT, Aragona BJ (2014) Rapid dopamine transmission within the nucleus accumbens: dramatic difference between morphine and oxycodone delivery. Eur J Neurosci 40(7):3041–3054. https://doi.org/10.1111/ejn.12709
Vaysse PJ, Gardner EL, Zukin RS (1987) Modulation of rat brain opioid receptors by cannabinoids. J Pharmacol Exp Ther 241(2):534–539
Ventola CL (2011) Direct-to-consumer pharmaceutical advertising: therapeutic or toxic? P T 36(10):669–684
Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S, Soergel DG, Subach RA, Cook E, Skobieranda F (2016) A randomized, phase 2 study investigating TRV130, a biased ligand of the mu-opioid receptor, for the intravenous treatment of acute pain. Pain 157(1):264–272. https://doi.org/10.1097/j.pain.0000000000000363
Viscusi ER, Skobieranda F, Soergel DG, Cook E, Burt DA, Singla N (2019) APOLLO-1: a randomized placebo and active-controlled phase III study investigating oliceridine (TRV130), a G protein-biased ligand at the micro-opioid receptor, for management of moderate-to-severe acute pain following bunionectomy. J Pain Res 12:927–943. https://doi.org/10.2147/JPR.S171013
Wade CL, Vendruscolo LF, Schlosburg JE, Hernandez DO, Koob GF (2015) Compulsive-like responding for opioid analgesics in rats with extended access. Neuropsychopharmacology 40(2):421–428. https://doi.org/10.1038/npp.2014.188
Walentiny DM, Moisa LT, Beardsley PM (2019) Oxycodone-like discriminative stimulus effects of fentanyl-related emerging drugs of abuse in mice. Neuropharmacology 150:210–216. https://doi.org/10.1016/j.neuropharm.2019.02.007
Watson CP, Babul N (1998) Efficacy of oxycodone in neuropathic pain: a randomized trial in postherpetic neuralgia. Neurology 50(6):1837–1841. https://doi.org/10.1212/wnl.50.6.1837
Watson CP, Moulin D, Watt-Watson J, Gordon A, Eisenhoffer J (2003) Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain 105(1–2):71–78. https://doi.org/10.1016/s0304-3959(03)00160-x
Webster LR, Johnson FK, Stauffer J, Setnik B, Ciric S (2011) Impact of intravenous naltrexone on intravenous morphine-induced high, drug liking, and euphoric effects in experienced, nondependent male opioid users. Drugs R D 11(3):259–275. https://doi.org/10.2165/11593390-000000000-00000
Webster LR, Bath B, Medve RA, Marmon T, Stoddard GJ (2012) Randomized, double-blind, placebo-controlled study of the abuse potential of different formulations of oral oxycodone. Pain Med 13(6):790–801. https://doi.org/10.1111/j.1526-4637.2012.01380.x
Webster L, Henningfield J, Buchhalter AR, Siddhanti S, Lu L, Odinecs A, Di Fonzo CJ, Eldon MA (2018) Human abuse potential of the new opioid analgesic molecule NKTR-181 compared with oxycodone. Pain Med 19(2):307–318. https://doi.org/10.1093/pm/pnw344
Werling LL, Brown SR, Puttfarcken P, Cox BM (1986) Sodium regulation of agonist binding at opioid receptors. II. Effects of sodium replacement on opioid binding in guinea pig cortical membranes. Mol Pharmacol 30(2):90–95
Wightman R, Perrone J, Portelli I, Nelson L (2012) Likeability and abuse liability of commonly prescribed opioids. J Med Toxicol 8(4):335–340. https://doi.org/10.1007/s13181-012-0263-x
Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ (2013) Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65(1):223–254. https://doi.org/10.1124/pr.112.005942
Willuhn I, Burgeno LM, Groblewski PA, Phillips PE (2014) Excessive cocaine use results from decreased phasic dopamine signaling in the striatum. Nat Neurosci 17(5):704–709. https://doi.org/10.1038/nn.3694
Wirz S, Wartenberg HC, Nadstawek J (2005) Pain management procedures used by dental and maxillofacial surgeons: an investigation with special regard to odontalgia. Head Face Med 1:14. https://doi.org/10.1186/1746-160X-1-14
Wise RA (1985) The anhedonia hypothesis: Mark III. Behav Brain Sci 8:178–186. https://doi.org/10.1017/s0140525x00020306
Witteman J, Post H, Tarvainen M, de Bruijn A, Perna Ede S, Ramaekers JG, Wiers RW (2015) Cue reactivity and its relation to craving and relapse in alcohol dependence: a combined laboratory and field study. Psychopharmacology (Berl) 232(20):3685–3696. https://doi.org/10.1007/s00213-015-4027-6
Woolley JD, Lee BS, Fields HL (2006) Nucleus accumbens opioids regulate flavor-based preferences in food consumption. Neuroscience 143(1):309–317. https://doi.org/10.1016/j.neuroscience.2006.06.067
Wu J, Lu Y, Cao X (2019) Different effects of oxycodone and remifentanil in patients undergoing ultrasound-guided percutaneous radiofrequency ablation of hepatic cancer: a randomized trial. Drug Des Devel Ther 13:365–372. https://doi.org/10.2147/DDDT.S188728
Yager LM, Garcia AF, Wunsch AM, Ferguson SM (2015) The ins and outs of the striatum: role in drug addiction. Neuroscience 301:529–541. https://doi.org/10.1016/j.neuroscience.2015.06.033
Yang PP, Yeh GC, Yeh TK, Xi J, Loh HH, Law PY, Tao PL (2016) Activation of delta-opioid receptor contributes to the antinociceptive effect of oxycodone in mice. Pharmacol Res 111:867–876. https://doi.org/10.1016/j.phrs.2016.05.034
Yang H, de Jong JW, Tak Y, Peck J, Bateup HS, Lammel S (2018) Nucleus accumbens subnuclei regulate motivated behavior via direct inhibition and disinhibition of VTA dopamine subpopulations. Neuron 97(2):434–449. https://doi.org/10.1016/j.neuron.2017.12.022
Yang PP, Yeh TK, Loh HH, Law PY, Wang Y, Tao PL (2019) Delta-opioid receptor antagonist naltrindole reduces oxycodone addiction and constipation in mice. Eur J Pharmacol 852:265–273. https://doi.org/10.1016/j.ejphar.2019.04.009
Young AM, Havens JR (2012) Transition from first illicit drug use to first injection drug use among rural Appalachian drug users: a cross-sectional comparison and retrospective survival analysis. Addiction 107(3):587–596. https://doi.org/10.1111/j.1360-0443.2011.03635.x
Yu Y, Li D, Duan J, Xu H, Li L, Tan D, Yan H (2020) The pro- and anti-cancer effects of oxycodone are associated with epithelial growth factor receptor level in cancer cells. Biosci Rep 40:2
Zacny JP, Gutierrez S (2003) Characterizing the subjective, psychomotor, and physiological effects of oral oxycodone in non-drug-abusing volunteers. Psychopharmacology (Berl) 170(3):242–254. https://doi.org/10.1007/s00213-003-1540-9
Zacny JP, Gutierrez S (2008) Subjective, psychomotor, and physiological effects profile of hydrocodone/acetaminophen and oxycodone/acetaminophen combination products. Pain Med 9(4):433–443. https://doi.org/10.1111/j.1526-4637.2007.00359.x
Zacny JP, Gutierrez S (2009) Within-subject comparison of the psychopharmacological profiles of oral hydrocodone and oxycodone combination products in non-drug-abusing volunteers. Drug Alcohol Depend 101(1–2):107–114. https://doi.org/10.1016/j.drugalcdep.2008.11.013
Zacny JP, Lichtor SA (2008) Within-subject comparison of the psychopharmacological profiles of oral oxycodone and oral morphine in non-drug-abusing volunteers. Psychopharmacology (Berl) 196(1):105–116. https://doi.org/10.1007/s00213-007-0937-2
Zacny J, Bigelow G, Compton P, Foley K, Iguchi M, Sannerud C (2003) College on Problems of Drug Dependence taskforce on prescription opioid non-medical use and abuse: position statement. Drug Alcohol Depend 69(3):215–232. https://doi.org/10.1016/s0376-8716(03)00003-6
Zech DF, Grond S, Lynch J, Hertel D, Lehmann KA (1995) Validation of World Health Organization Guidelines for cancer pain relief: a 10-year prospective study. Pain 63(1):65–76
Zhang M, Gosnell BA, Kelley AE (1998) Intake of high-fat food is selectively enhanced by mu opioid receptor stimulation within the nucleus accumbens. J Pharmacol Exp Ther 285(2):908–914
Zhang L, Zhang JT, Hang L, Liu T (2020) Mu opioid receptor heterodimers emerge as novel therapeutic targets: recent progress and future perspective. Front Pharmacol 11:1078. https://doi.org/10.3389/fphar.2020.01078
Zheng H, Loh HH, Law PY (2008) Beta-arrestin-dependent mu-opioid receptor-activated extracellular signal-regulated kinases (ERKs) Translocate to Nucleus in Contrast to G protein-dependent ERK activation. Mol Pharmacol 73(1):178–190. https://doi.org/10.1124/mol.107.039842
Zheng H, Chu J, Zhang Y, Loh HH, Law PY (2011) Modulating micro-opioid receptor phosphorylation switches agonist-dependent signaling as reflected in PKCepsilon activation and dendritic spine stability. J Biol Chem 286(14):12724–12733. https://doi.org/10.1074/jbc.M110.177089
Zhou Y, Li X, Zhang M, Zhang F, Zhu C, Shen M (2012) Behavioural approach tendencies to heroin-related stimuli in abstinent heroin abusers. Psychopharmacology (Berl) 221(1):171–176. https://doi.org/10.1007/s00213-011-2557-0
Acknowledgements
Tribute to Dr. Gavril Pasternak: Our manuscript is submitted as a part of the Special Issue to honor Dr. Pasternak. In memory of Gavril (Gav) Pasternak who pushed the boundaries of opioid pharmacology and through his research career revealed new insights into the complexity of drug interactions with opioid receptors. It is well acknowledged in clinical practice that there is a lack of complete cross-tolerance between opioid analgesic drugs. While there are likely various explanations for this phenomenon, Gav contributed to the understanding of how different opioid analgesics have unique pharmacological profiles and exhibit synergistic interactions between opioids or when combined with other non-opioid analgesic drugs.
Funding
Financial support is gratefully provided by the Shirley Hatos Neuroscience Research Foundation (CJE, CK, and CMC), National Institutes of Health (NIH) R01DA041781 (CMC), NIH 1UG3TR003148-01 (CMC), NIH 2P50 DA005010 (CMC, CJE, CK), and the Department of Defense W81XWH-15-1-0435 (CMC).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kibaly, C., Alderete, J.A., Liu, S.H. et al. Oxycodone in the Opioid Epidemic: High ‘Liking’, ‘Wanting’, and Abuse Liability. Cell Mol Neurobiol 41, 899–926 (2021). https://doi.org/10.1007/s10571-020-01013-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-020-01013-y