
Abstract
Caloric restriction (CR) has anti-epileptic effects in different animal models, at least partially due to inhibition of the mechanistic or mammalian target of rapamycin (mTOR) signaling pathway. Adenosine monophosphate-activated protein kinase (AMPK) inhibits mTOR cascade function if energy levels are low. Since hyper-activation of mTOR participates in epilepsy, its inhibition results in beneficial anti-convulsive effects. A way to attain this is to activate AMPK with metformin. The effects of metformin, alone or combined with CR, on the electrical kindling epilepsy model and the mTOR cascade in the hippocampus and the neocortex were studied. Combined metformin plus CR beneficially affected many kindling aspects, especially those relating to generalized convulsive seizures. Therefore, metformin plus CR could decrease measures of epileptic activity in patients with generalized convulsive seizures. Patients that are obese, overweight or that have metabolic syndrome in addition to having an epileptic disease are an ideal population for clinical trials to test the effectiveness of metformin plus CR.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
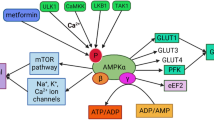
Hyper-activation of the mechanistic target of rapamycin (mTOR) intracellular signaling pathway plays a key role in the pathophysiology of genetic and acquired epilepsies (Citraro et al. 2016; Crino 2016). In most models (Cybulski and Hall 2009; Eltschinger and Loewith 2016; Gaubitz et al. 2016), this cascade responds to diverse extra- and intra-cellular cues. Low energy levels stimulate adenosine monophosphate-activated protein kinase (AMPK), which then activates tuberin protein (encoded by the tuberous sclerosis 2 gene, TSC2). Tuberin plus hamartin (encoded by TSC1) inhibits the action of a complex (mTORC1) that includes the mTOR kinase (encoded by MTOR). Quite the opposite, insulin and growth factors activate protein kinase B (PKB/Akt), which inhibits tuberin and activates mTORC1. This triggers ribosomal S6 kinase (encoded by S6K), which then turns on ribosomal protein S6 (S6). In this manner, the mTOR cascade regulates protein production, cell and body growth, as well as other important cell processes (Citraro et al. 2016; Crino 2016; Cybulski and Hall 2009; Eltschinger and Loewith 2016; Gaubitz et al. 2016).
Since excess mTOR cascade function contributes to epilepsy, its inhibition has shown beneficial anti-epileptic effects (Citraro et al. 2016; Crino 2016). Nutritional maneuvers such as the ketogenic diet and caloric restriction (CR) show anti-convulsive actions in several different animal models (Azarbar et al. 2010; Bough et al. 2003; Gasior et al. 2006; Greene et al. 2001; Kossoff and Rho 2009), at least partially caused by mTOR pathway inhibition (McDaniel et al. 2011; Phillips-Farfán et al. 2015). The anti-epileptic effects of CR include a delayed seizure onset and diminished seizure incidence in epileptic inbred mice (Greene et al. 2001), increased paired-pulse inhibition, higher threshold of maximal dentate gyrus activation and no spreading depression-like events (Bough et al. 2003), delayed kindling rate in seizure-prone rats (Azarbar et al. 2010), and increased after-discharge threshold in the electric kindling epilepsy model (Phillips-Farfán et al. 2015).
The traditional manner to investigate the mTOR cascade is to use its inhibitor rapamycin or related drugs. But this is by no means the only way to hinder the activity of this pathway. When AMPK is activated, it phosphorylates tuberin and raptor (mTOR cascade proteins), resulting in a rapid repression of pathway activity (Shaw 2009). Thus, a way to down-regulate mTOR cascade function is to activate AMPK. In this line, the anti-diabetic drug metformin indirectly activates AMPK (Foretz et al. 2014). Although still controversial, the mechanism is believed to be specific inhibition of complex I of the mitochondrial respiratory chain and an ensuing decrease in ATP levels (Adak et al. 2018; Foretz et al. 2014; Rena et al. 2017).
Thus, the main goal of this report was to investigate the effects of metformin on a model of temporal lobe epilepsy and whether its actions were due to mTOR pathway inhibition. The rationale was that metformin would activate AMPK, hindering mTOR cascade function and thus having beneficial anti-epileptic effects. Since CR had exactly these effects, the action of metformin combined with CR was also investigated. Our hypothesis was that metformin alone would have positive anti-seizure effects by inhibiting the mTOR pathway. In addition, we hypothesized that metformin combined with CR would work even better. If these hypotheses are true, these treatments should be very advantageous for patients suffering from temporal lobe epilepsy. This is especially true given that metformin, with or without CR, is expected to have a better side-effect profile (Kaplan et al. 2014) and is likely more cost-effective than other molecules that hinder the mTOR cascade, such as sirolimus (rapamycin) and its derived drugs.
Electrical kindling of the amygdala was used because it is a very useful model of temporal lobe epilepsy. On the first day, electrical stimulations of increasing magnitude are applied until an electrographic seizure, the after-discharge (AD), is obtained at a threshold level. On subsequent days, facilitation occurs although electrical stimulation of the same threshold degree is always applied. Partial convulsive motor seizures (stages 1–3), followed by generalized tonic-clonic paroxysmal attacks (stages 4–5), are seen (Gorter et al. 2016; Kandratavicius et al. 2014; Racine 1972).
mTOR cascade function was assessed by measuring the phosphorylation levels of AMPK, PKB, and S6, as well as the expression of several genes encoding for participating proteins in the hippocampus and the temporal neocortex. These brain areas were studied due to their importance for temporal lobe epilepsy as well as their connectivity with the amygdala. mTOR pathway activity is reflected in the body weight (Hebert et al. 2014; Rovira et al. 2008) and is regulated by energy levels as well as insulin concentrations (Crino 2016; Cybulski and Hall 2009). Thus, rat body weights were recorded in addition to measuring blood glucose and β-hydroxybutyrate (β-HB) concentrations, as well as plasma insulin levels. Weanling rats were used since dietary treatment for epilepsy is more effective in the younger the animal (Bough et al. 1999). Mild CR (of 15%, achieved by total food restriction without addition of vitamins or minerals) was performed to reduce potential under-nutrition. Metformin was administered at a dose of 100 mg/kg/day given that young weanling animals were used.
Materials and Methods
Every effort was made to match as much as possible the previous study including animals, reagents, and experimental protocols.
Animals
Following the three Rs principles, the number of rats used was reduced to a minimum. 25 male Wistar rats aged 21 days post-natal were acquired (Envigo) and fasted for 4 h such that they all started the procedures at a comparable metabolic set point. All animals were weight-matched and divided into 3 groups: 10 were fed ad libitum (AL), 8 rats fed ad libitum were treated with metformin (AM), and 7 were subjected to 15% CR plus administration of metformin (CM). Due to space constraints, not all the animals were used for the kindling procedure where the groups were as follows: 8 AL, 4 AM, and 7 CM (timing and design of the study are shown in Fig. 1).

Experimental timing and design. 25 male Wistar rats aged 21 days post-natal were acquired, fasted for 4 h, weight-matched, and divided into 3 groups. 10 rats were fed ad libitum (AL), 8 rats fed ad libitum were treated with metformin (AM), and 7 rats were subjected to 15% CR plus administration of metformin (CM). For kindling, 8 AL, 4 AM, and 7 CM animals were used. Metformin was administered orally at a dose of 100 mg/kg/day, 5 days each week. CR was done by restricting food by 15% of the daily caloric needs, based on the body weight of the AM group. Rats were kept on their respective diets and treated with metformin during the entire procedure (31 days before surgery, 1 surgery day, 9 recovery days, 37 kindling days, and the next day for sacrifice; 79 days total). ed experimental day, pd postnatal day
Metformin was administered orally at a dose of 100 mg/kg/day, 5 days a week. In the CM group, CR was done by restricting food by 15% (giving 85%) of the daily caloric needs (Rogers 1979), based on the body weight of rats in the AM group. All animals were kept on their respective diets and treated with metformin during the entire procedure (31 days before surgery, 1 surgery day, 9 recovery days, 37 kindling days, and the next day for sacrifice; 79 days in total) and were housed in groups of 2–5 under standard conditions. All possible measures were taken to minimize pain and discomfort of rats. All procedures followed both international (Guide for the Care and Use of Laboratory Animals, NIH publication No. 86-23, revised 1987) and national (NOM-062-ZOO-1999) guidelines. The protocol was approved by our institutional animal ethics committee.
Stereotaxic Surgery
The animals were subjected to surgery 1 month after starting their respective diets and treatments. Under deep ketamine (40 mg/kg) anesthesia, bipolar electrodes were implanted into the left baso-lateral amygdala and the right sensory cortex (coordinates in mm from bregma and inter-aural line, respectively: anterior 2.8, 6.7; lateral 5, 2.5; and ventral 8.5, 9). The electrodes consisted of two twisted stainless steel wires (0.005′ diameter), Teflon-coated except the tips (separated by 0.02′). They were soldered to connectors and fastened to the rat skull with acrylic cement and four screws, one screw served as an indifferent source of isoelectric reference. Electrode positioning was later verified with histological staining techniques in some animals after sacrifice.
Electrical Kindling of the Amygdala
An AD was defined as an amygdala discharge lasting at least 5 s; its duration was defined by the length of time that the electrographic seizure was observed (Online Resource 1). The AD threshold was defined as the lowest electrical current stimulus that elicited an AD in the amygdala; it was determined only on day one of the kindling procedure. An electronic circuit breaker device, switching either to a polygraph (Grass 78D, Grass Technologies) or a stimulator (Grass S88, Grass Technologies), connected to the amygdala electrode (1.8 cm long) was used, allowing stimulation and recording from the same electrode (Ferrer et al. 1978). The electrode (1.5 cm long) in the sensory cortex served to verify propagation of electric activity. The electrical signal was amplified 1000 × and processed with a 16-bit sigma delta converter at a 256 Hz sampling frequency. Records were monitored and saved for off-line analysis.
After a 2-min basal polygraph record, the amygdala was stimulated for 1 s with 1 ms rectangular pulses at 60 Hz (Paz et al. 1991). The stimulation intensity started at 50 µA at first, followed by increments of 20 µA until an AD was obtained. The obtained threshold current was then applied once every day, until at least 5 consecutive generalized convulsive seizures (stage 5) were observed. Seizure severity was scored based on behavioral criteria: stage 1-mouth and facial movements, stage 2-head nodding, stage 3-limb clonus, stage 4-rearing or stage 5-rearing and falling (Racine 1972). The following kindling parameters were measured: AD threshold (first day) and duration (every day), seizure stage each day, latency to each seizure stage, number of stimulations/days the rats were in each seizure stage, number of stimulations/days animals had either focal (1–3) or generalized (4–5) convulsive seizures, cumulative seizure duration to each seizure stage, total cumulative seizure duration and seizure stage across time for each group (mean and median seizure stage). Electrographic activity was recorded with signal capture software (Galileo, Stellate Systems) and merged with digital video footage (Diva, Stellate Systems). All rats were sacrificed 1 day after the last stimulation.
Blood and Plasma Measurements
The first day of procedures and day of sacrifice, blood glucose, and β-hydroxybutyrate levels were measured with a digital monitor system (Optium Xceed, Abbott Laboratories) after a 4 h fast, using samples obtained from the tail vein (Lee and Goosens 2015). Plasma was obtained by centrifugation at 2500 rpm for 20 min at 4 °C, kept at − 70 °C, and used to measure insulin levels with an enzyme-linked immunosorbent assay (Alpco Diagnostics) following the procedures recommended by the manufacturer.
Tissue Processing
After quickly obtaining the brain, the hippocampus and adjacent temporal neocortex were rapidly micro-dissected. They were frozen in liquid nitrogen and stored at − 70 °C. Randomly, either the left or right hippocampus was homogenized at 4 °C using a buffer (pH 7) with 150 mM NaCl, 50 mM Tris, 1 mM EGTA, 0.05% Triton X-100, as well as protease and phosphatase inhibitors. The same procedure was done separately for the left or right (at random) temporal neocortex. After 30 min of incubation at 4 °C, the homogenate was centrifuged at 16,000×g for 10 min at 4 °C and the supernatant was obtained. The concentration of the total protein was measured using the Lowry technique.
Randomly, the left or right part of each brain area was separately homogenized with Trizol at 4 °C. RNA was obtained by adding chloroform, centrifugation at 12,000×g for 15 min at 4 °C, the addition of isopropanol, centrifugation at 12,000×g for 10 min at 4 °C, adding ethanol, and lastly centrifugation at 7500×g for 10 min at 4 °C. In some cases, the rest of the brain was post-fixed using 4% paraformaldehyde, cryo-protected in a 30% sucrose solution, frozen, and cut. The sections were stained using histological techniques to verify the electrode location.
Western Blots
The phosphorylated protein to total protein relationship was used as an indirect measurement of protein activity and hence mTOR cascade function. Total protein homogenates (50 µg/µl) were separated in 10% acrylamide gels using SDS-PAGE and transferred to PVDF membranes, which were then probed with the following antibodies: AMPK alpha catalytic subunits, Thr172 phospho-AMPK alpha catalytic subunits, protein kinase B (PKB), Thr308 phospho-PKB, ribosomal protein S6, Ser235/236 phospho-S6 (all were from Cell Signaling Technologies) or actin (Sigma–Aldrich Corporation), used as a loading control. The proteins were revealed by chemiluminescence (Online Resource 2) and analyzed with Quantity One software (Bio-Rad Laboratories).
Polymerase Chain Reaction
From the total RNA obtained from each brain area, complementary DNA was obtained by retro-transcription using the M-MLV reverse transcriptase enzyme plus random hexamers (Applied Biosystems) with the 2400 Geneamp PCR system (Perkin Elmer Incorporated). Quantitative real-time polymerase chain reactions were performed to analyze relative gene expression levels using TaqMan® gene expression assays (Applied Biosystems, see Online Resource 3) and standard methods. The following genes were analyzed: α1 and α2 catalytic subunits of AMPK, MTOR, tuberous sclerosis 1 and 2 (TSC1, TSC2), and ribosomal protein S6 kinase (S6K, poly-peptide 1). Eukaryotic 18S rRNA gene expression was used as an endogenous control to normalize the other gene expression levels using the difference in cycle threshold method with a StepOne real-time PCR system (Applied Biosystems).
Statistical Comparisons
Continuous quantitative data were first analyzed for normality and equal variance with Shapiro–Wilk and Levene’s tests, respectively. Other data types and results that did not pass either test were explored using non-parametric tests, whereas results that passed both tests were analyzed by parametric tests. Thus, analysis of variance (ANOVA) tests followed by Holm–Sidak or Kruskall–Wallis ANOVAs on ranks followed by Dunn’s tests were used for most results. Wilcoxon signed rank or paired t tests were used for comparisons across time (before versus after) of β-HB and glucose blood levels. Mann–Whitney rank sum or t tests were used to compare AD durations when one of the groups dropped out, leaving only two. Results are shown as mean ± standard error. All figures were created using Adobe Photoshop (Adobe Systems).
Results
Body Weights, β-HB, Glucose, and Insulin Levels
All groups gained body weight (Fig. 2); however, the CM group showed a significantly reduced weight gain (from days 4, 8 versus AM, AL, respectively). The AM group also showed diminished weight gain compared to the AL group from day 29 after its initial administration. We previously observed that the differences in body weight gain were maintained throughout all the procedures (Phillips-Farfán et al. 2015); thus, body weights were recorded only until experimental day 31 (postnatal day 51).

Body weight gain in male Wistar rats fed ad libitum (AL, n = 10, circles), fed ad libitum and treated with 100 mg/kg metformin (AM, n = 8, diamonds) or subjected to 15% caloric restriction and administered metformin at 100 mg/kg (CM, n = 7, squares), postnatal days 21–51. Data points were fitted with logistic growth curves. Kruskall–Wallis ANOVA on ranks, Dunn’s: days 1–27. ANOVA, Holm–Sidak: days 28–31. &Start of statistical differences among AM versus CM groups (p < 0.05, all subsequent comparisons met the criterion), *start of statistical disparities between AL versus CM groups (p < 0.05, all successive contrasts met this criterion)
In contrast, fasting levels of blood β-HB and glucose (Fig. 3a, b) were similar between all the groups at the start and end of the treatments. Similarly, the fasting levels of plasma insulin at the end of the experiments (Fig. 3c) were not significantly different between the groups. But the fasting concentrations of blood glucose increased and the fasting levels of blood β-HB decreased across time within all the groups.

Systemic concentration of energy substrates and the hormone insulin in male rats fed ad libitum (AL, n = 10), fed ad libitum + metformin (AM, n = 8, crosshatched) or subjected to 15% caloric restriction + metformin (CM, n = 7, checkerboard). a Blood β-hydroxybutyrate (β-HB) levels (mmol/l) at the start and end of the procedures (postnatal days 21 and 99, respectively). b Blood glucose levels (mg/dl) at the beginning and conclusion of experiments. c Plasma insulin levels (ng/ml) at the termination of the procedures. ANOVA, Holm–Sidak: initial β-HB and glucose. Kruskall–Wallis ANOVA on ranks, Dunn’s: final β-HB, final glucose and insulin. Wilcoxon signed rank: β-HB AL, glucose AL and AM. Paired t: β-HB AM and CM, glucose CM. #p < 0.05, ##p < 0.01, ###p < 0.001, initial versus final
Electrical Kindling of the Amygdala
The AD threshold (Fig. 4) was increased in the CM group, compared to the AL group. All the differences in AD durations (Fig. 5) involved the CM group: it showed lower AD lengths in all cases. The latencies to all convulsive seizure stages were similar between the groups (Fig. 6a). However, the CM group remained in convulsive seizure stage 5 for a shorter period (vs. AL and AM groups, Fig. 6b). Likely as a result, metformin plus CR also provoked a lower number of generalized convulsive seizures (vs. AM, Fig. 6c). Cumulative AD durations for seizure stages 1–4 were comparable between the groups (Fig. 7a), but the CM group showed a lower cumulative AD length for seizure stage 5 (Fig. 7b). Likely as a result, metformin plus CR also diminished the total cumulative AD duration (Fig. 7b). There were no differences between the groups in the median seizure stage in each kindling day (Fig. 8). However, metformin plus CR showed a clear trend for lowering and delaying seizure progression (i.e., it shifted the curve down and to the right).

% After-discharge threshold measured the first day of kindling in male rats fed ad libitum (AL, n = 8), fed ad libitum + metformin (AM, n = 4, crosshatched), or subjected to 15% caloric restriction + metformin treatment (CM, n = 7, checkerboard). Kruskall–Wallis ANOVA on ranks, Dunn’s; **p < 0.01 versus AL group

After-discharge durations (seconds) during the kindling experiment in rats fed ad libitum (AL, n = 8, circles), fed ad libitum + metformin (AM, n = 4, diamonds), or subjected to caloric restriction + metformin (CM, n = 7, squares). AD durations were scored as zero when none of the amygdala discharges lasted at least 5 s, although motor patterns-behaviors characteristic of generalized convulsive seizures (stages 4–5) were clearly observed. Seizure stage 5 results (from day 25) were calculated and analyzed separately, since CR + metformin mostly affected this stage. Kruskall–Wallis ANOVA on ranks, Dunn’s: days 1–4, 6–7, 26–28 and 30. ANOVA, Holm–Sidak: days 5, 8–10, 25, 29, and 31–33. Mann–Whitney rank sum: days 11–13, 35; t test: day 34. The comparisons reaching statistical significance are shown above the error bars: &p < 0.05, &&p < 0.01 versus AM group; *p < 0.05, **p < 0.01, ***p < 0.001 versus AL group

Seizure stage periods (days) in male rats fed ad libitum (AL, n = 8), fed ad libitum + metformin (AM, n = 4, crosshatched), or subjected to 15% caloric restriction + metformin (CM, n = 7, checkerboard). a Latencies to seizure stages. b Time in seizure stages. c Period in partial (1–3) or generalized (4–5) convulsive seizure stages. Kruskall–Wallis ANOVA on ranks, Dunn’s; &p < 0.05, &&p < 0.01 versus AM group; *p < 0.05 versus AL group

Cumulative after-discharge durations (seconds) in male rats fed ad libitum (AL, n = 8), fed ad libitum + metformin (AM, n = 4, crosshatched), or subjected to caloric restriction + metformin (CM, n = 7, checkerboard). a Collected after-discharge intervals to seizure stages 1–4. b Combined after-discharge time to seizure stage 5 and total pooled after-discharge periods. Kruskall–Wallis ANOVA on ranks, Dunn’s: stages 1–4, total. ANOVA, Holm–Sidak: stage 5. &&&p < 0.001 versus AM group; *p < 0.05, ***p < 0.001 versus AL group

Mean seizure stage up to day 25 of the kindling process in animals fed ad libitum (AL, n = 8, circles), fed ad libitum + metformin (AM, n = 4, diamonds), or subjected to 15% caloric restriction + metformin (CM, n = 7, squares). Although the comparisons did not reach statistical significance, CM had a clear trend for a lower and delayed seizure progression (i.e., the curve is shifted down and to the right)
mTOR Cascade
AMPK phosphorylation (Figs. 9b, 10a) increased in the temporal neocortex and hippocampus of the CM group (vs. AL, AM; vs. AL, respectively). AMPK phosphorylation was also augmented in the AM group compared to the AL group in the neocortex. Conversely, PKB phosphorylation was diminished (Figs. 9b, 10b) in AM and CM groups in both the neocortex and the hippocampus (compared to the AL group). The phosphorylation of ribosomal protein S6 (Figs. 9b, 10c) declined in the neocortex of the AM group compared to the AL group and diminished in the hippocampus of the AM and CM groups compared to the AL group. mTOR gene expression (Fig. 11a) was augmented in the neocortex and hippocampus of the AM and CM groups compared to the AL group. The expression of all other genes studied (Fig. 11b, c) was comparable in the neocortex and hippocampus.

a Brief diagram of the mechanistic target of rapamycin (mTOR) cascade, analyzed proteins, or genes are shown (AMPK adenosine monophosphate-activated protein kinase, PKB protein kinase B, TSC1, TSC2 tuberous sclerosis 1 and 2 genes, MTOR mechanistic target of rapamycin gene, S6K ribosomal protein S6 kinase gene, S6 ribosomal protein S6; arrow lines: activation, hyphens ending in circles: inhibition). b Representative western blot results showing phosphorylated (p) and total AMPK, PKB, and S6 in the neocortex (Cx, green) and the hippocampus (Hp, purple) of rats fed ad libitum + metformin (AM, blue) or subjected to mild caloric restriction + metformin (CM, orange). Groups and brain regions are shown above each pair of images. Western blots corresponding to AL and CR groups were previously published (Phillips-Farfán et al. 2015). The molecular weight (MW) in kilodaltons (kDa) is shown to the right of each pair of protein images

Phosphorylated to total protein ratio of proteins participating in the mechanistic target of rapamycin (mTOR) pathway in the neocortex and hippocampus of male animals fed ad libitum (AL, n = 10), fed ad libitum + metformin (AM, n = 8, crosshatched), or subjected to mild caloric restriction + metformin (CM, n = 7, checkerboard). a Phospho-AMPK to total AMPK quotient. b Phospho-PKB proportion to total PKB. c Phospho-ribosomal protein S6 to total fraction. ANOVA, Holm–Sidak: AMPK Cx, PKB Cx and Hp, S6 Hp. Kruskall–Wallis ANOVA on ranks, Dunn’s: AMPK Hp, S6 Cx. &&p < 0.01 versus AM group; *p < 0.05, **p < 0.01, ***p < 0.001 versus AL group

Relative gene expression in the neocortex and hippocampus of rats fed ad libitum (AL, n = 10), fed ad libitum + metformin (AM, n = 8, crosshatched), or subjected to caloric restriction + metformin (CM, n = 7, checkerboard). a Comparative expression of the α1 catalytic subunit of adenosine monophosphate-activated protein kinase (AMPK α1), mechanistic target of rapamycin (MTOR), ribosomal protein S6 kinase poly-peptide 1 (S6K), or tuberous sclerosis 1 (TSC1) genes. b Semi-quantitative expression of the α2 AMPK catalytic subunit (AMPKα2) gene. c Relative expression of tuberous sclerosis 2 gene (TSC2). ANOVA, Holm–Sidak: AMPKα1 Cx and Hp, S6K Cx, TSC1 Cx, TSC2 Hp. Kruskall–Wallis ANOVA on ranks, Dunn’s: AMPKα2 Cx and Hp, MTOR Cx and Hp, S6K Hp, TSC1 Hp, TSC2 Cx. *p < 0.05, **p < 0.01, ***p < 0.001 versus AL group
Discussion
Body Weights, β-HB, Glucose, and Insulin Levels
All groups gained body weight (Fig. 2) according to their natural development, but both the AM group and CM group showed reduced body weight gain. Since both groups were treated with metformin, any differences between them are solely due to CR. Consistent with our previous report (Phillips-Farfán et al. 2015), CR showed a larger and earlier action than metformin on this group. This suggests an independent early effect and an additive action later. The action of metformin on body weight gain in normal healthy rats was somewhat surprising, since no adverse effects were seen up to a dose of 200 mg/kg/day for a longer period in older animals of a different strain (Quaile et al. 2010). Thus, this adverse action of metformin on developmental growth was likely caused by the younger age of rats at the start of treatment, although a strain difference may also account for the discrepancy. Use of metformin causes a loss of body weight, but only in obese or overweight patients whether or not they have diabetes (Luciano-Mateo et al. 2017; Malin and Kashyap 2014) and in animal models of these conditions (de Oliveira Santana et al. 2016; Kim et al. 2013; Matsui et al. 2010). Of note, mTOR cascade inhibition is consistent with a reduced body weight gain or loss of body weight (Hebert et al. 2014; Rovira et al. 2008). We do not know any previous reports regarding the effect of metformin on body weight gain in young rats; however, this reduction was much less than that evoked by CR.
Levels of energy substrates (glucose or β-HB) and plasma insulin concentrations (Fig. 3) were similar among the groups. As previously discussed (Phillips-Farfán et al. 2015), β-HB levels seem independent of CR and CR longer than a week has no effect on glucose concentrations in rats. Because “normal healthy” animals were used, the dose of metformin chosen (100 mg/kg/ day) was not expected to affect glucose, β-HB, or insulin concentration (Klepser and Kelly 1997) in the long term. This is because the beneficial effect of chronic metformin administration (i.e., the decrease in blood glucose) has only been observed in hyperglycemic subjects (Klepser and Kelly 1997), while long-term metformin treatment has no adverse side effects up to a dose of 200 mg/kg/day in rats (Quaile et al. 2010). Most studies concerning metformin effects have been done in adult animals; we thus showed that metformin has no deleterious effect on glucose metabolism in weanling young rats. It is crucial to note that metformin plus CR did not affect glucose, β-HB, or insulin levels. Thus, the actions of the treatments on the kindling experiment were totally independent of changes in glucose, β-HB, or insulin concentrations. Most likely the effects of CR and metformin were due to mTOR pathway inhibition.
Electrical Kindling of the Amygdala
Metformin by itself did not affect any kindling parameter. However, metformin has protective actions in chemically induced epilepsy models, such as acute and chronic seizures induced by pentylenetetrazol (Sanchez-Elexpuru et al. 2017; Yang et al. 2017; Zhao et al. 2014), as well as spontaneous recurrent seizures after status epilepticus induced by kainic acid (Yang et al. 2017) or pilocarpine (Mehrabi et al. 2018). Metformin reduced the mean seizure stage due to chronic treatment with pentylenetetrazol (Zhao et al. 2014). Metformin decreased mortality caused by acute pentylenetetrazol injection, lowered generalized tonic-clonic seizures intervals, increased post-ictal depression, and also decreased the length of seizure-like events induced by kainic acid (Yang et al. 2017). Metformin diminished mortality due to acute PTZ injection in mice lacking malin (a protein involved in Lafora disease). Metformin reduced the number of mice showing generalized seizures, increased seizure latency, and reduced seizure duration (Sanchez-Elexpuru et al. 2017). Metformin also had beneficial effects on the pilocarpine epilepsy model (Mehrabi et al. 2018). However, metformin by itself had no effects herein; likely due to the lower dose: 100 versus ~ 300 mg/kg in previous publications (Ma et al. 2007), though a species difference (rats vs. C57BL/6 mice) and the model used could also contribute to the discrepant findings. In this regard, we chose to administer metformin orally at a dose of 100 mg/kg/day since young weanling animals were used. As seen before, this dose of metformin had an adverse effect on body weight gain at this age.
Metformin plus CR augmented significantly the AD threshold (Fig. 4) and decreased its length (Fig. 5); it thus also lowered the total cumulative AD duration (Fig. 7) and the cumulative AD duration in which the animals were in seizure stage 5. Additionally, metformin plus CR decreased the time spent in seizure stage 5 (Fig. 6) and hence reduced the time spent in generalized convulsive seizures (stages 4–5). These positive anti-epileptic actions mostly affected generalized convulsive seizures instead of focal-partial convulsive seizures. Thus, metformin plus CR likely would be helpful for all measures of epileptic activity (seizure number, duration, and severity) in patients showing generalized convulsive seizures. Comparing among the AM group and CM group, since the only difference was that the latter group was subjected to 15% CR, it appears that CR contributes more than metformin on the beneficial effects of metformin plus CR. The advantages of metformin plus CR are that lower levels of CR could be used not to induce malnutrition or higher doses of metformin could be avoided to reduce adverse side effects.
mTOR Cascade
Metformin alone significantly increased AMPK phosphorylation in the neocortex, whereas metformin plus CR increased AMPK phosphorylation in the neocortex and hippocampus (Figs. 9, 10). Metformin by itself did not significantly augment AMPK phosphorylation in the hippocampus but there was a trend for this effect.
The AM and CM groups had reduced PKB phosphorylation in the neocortex and hippocampus. Since both groups were metformin-treated, the results indicate an unexpected effect of metformin on PKB. The link between metformin and PKB has been recently investigated (Ge et al. 2017; Mostafa et al. 2016). In the scopolamine-induced amnesia model, metformin augmented PKB phosphorylation and diminished total PKB in the cortex and the hippocampus. Metformin prevented scopolamine-induced changes by normalizing PKB levels (Mostafa et al. 2016). Metformin also increased PKB phosphorylation in the hippocampus, without changing total PKB after ischemia–reperfusion (Ge et al. 2017). Presently, metformin alone or combined with CR decreased PKB phosphorylation without changes to total PKB. This opposite result is likely due to the age of the rats—prior reports used adults (Ge et al. 2017; Mostafa et al. 2016) and young weanlings were used in this report. Of note, all these effects were beneficial; metformin prevented scopolamine-induced memory deficits (Mostafa et al. 2016), reduced apoptosis in the hippocampus after ischemia–reperfusion (Ge et al. 2017), and showed anti-epileptic effects by inhibiting the mTOR pathway. Of note, metformin use often results in conflicting results (Ge et al. 2017; Mostafa et al. 2016). Interestingly, a different anti-diabetic drug with a distinct mechanism of action (liraglutide) also seems to activate AMPK and thus inhibits the mTOR pathway (Chen et al. 2017; Palleria et al. 2017; Kong et al. 2018).
S6 phosphorylation was reduced in the neocortex of the AM group, while in the hippocampus it was diminished in both AM and CM groups. The actions of metformin on AMPK (Foretz et al. 2014) and mTOR cascade function, usually assessed by phosphorylation of ribosomal S6 kinase or its target, are known (Mehrabi et al. 2018; Shaw 2009). Metformin plus CR were used for the first time; however, the actions of this combination are consistent with the identified effects of each component (Phillips-Farfán et al. 2015; Shaw 2009; Yang et al. 2014).
AM and CM groups had increased mTOR gene expression in the neocortex and the hippocampus (Fig. 11). The significance of this finding is not apparent since the mTOR protein participates in two different protein complexes (Cybulski and Hall 2009; Eltschinger and Loewith 2016; Gaubitz et al. 2016). This result likely is a compensatory mechanism due to mTOR pathway inhibition, as suggested before (de Kreutzenberg et al. 2015). On the other hand, since the expression of all the other genes was unaffected, the anti-epileptic effects of metformin and CR were mostly independent from changes in gene expression.
Conclusions
Metformin combined with CR showed many protective anti-convulsive actions on generalized convulsive seizures. It augmented the AD threshold and reduced the following: AD duration, time in seizure stage 5, number of generalized convulsive seizures, cumulative AD interval in seizure stage 5, and total cumulative AD length. Thus, metformin plus CR would likely be beneficial for generalized convulsive seizure treatment. Patients that are overweight, obese, or have metabolic syndrome in addition to having an epileptic disease are an ideal population for clinical trials to test the effectiveness of this therapeutic scheme.
Abbreviations
- AD:
-
After-discharge
- AL:
-
Ad libitum fed group
- AM:
-
Ad libitum fed group metformin-treated
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- AMPKα1 :
-
Adenosine monophosphate-activated protein kinase alpha 1 catalytic subunit gene
- AMPKα2 :
-
Adenosine monophosphate-activated protein kinase alpha 2 catalytic subunit gene
- ANOVA:
-
Analysis of variance
- β-HB:
-
β-Hydroxybutyrate
- CM:
-
Group subjected to caloric restriction and metformin-treated
- CR:
-
Caloric restriction
- Cx:
-
Neocortex
- ed:
-
Experimental day
- Hp:
-
Hippocampus
- mTOR:
-
Mechanistic target of rapamycin
- MTOR :
-
Mechanistic target of rapamycin gene
- mTORC1:
-
Mechanistic target of rapamycin complex 1
- pAMPK:
-
Phosphorylated adenosine monophosphate-activated protein kinase
- pd:
-
Postnatal day
- PKB:
-
Protein kinase B
- pPKB:
-
Phosphorylated protein kinase B
- pS6:
-
Phosphorylated ribosomal protein S6
- S6:
-
Ribosomal protein S6
- S6K :
-
Ribosomal S6 kinase gene
- TSC1 :
-
Tuberous sclerosis 1 gene
- TSC2 :
-
Tuberous sclerosis 2 gene
References
Adak T, Samadi A, Ünal AZ, Sabuncuoğlu S (2018) A reappraisal on metformin. Regul Toxicol Pharmacol 92:324–332. https://doi.org/10.1016/j.yrtph.2017.12.023
Azarbar A, McIntyre DC, Gilby KL (2010) Caloric restriction alters seizure disposition and behavioral profiles in seizure-prone (fast) versus seizure-resistant (slow) rats. Behav Neurosci 124:106–114. https://doi.org/10.1037/a0018307
Bough KJ, Valiyil R, Han FT, Eagles DA (1999) Seizure resistance is dependent upon age and calorie restriction in rats fed a ketogenic diet. Epilepsy Res 35:21–28. https://doi.org/10.1016/S0920-1211(98)00125-9
Bough KJ, Schwartzkroin PA, Rho JM (2003) Calorie restriction and ketogenic diet diminish neuronal excitability in rat dentate gyrus in vivo. Epilepsia 44:752–760. https://doi.org/10.1046/j.1528-1157.2003.55502.x
Chen J, Wang Z, Mao Y, Zheng Z, Chen Y, Khor S et al (2017) Liraglutide activates autophagy via GLP-1R to improve functional recovery after spinal cord injury. Oncotarget 8:85949–85968. https://doi.org/10.18632/oncotarget.20791
Citraro R, Leo A, Constanti A, Russo E, De Sarro G (2016) mTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol Res 107:333–343. https://doi.org/10.1016/j.phrs.2016.03.039
Crino PB (2016) The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol 12:379–392. https://doi.org/10.1038/nrneurol.2016.81
Cybulski N, Hall MN (2009) TOR complex 2: a signaling pathway of its own. Trends Biochem Sci 34:620–627. https://doi.org/10.1016/j.tibs.2009.09.004
de Kreutzenberg SV, Ceolotto G, Cattelan A, Pagnin E, Mazzucato M, Garagnani P et al (2015) Metformin improves putative longevity effectors in peripheral mononuclear cells from subjects with prediabetes. A randomized controlled trial. Nutr Metab Cardiovasc Dis 25:686–693. https://doi.org/10.1016/j.numecd.2015.03.007
de Oliveira Santana KN, Lelis DF, Mendes KL, Lula JF, Paraíso AF, Andrade JM et al (2016) Metformin reduces lipogenesis markers in obese mice fed a low-carbohydrate and high-fat diet. Lipids 51:1375–1384. https://doi.org/10.1007/s11745-016-4209-y
Eltschinger S, Loewith R (2016) TOR complexes and the maintenance of cellular homeostasis. Trends Cell Biol 26:148–159. https://doi.org/10.1016/j.tcb.2015.10.003
Ferrer AZ, Fernández-Guardiola A, Solís H (1978) Electronic circuit breaker for recording and stimulation from same electrode. Electroencephalogr Clin Neurophysiol 45:299–301. https://doi.org/10.1016/0013-4694(78)90016-0
Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B (2014) Metformin: from mechanisms of action to therapies. Cell Metab 20:953–966. https://doi.org/10.1016/j.cmet.2014.09.018
Gasior M, Rogawski MA, Hartman AL (2006) Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol 17:431–439
Gaubitz C, Prouteau M, Kusmider B, Loewith R (2016) TORC2 structure and function. Trends Biochem Sci 41:532–545. https://doi.org/10.1016/j.tibs.2016.04.001
Ge XH, Zhu GJ, Geng DQ, Zhang HZ, He JM, Guo AZ et al (2017) Metformin protects the brain against ischemia/reperfusion injury through PI3K/Akt1/JNK3 signaling pathways in rats. Physiol Behav 170:115–123. https://doi.org/10.1016/j.physbeh.2016.12.021
Gorter JA, van Vliet EA, Lopes da Silva FH (2016) Which insights have we gained from the kindling and post-status epilepticus models? J Neurosci Methods 260:96–108. https://doi.org/10.1016/j.jneumeth.2015.03.025
Greene AE, Todorova MT, McGowan R, Seyfried TN (2001) Caloric restriction inhibits seizure susceptibility in epileptic EL mice by reducing blood glucose. Epilepsia 42:1371–1378. https://doi.org/10.1046/j.1528-1157.2001.17601.x
Hebert M, Licursi M, Jensen B, Baker A, Milway S, Malsbury C et al (2014) Single rapamycin administration induces prolonged downward shift in defended body weight in rats. PLoS ONE 9:e93691. https://doi.org/10.1371/journal.pone.0093691
Kandratavicius L, Balista PA, Lopes-Aguiar C, Ruggiero RN, Umeoka EH, Garcia-Cairasco N et al (2014) Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat 10:1693–1705. https://doi.org/10.2147/NDT.S50371
Kaplan B, Qazi Y, Wellen JR (2014) Strategies for the management of adverse events associated with mTOR inhibitors. Transplant Rev 28:126–133. https://doi.org/10.1016/j.trre.2014.03.002
Kim HJ, Park EY, Oh MJ, Park SS, Shin KH, Choi SH et al (2013) Central administration of metformin into the third ventricle of C57BL/6 mice decreases meal size and number and activates hypothalamic S6 kinase. Am J Physiol Regul Integr Comp Physiol 305:R499–R505. https://doi.org/10.1152/ajpregu.00099.2013
Klepser TB, Kelly MW (1997) Metformin hydrochloride: an antihyperglycemic agent. Am J Health Syst Pharm 54:893–903
Kong FJ, Wu JH, Sun SY, Ma LL, Zhou JQ (2018) Liraglutide ameliorates cognitive decline by promoting autophagy via the AMP-activated protein kinase/mammalian target of rapamycin pathway in a streptozotocin-induced mouse model of diabetes. Neuropharmacology 131:316–325. https://doi.org/10.1016/j.neuropharm.2018.01.001
Kossoff EH, Rho JM (2009) Ketogenic diets: evidence for short- and long-term efficacy. Neurotherapeutics 6:406–414. https://doi.org/10.1016/j.nurt.2009.01.005
Lee G, Goosens KA (2015) Sampling blood from the lateral tail vein of the rat. J Vis Exp 99:e52766. https://doi.org/10.3791/52766
Luciano-Mateo F, Hernández-Aguilera A, Cabre N, Camps J, Fernández-Arroyo S, Lopez-Miranda J et al (2017) Nutrients in energy and one-carbon metabolism: learning from metformin users. Nutrients 9:121. https://doi.org/10.3390/nu9020121
Ma TC, Buescher JL, Oatis B, Funk JA, Nash AJ, Carrier RL et al (2007) Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci Lett 411:98–103. https://doi.org/10.1016/j.neulet.2006.10.039
Malin SK, Kashyap SR (2014) Effects of metformin on weight loss: potential mechanisms. Curr Opin Endocrinol Diabetes Obes 21:323–329. https://doi.org/10.1097/MED.0000000000000095
Matsui Y, Hirasawa Y, Sugiura T, Toyoshi T, Kyuki K, Ito M (2010) Metformin reduces body weight gain and improves glucose intolerance in high-fat diet-fed C57BL/6J mice. Biol Pharm Bull 33:963–970. https://doi.org/10.1248/bpb.33.963
McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M (2011) The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 52:e7–e11. https://doi.org/10.1111/j.1528-1167.2011.02981.x
Mehrabi S, Sanadgol N, Barati M, Shahbazi A, Vahabzadeh G, Barzroudi M et al (2018) Evaluation of metformin effects in the chronic phase of spontaneous seizures in pilocarpine model of temporal lobe epilepsy. Metab Brain Dis 33:107–114. https://doi.org/10.1007/s11011-017-0132-z
Mostafa DK, Ismail CA, Ghareeb DA (2016) Differential metformin dose-dependent effects on cognition in rats: role of Akt. Psychopharmacology 233:2513–2524. https://doi.org/10.1007/s00213-016-4301-2
Palleria C, Leo A, Andreozzi F, Citraro R, Iannone M, Spiga R, Sesti G, Constanti A, De Sarro G, Arturi F, Russo E (2017) Liraglutide prevents cognitive decline in a rat model of streptozotocin-induced diabetes independently from its peripheral metabolic effects. Behav Brain Res 321:157–169. https://doi.org/10.1016/j.bbr.2017.01.004
Paz C, Gutiérrez-Baeza F, Bazán-Perkins B (1991) Transection of the superior cerebellar peduncle interferes with the onset and duration of generalized seizures induced by amygdaloid kindling. Brain Res 558:90–92. https://doi.org/10.1016/0006-8993(91)90718-B
Phillips-Farfán BV, Rubio Osornio M del C, Custodio Ramírez V, Paz Tres C, Carvajal Aguilera KG (2015) Caloric restriction protects against electrical kindling of the amygdala by inhibiting the mTOR signaling pathway. Front Cell Neurosci 9:90. https://doi.org/10.3389/fncel.2015.00090
Quaile MP, Melich DH, Jordan HL, Nold JB, Chism JP, Polli JW et al (2010) Toxicity and toxicokinetics of metformin in rats. Toxicol Appl Pharmacol 243:340–347. https://doi.org/10.1016/j.taap.2009.11.026
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294. https://doi.org/10.1016/0013-4694(72)90177-0
Rena G, Hardie DG, Pearson ER (2017) The mechanisms of action of metformin. Diabetologia 60:1577–1585. https://doi.org/10.1007/s00125-017-4342-z
Rogers AE (1979) Nutrition. In: Baker HJ, Lindsey JR, Weisbroth SH (eds) The laboratory rat, volume I biology and diseases. Academic Press, New York, pp 123–152
Rovira J, Marcelo Arellano E, Burke JT, Brault Y, Moya-Rull D, Bañón-Maneus E et al (2008) Effect of mTOR inhibitor on body weight: from an experimental rat model to human transplant patients. Transpl Int 21:992–998. https://doi.org/10.1111/j.1432-2277.2008.00710.x
Sánchez-Elexpuru G, Serratosa JM, Sanz P, Sánchez MP (2017) 4-Phenylbutyric acid and metformin decrease sensitivity to pentylenetetrazol-induced seizures in a malin knockout model of Lafora disease. Neuroreport 28:268–271. https://doi.org/10.1097/WNR.0000000000000751
Shaw RJ (2009) LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol 196:65–80. https://doi.org/10.1111/j.1748-1716.2009.01972.x
Yang F, Chu X, Yin M, Liu X, Yuan H, Niu Y et al (2014) mTOR and autophagy in normal brain aging and caloric restriction ameliorating age-related cognition deficits. Behav Brain Res 264:82–90. https://doi.org/10.1016/j.bbr.2014.02.005
Yang Y, Zhu B, Zheng F, Li Y, Zhang Y, Hu Y et al (2017) Chronic metformin treatment facilitates seizure termination. Biochem Biophys Res Commun 484:450–455. https://doi.org/10.1016/j.bbrc.2017.01.157
Zhao RR, Xu XC, Xu F, Zhang WL, Zhang WL, Liu LM et al (2014) Metformin protects against seizures, learning and memory impairments and oxidative damage induced by pentylenetetrazole-induced kindling in mice. Biochem Biophys Res Commun 448:414–417. https://doi.org/10.1016/j.bbrc.2014.04.130
Acknowledgements
We thank Reyna Hernández Morales, María de la Luz Correa Gómez, and Patricia Zárate Grajales for their technical assistance.
Funding
This study was supported by the Instituto Nacional de Pediatría.
Author information
Authors and Affiliations
Contributions
MdelCRO, VCR, and DCG were responsible for performing all the experimental procedures, as well as obtaining all the resulting data and contributing to its analysis. CPT, KGCA, and BVPF contributed to the plan and design of the experiments, as well as to the writing and revising of the manuscript. BVPF performed the original plan and design of the experiments, contributed to the analysis of all the resulting data, as well as to the writing and revising of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10571_2018_611_MOESM1_ESM.tif
Representative electrographic recordings taken from one animal from each of the groups: fed ad libitum (AL), fed ad libitum + metformin (AM) or subjected to caloric restriction + metformin (CM). Epileptic activity is observed, characterized by spikes of great amplitude and low frequency. The horizontal lines under the electrographic records indicate the AD duration. Scale bar 1 s. Supplementary material 1 (TIF 4157 KB)
10571_2018_611_MOESM2_ESM.tif
Taqman® gene expression assays that were used to perform the real-time PCR. The catalog number, gene, NCBI reference sequences and amplicon lengths are shown. Supplementary material 2 (TIF 4625 KB)
10571_2018_611_MOESM3_ESM.pdf
Representative full western blots showing phosphorylated (p) and total AMPK and PKB in the neocortex (Cx) and hippocampus (Hp) of rats fed ad libitum + metformin (AM) or subjected to 15 % caloric restriction + metformin (CM). A protein ladder showing the relative molecular weight (in kilodaltons) is shown to the left of the bands, while the groups and brain regions are shown above the bands. Supplementary material 3 (PDF 17 KB)
Rights and permissions
About this article

Cite this article
Rubio Osornio, M.C., Custodio Ramírez, V., Calderón Gámez, D. et al. Metformin Plus Caloric Restriction Show Anti-epileptic Effects Mediated by mTOR Pathway Inhibition. Cell Mol Neurobiol 38, 1425–1438 (2018). https://doi.org/10.1007/s10571-018-0611-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-018-0611-8