
Abstract
An increase of stroke incidence occurs in women with the decline of estrogen levels following menopause. This ischemic damage may recur, especially soon after the first insult has occurred. We evaluated the effects of estrogen and phytoestrogen treatment on an in vitro recurrent stroke model using the HT22 neuronal cell line. HT22 cells were treated with 17β-estradiol or genistein 1 h after the beginning of the first of two oxygen and glucose deprivation/reoxygenation (OGD/R) cycles. During the second OGD, there was a deterioration of some components of the electron transport chain, such as cytochrome c oxidase subunit 1 with a subsequent increase of reactive oxygen species (ROS) production. Accordingly, there was also an increase of apoptotic phenomena demonstrated by poly(ADP-ribose) polymerase 1 cleavage, Caspase-3 activity, and Annexin V levels. The recurrent ischemic injury also raised the hypoxia-inducible factor 1α and glucose transporter 1 levels, as well as the ratio between the lipidated and cytosolic forms of microtubule-associated protein 1A/1B-light chain 3 (LC3-II/LC3-I). We found a positive effect of estradiol and genistein treatment by partially preserving the impaired cell viability after the recurrent ischemic injury; however, this positive effect does not seem to be mediated neither by blocking apoptosis processes nor by decreasing ROS production. This work contribute to the better understanding of the molecular mechanisms triggered by recurrent ischemic damage in neuronal cells and, therefore, could help with the development of an effective treatment to minimize the consequences of this pathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is one of the leading causes of death worldwide. During ischemic damage, there is a reduction in cerebral blood flow, and the resulting lack of oxygen and nutrients can lead to cell death (Malagelada et al. 2005). Ischemia/reperfusion (I/R) injury triggers malfunction of the electron transport chain (ETC) and excessive production of reactive oxygen species (ROS) (Huttemann et al. 2012). These ROS induce DNA fragmentation, leading to overactivation of nuclear poly(ADP-ribose) polymerase 1 (PARP-1), involving excess demand for energy metabolism, which can lead to cell death (van Wijk et al. 2005).
Furthermore, ischemic damage can activate autophagic mechanisms (Qin et al. 2010), which are involved in the regulation of multiple cellular processes, including survival in a stress situation (Lopez-Alonso et al. 2013). The complex molecular machinery of autophagy leads to the lipidation of microtubule-associated protein 1A/1B light chain 3 (LC3) (Lopez-Alonso et al. 2013). Cytosolic LC3 (LC3-I) is conjugated with phosphatidylethanolamine, resulting in the lipidated form (LC3-II), which is a specific element of the autophagosomal membrane (Kabeya et al. 2000) and whose increase corresponds to an increase of autophagic activity (Marino et al. 2008).
Hypoxia-inducible factor 1 (HIF-1) is a highly conserved transcription factor consisting of α and β subunits (Wang et al. 1995). The expression and stability of HIF-1α is negatively regulated by oxygen levels (Adamcio et al. 2010). In normoxia, α subunits are hydroxylated in proline residues by prolyl hydroxylases (PHs), allowing their recognition and ubiquitination by ubiquitin ligases for subsequent degradation by the proteasome (Maxwell et al. 1999). However, under hypoxic conditions, HIF-PHs are inhibited because they need oxygen to carry out their action (Semenza 2004). Therefore, the heterodimer is stable under low oxygen levels and it is translocated into the nucleus, where it binds with hypoxia response elements (HREs) of promoter regions enhancing the transcription of several genes that counteract the effects of hypoxia (Adamcio et al. 2010). Some of the proteins that are under HIF-1 control are glucose transporters (GLUTs) (Sandau et al. 2000). Specifically, glucose transporter 1 (GLUT1) and GLUT3 are widely expressed in the cerebral tissue, and they are responsible for the majority of glucose utilization and uptake in the brain (Duelli and Kuschinsky 2001; McEwen and Reagan 2004).
Regarding stroke etiology, there is a strong increase in stroke incidence in postmenopausal women, which makes them more susceptible than men to stroke during aging (Towfighi et al. 2007). Studies in rodents have shown that ovariectomy breaks the protection against brain ischemic insult of intact female rats, which in turn may be reversed with estrogen replacement (Simpkins et al. 1997). Likewise, physiological doses of estradiol improve vasodilation capacity and increased cerebral blood flow in ovariectomized rats exposed to ischemic injury (Pelligrino et al. 1998). However, sometimes medium and long-term estrogen treatment can act as promoter of breast cancer (Colditz 1999) and venous thromboembolism (Daly et al. 1996). There is therefore a growing interest in the study of alternative compounds, such as phytoestrogen derived from plants. These molecules have structural similarities to 17β-estradiol, so they can bind with estrogen receptors leading to estrogenic or antiestrogenic effects (Kuiper et al. 1998). Phytoestrogen are natural selective estrogen receptor modulators (SERMs) (Baker et al. 2000), which are able to act as ER agonists or antagonists depending on the tissue (Beck et al. 2005). In this sense, genistein, one of the best known phytoestrogens, has antioxidant properties (Liang et al. 2008), effects on the circulatory function (Cortina et al. 2013) and anti-apoptotic actions (Schreihofer and Redmond 2009), which has led it to become a potential treatment against ischemic damage.
Despite many studies reporting the beneficial effects of estrogens and phytoestrogens against ischemic injury during female aging, little is known about their effects in consecutive stroke situations. It is worth noting that sometimes stroke can be recurrent, especially soon after the first insult has occurred (Kaplan et al. 2005). Therefore, the aim of this work was to study the potential neuroprotective role of estrogens and genistein in an in vitro model of recurrent stroke.
Experimental
Cell Culture
HT22 neuronal line of mouse hippocampus was kindly provided by Dr. Juan Carlos Mayo from the University of Oviedo, Spain. HT22 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with fetal bovine serum (FBS) 10 % v/v, glucose 4.5 g/l, glutamine 0.86 g/l, pyruvate 0.11 g/l, penicillin 100 IU/ml, streptomycin 100 µg/ml, at 37 °C in a fully humidified 5 % CO2 atmosphere. Culture medium was renewed every three days. Seeding density is specified at each experiment.
Oxygen and Glucose Deprivation/Reoxygenation Protocol
Oxygen and glucose deprivation/reoxygenation (OGD/R) is a well-established in vitro model to study the pathology and pharmacology of ischemic damage, since it allows mimicking the conditions occurring in an ischemic/reperfusion damage (Gu et al. 2013; Guo et al. 2013). There are already published a number of works about the effects of different drugs on the outcome of an ischemic injury; however, little is known about consecutive stroke situations. In this regard, it is worth noting that sometimes stroke can be recurrent, especially soon after the first insult has occurred (Kaplan et al. 2005). In order to study the potential neuroprotective role of estrogens and genistein in an in vitro model of recurrent stroke, we generated two OGD/R successive situations. First of all, cells were washed three times with Dulbecco’s PBS (Gentaur, Belgium). Then, cells were incubated in culture medium without FBS and with low glucose (DMEM with glucose 1 g/l, glutamine 2 mM, pyruvate 0.11 g/l, penicillin 100 IU/ml, streptomycin 100 µg/ml) in a hypoxic chamber (TEB1000 Flow Bioreactor, EBERS Medical Technology, Spain) at 1 % O2, 5 % CO2, and 37 °C for 18 h. After the OGD period, cells were washed three times with PBS and incubated with complete culture medium under normoxic conditions for 24 h. Once finished the reoxygenation phase, surviving cells were subjected to a new OGD/R cycle under the same conditions. These OGD/R protocol times were chosen according to the literature (Gu et al. 2013). In addition, cells were treated with 17β-estradiol 10 nM, genistein 1 µM, or vehicle (DMSO) from 1 h after starting first OGD period until the end of first reoxygenation. Furthermore, during first reoxygenation FBS was replaced by FBS treated with active carbon to remove serum sex hormones and to avoid potential interference with treatments. This 17β-estradiol concentration is considered as a physiological dose (Garrido et al. 2013), and it has been used previously by other authors in this cellular model (Sheldahl et al. 2008). Moreover, this genistein dose corresponds to circulating phytoestrogen levels in humans, and it has been previously shown to be neuroprotective in in vitro models of ischemic injury (Schreihofer and Redmond 2009). Throughout the experiment, a cell control group has been subjected to the same washing and medium changes, but it has been always maintained in complete culture medium and normoxia. These cells have been also treated for the same time with vehicle (DMSO). Therefore, the experiment consisted of four treatment groups: cells under normoxia and treated with DMSO (N); and cells subjected to recurrent OGD/R damage and treated with DMSO (V), 17β-estradiol (E), or genistein (G).
Cell Viability Assay
The reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) was carried out to evaluate cell viability (Gu et al. 2013). Cells were seeded at 5 × 103 seeding density in 96-well plates and incubated with culture medium containing MTT reagent 0.5 mg/ml for 4 h incubation under normal growth conditions (37 °C, 5 % CO2). Then, cells were lysed at room temperature by adding a solution containing SDS 20 % w/v and N,N-Dimethylformamide 50 % v/v for 24 h. Finally, the reduced reagent was quantified by optical density at 540 nm in a microplate reader (KC junior, BioTek, USA). Each treatment was assayed in octuplicate, and data were represented as the percentage of normoxic group mean value. MTT assays were performed at the end of the second reoxygenation period, in order to study the effect of a recurrent OGD/R injury on cell viability.
Cell Crude Extracts Collection and Western Blot Analysis
Cells were seeded at 2 × 105 seeding density in 6-well plates in order to carry out protein studies by Western blot. Cell lysates were obtained at two different times, at the end of the second OGD and at the end of the second reoxygenation period, in order to evaluate the molecular mechanisms underlying a recurrent OGD/R injury. For this purpose, cells were washed three times with PBS and lysed in 10 µl/cm2 RIPA buffer containing Tris–HCl 50 mM pH 7.4, NaCl 150 mM, Nonidet P-40 0.5 % v/v, sodium deoxycholate 0.5 % w/v, SDS 0.1 % w/v, EDTA 1 mM, PMSF 1 mM, sodium orthovanadate 1 mM, sodium glycerophosphate 1 mM, and pyrophosphate 1 mM. Then, samples were sonicated and centrifuged at 12,000×g for 30 min at 4 °C to remove non-soluble fractions. Finally, samples were stored at −20 °C until protein content determination by the Bradford dye-binding method (Bradford 1976).
To carry out Western blot analysis, equal amounts of protein (30 µg) were resolved by 8–13 % SDS-PAGE gel and electro-transferred from the gel to PVDF membranes (Immobilon-P Transfer Membrane, Millipore, USA) by the Towbin’s method (Towbin et al. 1979). Non-specific protein binding to the PVDF membranes was decreased by incubation for 1 h at room temperature (RT) with blocking buffer containing BSA 5 % w/v diluted in Tris–HCL 20 mM pH 7.5, NaCl 150 mM, Tween-20 0.1 % v/v (TBS-T). Then, membranes were incubated overnight at 4 °C with a primary antibody against the target protein: PARP-1 (diluted 1:1000, #9542, Cell Signaling, USA), LC3 (diluted 1:200, 0260S, Nanotools, Germany), cytochrome c oxidase subunit 1 (CO-1) (diluted 1:3000, Invitrogen, USA), HIF-1α (diluted 1:2000, AF1935, R&D Systems, USA), GLUT3 (diluted 1:2000, sc-7582, Santa Cruz, USA), and GLUT1 (diluted 1:1000, sc-7903, Santa Cruz, USA). After incubation with the primary antibody, membranes were washed with TBS-T and incubated for 1 h at RT with their respective horseradish peroxidase (HRP) coupled secondary antibody (diluted 1:20000 in TBS-T). The immunoreactive band of each protein was detected using an enhanced chemiluminescence (ECL) system (Chemiluminescent HRP Substrate, Millipore, USA) in a chemiluminescence revealed device (Chemic Doc-It Imaging System, UVP, USA). The densitometric values of each band area were quantified using the analysis and image processing program Quantity One 4.6.6. (Bio-Rad, USA). Later, membranes were incubated for 35 min at 65 °C with stripping buffer containing glycine 1.5 % w/v, Tween 20 0.5 % v/v, sodium dodecyl sulfate 0.1 % w/v, and pH 2.5, and probed with a HRP coupled anti-β-actin antibody (diluted 1:15000, sc-1616, Santa Cruz, USA). Then, all values were normalized with its respective loading control (β-actin), and they were represented as the percentage of normoxic group mean value to standardize all data. Each treatment was assayed in triplicate in two independent experiments.
Reactive Oxygen Species (ROS) Detection
Cells were seeded at 1 × 105 seeding density in 12-well plates, and cellular ROS were detected by using the cell permeant reagent 2′,7′-dichlorofluorescein diacetate (DCFDA), a fluorogenic dye that measures hydroxyl, peroxyl, and other ROS activity within the cell (Abcam, United Kingdom). At the end of the second OGD, cells were harvested and stained with 20 µM DCFDA for 30 min at 37 °C before analysis on flow cytometer. Signaling was detected with an excitation wavelength at 488 nm and emission wavelength at 535 nm. Each treatment was assayed in octuplicate. Data were represented as the percentage of viable cells with DCFDA fluorescence.
Apoptosis Assay
Cells were seeded at 1 × 105 seeding density in 12-well plates and apoptotic cells were detected by measuring both Annexin V and activated Caspase-3 levels. Annexin V is a calcium-dependent phospholipids binding protein that has a high affinity for phosphatidylserine (PS). Changes in PS asymmetry, which is analyzed by measuring Annexin V binding to the cell membrane, are detected before apoptosis-related morphological changes have occurred, and membrane integrity has been lost. At the end of the second OGD, cells were harvested, washed twice in PBS, and re-suspended in Annexin-binding buffer. Then, Annexin V labeled with DY-634 and the non-vital dye propidium iodide (PI) (Immunostep, Spain) were added to cell suspension and incubated for 15 min at RT in the dark. After the incubation period, Annexin-binding buffer was added, and the cell suspension was analyzed by flow cytometry within one hour. The simultaneous staining of cells with Annexin V and PI allows the discrimination of intact cells (Annexin V− and PI−), early apoptotic (Annexin V+ and PI−), and late apoptotic or necrotic cells (Annexin V+ and PI+). Each treatment was assayed in octuplicate. Data were represented as the percentage of Annexin V+ PI− cells.
In addition, the Caspase-3 inhibitor DEVD-FMK conjugated to FITC (Abcam, United Kingdom) was used as a marker to detect activated Caspase-3 in living cells, which plays a central role in apoptosis. FITC-DEVD-FMK is cell permeable, is non-toxic, and irreversibly binds to activated Caspase-3 in apoptotic cells. At the end of the second OGD, cells were harvested and re-suspended at 1 × 106 cells/ml. Then, cells were incubated with FITC-DEVD-FMK for 1 h at 37 °C and 5 % CO2. After this, cells were centrifuged at 3000 rpm for 5 min, and the supernatant was removed. Finally, cells were washed twice with 500 μl wash buffer and finally re-suspended in 300 μl of wash buffer and kept on ice until flow cytometry analysis. Each treatment was assayed in octuplicate, and data were represented as the percentage of cells with activated Caspase-3.
Statistical Analysis
First of all, the Gaussian distribution of our data was checked through a Kolmogorov–Smirnov test, as well as the variance homogeneity was studied by a Levene Test. Secondly, One Way Analysis of Variance (ANOVA) was carried out to compare the different treatment groups. Moreover, a Tukey Test has been done to carry out all pairwise multiple comparisons. A non-parametric Kruskal–Wallis Test followed by the Dunn’s Method was performed when normality or equal variances failed. Data were expressed as mean ± SEM. A p value <0.05 was considered statistically significant. Data were analyzed with Sigma-Stat 3.5 (Systat Software, USA) and GraphPad Prism 5.03 (GraphPad Software Inc., USA).
Results
Cell Viability and HIF-1α Levels
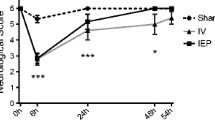
Cell viability was significantly reduced after two consecutive cycles of OGD/R in comparison to N group. However, both estradiol and genistein-treated cells showed significantly higher viability values than V group (Fig. 1a).

MTT assay of cell viability in HT22 cells at the end of two consecutive cycles of oxygen and glucose deprivation/reoxygenation (OGD/R) (n = 8 per group) (a). HIF-1α levels in HT22 cells subjected to a recurrent OGD/R injury (n = 6 per group) (b). Values represent mean ± SEM and are shown as the relative percentage of the normoxic group value (N). p < 0.05 versus normoxic group (asterisk), versus vehicle group (hash). HIF-1α: Hypoxia-inducible factor 1α; N: cells cultured in complete medium under normoxia; V, E, and G: cells subjected to the recurrent oxygen and glucose deprivation (OGD) or cells that were then also reoxygenated (OGD/R) and treated with vehicle, estradiol, and genistein, respectively
HIF-1α was studied as a marker of cells going through a hypoxic situation. This protein was significantly overexpressed in all groups exposed to the second OGD compared to the N group. Moreover, the levels of this protein also remained significantly higher in all groups compared to N cells after the second reoxygenation period (Fig. 1b).
Apoptosis Analysis
PARP-1 cleavage was evaluated as a marker of cells undergoing apoptosis. A significantly higher PARP-1 cleavage compared to normoxic group was observed at the end of the second OGD. However, both estradiol and genistein-treated cells showed a significant decrease of PARP-1 cleavage in comparison to the V group. When cells were reoxygenated, the PARP-1 cleavage decreased in all groups to the normoxic group levels (Fig. 2a).

PARP-1 cleavage in HT22 cells subjected to a recurrent glucose deprivation/reoxygenation (OGD/R) injury (n = 6 per group) (a). Activated caspase-3 (b) and Annexin V (c) detection at the end of the second oxygen and glucose deprivation (OGD) phase (n = 8 per group). Values represent mean ± SEM. p < 0.05 versus normoxic group (asterisk) versus vehicle group (hash). PARP-1: Poly (ADP-ribose) polymerase 1; AV: Annexin V; PI: propidium iodide; N: cells cultured in complete medium under normoxia; V, E, and G: cells subjected to the recurrent oxygen and glucose deprivation (OGD) or cells that were then also reoxygenated (OGD/R) and treated with vehicle, estradiol, and genistein, respectively
Moreover, apoptotic phenomena after the second cycle of OGD were studied by measuring both activated Caspase-3 and Annexin V levels. A significant increase of activated Caspase-3 (Fig. 2b) and Annexin V (Fig. 2c) positive cells was found in all groups compared to N group. No significant differences between treatments were found in any case.
LC3-II / LC3-I Ratio
LC3-II/LC3-I ratio was studied as a marker of cells undergoing autophagy. A significant increase of LC3-II/LC3-I ratio after the second cycle of OGD was found in all groups compared to N group. However, after the subsequent reoxygenation, the ratio decreased in all groups until the N group value (Fig. 3).

LC3-II / LC3-1 ratio in HT22 cells subjected to a recurrent glucose deprivation/reoxygenation (OGD/R) injury. The histogram shows the densitometric analysis of Western blot in two independent experiments assayed in triplicate (n = 6 per group). Values represent mean ± SEM and are shown as the relative percentage of the normoxic group value (N). p < 0.05 versus normoxic group (asterisk). LC3-I and LC3-II: cytosolic and lipidated form of microtubule-associated protein 1A/1B-light chain 3, respectively; N: cells cultured in complete medium under normoxia; V, E, and G: cells subjected to the recurrent oxygen and glucose deprivation (OGD) or cells that were then also reoxygenated (OGD/R) and treated with vehicle, estradiol, and genistein, respectively
Oxidative Metabolism Study
The main and catalytic subunit of cytochrome c oxidase (CO-1) was evaluated in order to detect possible changes of neuronal oxidative metabolism in response to the two consecutive OGD/R cycles. The levels of this protein decreased significantly below the N group value in all groups exposed to this second OGD. However, treatment with estradiol or genistein partially prevented this decrease compared to the V group. The subsequent reoxygenation increased CO-1 expression, reaching all groups a value close to that N group (Fig. 4a).

CO-1 levels in HT22 cells subjected to a recurrent oxygen and glucose deprivation/reoxygenation (OGD/R) injury (n = 6 per group) (a). Cellular ROS detection at the end of the second oxygen and glucose deprivation (OGD) phase (n = 8 per group) (b). Values represent mean ± SEM. p < 0.05 versus normoxic group (asterisk), versus vehicle group (hash). CO-1: Cytochrome c oxidase subunit 1; DCFDA: 2′,7′-dichlorofluorescein diacetate; N: cells cultured in complete medium under normoxia; V, E, and G: cells subjected to the recurrent oxygen and glucose deprivation (OGD) or cells that were then also reoxygenated (OGD/R) and treated with vehicle, estradiol, and genistein, respectively
Cellular ROS production was evaluated to complement the analysis of oxidative metabolism and as an indicator of the ETC viability. A significant increase of DCFDA positive cells after the second cycle of OGD was found in all groups compared to N group. No differences were found between treatments (Fig. 4b).
GLUT3 and GLUT1 Levels
The influence of two consecutive OGD/R cycles on GLUT3 levels, the main glucose transporter in neurons, was also evaluated. However, the expression of this protein was kept constant throughout the experiment, without showing statistically significant differences compared to N group, neither in groups subjected to a second OGD nor in those which were then reoxygenated. Similarly, no differences were found between the treatments assayed in any condition tested (Fig. 5a).

GLUT3 (a) and GLUT1 (b) levels in HT22 cells subjected to a recurrent oxygen and glucose deprivation/reoxygenation (OGD/R) injury. The histogram shows the densitometric analysis of Western blot in two independent experiments assayed in triplicate (n = 6 per group). Values represent mean ± SEM and are shown as the relative percentage of the normoxic group value (N). p < 0.05 versus normoxic group (asterisk). GLUT3 and GLUT1: Glucose transporter 3 and 1, respectively; N: cells cultured in complete medium under normoxia; V, E, and G: cells subjected to the recurrent oxygen and glucose deprivation (OGD) or cells that were then also reoxygenated (OGD/R) and treated with vehicle, estradiol, and genistein, respectively
The protein levels of another major glucose transporter in the CNS, GLUT1, were also checked. This transporter is found mostly in glial cells. Nevertheless, although in a very low amount, it is also expressed in neurons as shown by the normoxic group data. However, a second exposure to an OGD caused a statistically significant increase of GLUT1 amount in all groups compared to N group. After that, GLUT1 levels decreased during the reoxygenation period but remained significantly higher in all groups in comparison to the N group value (Fig. 5b).
Discussion
There is a marked increase in stroke incidence in women after menopause, who are more susceptible than men to stroke during aging (Towfighi et al. 2007). In fact, the protective effects of estrogen and phytoestrogen on several parameters deteriorated by an ischemic damage have been previously reported (Liang et al. 2008; Pelligrino et al. 1998; Simpkins et al. 1997). However, little is known about their roles in consecutive stroke situations. Taking into account that stroke can be recurrent, especially soon after once the first insult has occurred (Kaplan et al. 2005), we evaluated the potential protective role of estrogens and genistein in an in vitro model of recurrent stroke.
First, we evaluated cell viability after generating a recurrent oxygen and glucose deprivation/reoxygenation (OGD/R) injury. In agreement with other authors who have used different in vitro protocols of ischemia/reperfusion (I/R) (Mehta et al. 2012; Shimada et al. 2010), we found that the recurrent OGD/R caused a decrease in cell viability. The cascade of events leading to neuronal damage and death in I/R injury may include release of cytokines and free radicals, inflammatory processes, apoptosis, and excitotoxicity (Kuroda and Siesjo 1997). However, either treatment with estradiol or genistein was able to enhance cell viability in comparison to the vehicle group. Similarly, other authors found an increase in cell viability in response to acute treatment with estradiol, when administered shortly after generating an ischemic injury in ovariectomized female rats (Gulinello et al. 2006), as well as a beneficial role of genistein against ischemic damage has been previously found (Qian et al. 2012; Wang et al. 2014b). In this regard, it is known that estrogen can exert neuroprotective actions by inhibiting or activating pro- or anti-apoptotic pathways (Bagetta et al. 2004; Chiueh et al. 2003), decreasing excitotoxicity (Singer et al. 1996), interacting with growth factors (Azcoitia et al. 1999), or reducing inflammatory parameters (Suzuki et al. 2009).
In addition, crude cell extracts were obtained at the end of the second OGD, as well as at the end of subsequent reoxygenation, in order to analyze some molecular parameters underlying in vitro recurrent ischemic damage. First, we analyzed the expression of HIF-1α, which binds with the promoter regions of several genes to counteract the effects of oxygen deprivation, thereby promoting cell survival under hypoxic conditions (Sharp et al. 2001). HIF-1α levels remained low in normoxia, probably due to the action of prolyl hydroxylases (PHs), since it is known that their action allows the recognition and ubiquitination of HIF-1α by ubiquitin ligases for subsequent degradation by the proteasome (Maxwell et al. 1999). On the contrary, HIF-1α expression was enhanced during OGD, consistent with the previously described induction of this factor by hypoxia (Huang et al. 2012). Nevertheless, it is interesting that during reoxygenation, in which oxygen levels return to normality, HIF-1α is maintained at higher levels than under normoxia. This result is intriguing, because it is known that the presence of oxygen downregulates HIF-1α expression (Maxwell et al. 1999). This fact raises the question about whether factors other than oxygen may be involved in HIF-1α stabilization during reoxygenation. It has been suggested that PI3 K/Akt signaling can activate HIF-1α by direct phosphorylation or through prolyl hydroxylase 2 (PH2) inhibition (Jiang et al. 2001; Spinella et al. 2010). Thus, as previously suggested, signaling pathways inhibiting PHs activity could upregulate HIF-1α in spite of the presence of oxygen (Sun et al. 2013). Further studies about the involvement of the PI3 K/Akt pathway during reoxygenation following OGD are needed to understand better the potential regulatory role on HIF-1α.
PARP cleavage is a well-established marker of cells undergoing apoptosis (Oliver et al. 1998). PARP-1 cleavage increased because of the second OGD but decreased again to values close to that of the normoxic group during subsequent reoxygenation. Our data are therefore consistent with previous studies that showed induction of apoptosis due to oxygen/glucose deprivation in cortical neurons (Wang et al. 2014a). However, although the reoxygenation phase may also contribute to the damage by increasing ROS and the inflammatory response (van Wijk and Hageman 2005), we did not find increased apoptosis, at least at the level of PARP-1 cleavage. Moreover, we found a decrease of PARP-1 cleavage in estradiol- and genistein-treated groups after the second OGD period, suggesting a positive effect of both drugs. To further analyze these changes in apoptosis activity noticed after the second OGD, Caspase-3 activation has been detected by cytometry flow analysis. An increase of activated Caspase-3 positive cells was found in all treatment groups but, to our surprise, there were no differences between DMSO and estradiol- or genistein-treated cells. This means that estradiol and genistein do not seem to be mediating the decrease of PARP-1 cleavage through the blocking of one of its main upstream effectors such as Caspase-3. In this regard, it has been found that other caspases, such as Caspase-7, can also mediate PARP-1 cleavage during the apoptosis process (Cohen 1997). Moreover, Annexin V positive cells were also detected after this second OGD period, showing exactly the same pattern that Caspase-3 activation, i.e., an increase of apoptosis in response to the recurrent ischemic damage but any significant effect of the treatments. Taken all together, our data concerning apoptosis phenomena show that independently of the pathway used by estradiol and genistein to decrease PARP-1 cleavage, these treatments are not able to significantly decrease the elevated apoptosis rate after a recurrent ischemic injury.
We also analyzed the ratio between the lipidated and cytosolic forms of LC3 as a marker of autophagy. When cells activate the autophagic machinery, the cytosolic LC3 (LC3-I) is conjugated to phosphatidylethanolamine resulting in the lipidated form (LC3-II), which is a specific element of the autophagosomal membrane (Kabeya et al. 2000). LC3-II/LC3-I ratio increased during the second OGD and then decreased to the values of the normoxic group during reoxygenation. Neither estradiol nor genistein seemed to be involved in the autophagy activation/deactivation at any phase. This result agrees with previous works showing induction of autophagy by ischemic damage (Tian et al. 2010; Qin et al. 2010). In general, autophagy is considered as a survival mechanism, which provides cells with alternative energy sources under low-nutrient conditions, as well as helping them to eliminate deteriorated proteins or molecular waste (Aguirre et al. 2014). In contrast, there are also authors supporting the theory that autophagy can promote cell death, through both excessive self-digestion and degradation of essential cellular components (Reggiori and Klionsky 2002). This excessively increased autophagy could be involved in increasing neuronal death following cerebral ischemic damage (Ginet et al. 2009). This theory would fit with our data showing a similar pattern between apoptotic and autophagic activities. Some studies have shown a remarkable overlap between autophagy and apoptotic cell death, which may succeed each other or even coexist in the same cell (Bursch 2001; Canu et al. 2005). Therefore, the role of autophagy during recurrent brain ischemic damage seems ambiguous, and its pro-survival or pro-cell death potential remains to be elucidated.
The next step was to analyze the main and catalytic subunit (subunit 1) of cytochrome c oxidase (CO) as a marker of oxidative metabolism. CO is a key mitochondrial enzyme involved in cellular respiration that catalyzes the final step in the ETC (Moran et al. 2013). The brain consumes a lot of oxygen and glucose for the maintenance of neuronal activity (Bolanos et al. 1994). These substrates are mainly used by the aerobic metabolism to satisfy most brain energy requirements (Schurr and Rigor 1998). Neurons contain many mitochondria, suggesting high oxidative phosphorylation activity, and they have limited ability to metabolize substrates other than glucose (Fornazari et al. 2011). This indicates the importance of oxidative metabolism in neurons, and it largely explains the neuronal death occurring after ischemic damage. Our data show a strong decline in CO-1 levels after the second OGD period, suggesting a decrease of neuronal oxidative metabolism following the recurrent ischemic damage. However, both estradiol and genistein were able to partially prevent this decline. Moreover, cellular ROS production was evaluated at the end of the second OGD to further study the oxidative metabolism modulation by the recurrent ischemic injury. We found a high increase in cellular ROS production in all treatment groups with respect to the normoxic control cells, suggesting that although estradiol and genistein are able to raise the levels of CO-1, they are not able to compensate the general deterioration of the ETC function after the recurrent ischemic damage. Some studies regarding the effects of estradiol and genistein on lactate production by glial cells could be also advisable under this low-oxygen condition, because it has been shown that lactate may also be important for certain neuronal processes, such as differentiation, which requires a shift from oxidative to fermentative metabolism (Fornazari et al. 2011). Indeed, there is a hypothesis supporting the existence of a lactate shuttle between glial cells and neurons that could be important for post-ischemic neuronal survival (Schurr and Rigor 1998).
We finally studied the fluctuation of GLUT1 and GLUT3, which are the main glucose transporters in the central nervous system (Duelli and Kuschinsky 2001; McEwen and Reagan 2004), during the second OGD/R. Although both transporters have been detected in neuronal processes as well as in the neuronal cell body (Maher et al. 1991), GLUT3 is the main agent responsible for mediating glucose transport in neurons, whereas GLUT1 seems to play a more important role in glial cells (Maher et al. 1994). In spite of the fact that GLUT3 may increase its expression in response to hypoxia and hypoglycemia (Yu et al. 2012), our data showed high homogeneity of GLUT3 levels throughout the study, which were not modified by OGD or subsequent reoxygenation. In contrast, GLUT1 levels were low under normoxic conditions, agreeing with other studies supporting that this transporter is more expressed in glial cells, whereas glucose transport in neurons is primarily mediated by GLUT3 (Maher et al. 1991). However, during both the OGD and the reoxygenation phase GLUT1 expression was strongly increased. This result could suggest that these neurons, in order to optimize the limited supply of glucose received, are overexpressing a glucose transporter that has a secondary role under normal conditions. It is known that GLUT1 expression can be positively regulated by HIF-1α (Huang et al. 2012), which agrees with our data regarding the expression pattern of these proteins. It is also known that besides hypoxia, HIF-1α is sensitive to glucose levels. Thus, it has been observed that hyperglycemia leads to decreased HIF-1α transcriptional activity and levels (Catrina et al. 2004), as well as that the absence of glucose leads to transcription of several genes controlled by HIF-1α (Ryan et al. 1998). Nevertheless, GLUT3 upregulation by HIF-1α in response to I/R damage has also been demonstrated (Zhang et al. 2014), which suggests that cells have complex regulatory mechanisms to face a recurrent ischemic damage that are not exactly the same as those triggered by an isolated ischemic injury.
Taken all together, our data show that after a recurrent ischemic damage, there is a deterioration of some components of the ETC with a subsequent increase of ROS production. This excessive ROS partially mediate an increase of apoptotic phenomena leading to cell death. We found a positive effect of estradiol and genistein treatment by partially preserving the impaired cell viability after the recurrent ischemic injury; however, this positive effect does not seem to be mediated neither by blocking apoptosis processes nor by decreasing ROS production. Therefore, it would be advisable to assess the potential impact of estradiol and genistein on other important components of the I/R damage, such as excitotoxicity and inflammation. In addition, we must not forget that in vitro cultures have certain limitations. For example, repetitive ischemic injury not only selectively affects neurons, and it does not happen necessarily in the same brain area. Therefore, further studies with glial cells and in vivo models would help us to shed light on the molecular mechanisms involved in recurrent stroke, as well as to check the effectiveness of these treatments.
Conclusions
In summary, our results showed a protective role of 17β-estradiol and genistein in an in vitro model of recurrent stroke, by partially preserving the cell viability decrease generated as a result of the recurrent ischemic injury. However, more studies are needed to elucidate the molecular pathways involved in these neuroprotective effects, since neuronal cells seem to have complex regulatory mechanisms after a recurrent ischemic damage that partially differ from those triggered by an isolated ischemic injury. These data contribute to the better understanding of the molecular mechanisms triggered by recurrent ischemic damage in neuronal cells, and therefore, this work could help with the development of an effective treatment to minimize the consequences of this pathology.
Abbreviations
- OGD/R:
-
Oxygen and glucose deprivation/reoxygenation
- OGD:
-
Oxygen and glucose deprivation
- CO-1:
-
Cytochrome c oxidase subunit 1
- ROS:
-
Reactive oxygen species
- PARP-1:
-
Poly(ADP-ribose) polymerase 1
- HIF-1α:
-
Hypoxia-inducible factor 1α
- GLUT:
-
Glucose transporter
- LC3:
-
Microtubule-associated protein 1A/1B-light chain 3
- I/R:
-
Ischemia/reperfusion
- ETC:
-
Electron transport chain
- PH:
-
Prolyl hydroxylase
- HRE:
-
Hypoxia response element
- ER:
-
Estrogen receptor
- SERM:
-
Selective estrogen receptor modulator
- DMEM:
-
Dulbecco’s Modified Eagle’s Medium
- FBS:
-
Fetal bovine serum
- N:
-
Cells cultured in complete medium under normoxia
- V:
-
Cells subjected to OGD or OGD/R and treated with vehicle
- E:
-
Cells subjected to OGD or OGD/R and treated with estradiol
- G:
-
Cells subjected to OGD or OGD/R and treated with genistein
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
- TBS-T:
-
Tris-HCL 20 mM pH 7.5, NaCl 150 mM, Tween-20 0,1 % v/v
- HRP:
-
Horseradish peroxidase
- ECL:
-
Enhanced chemiluminescence
- DCFDA:
-
2′,7′-Dichlorofluorescein diacetate
- PI:
-
Propidium iodide
- AV:
-
Annexin V
- CNS:
-
Central nervous system
References
Adamcio B, Sperling S, Hagemeyer N, Walkinshaw G, Ehrenreich H (2010) Hypoxia inducible factor stabilization leads to lasting improvement of hippocampal memory in healthy mice. Behav Brain Res 208:80–84
Aguirre A, Lopez-Alonso I, Gonzalez-Lopez A, Amado-Rodriguez L, Batalla-Solis E, Astudillo A, Blazquez-Prieto J, Fernandez AF, Galvan JA, Dos Santos CC, Albaiceta GM (2014) Defective autophagy impairs ATF3 activity and worsens lung injury during endotoxemia. J Mol Med (Berl) 92:665–676
Azcoitia I, Sierra A, Garcia-Segura LM (1999) Neuroprotective effects of estradiol in the adult rat hippocampus: interaction with insulin-like growth factor-I signalling. J Neurosci Res 58:815–822
Bagetta G, Chiappetta O, Amantea D, Iannone M, Rotiroti D, Costa A, Nappi G, Corasaniti MT (2004) Estradiol reduces cytochrome c translocation and minimizes hippocampal damage caused by transient global ischemia in rat. Neurosci Lett 368:87–91
Baker VL, Leitman D, Jaffe RB (2000) Selective estrogen receptor modulators in reproductive medicine and biology. Obstet Gynecol Surv 55:S21–S47
Beck V, Rohr U, Jungbauer A (2005) Phytoestrogen derived from red clover: an alternative to estrogen replacement therapy? J Steroid Biochem Mol Biol 94:499–518
Bolanos JP, Peuchen S, Heales SJ, Land JM, Clark JB (1994) Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem 63:910–916
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Bursch W (2001) The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ 8:569–581
Canu N, Tufi R, Serafino AL, Amadoro G, Ciotti MT, Calissano P (2005) Role of the autophagic-lysosomal system on low potassium-induced apoptosis in cultured cerebellar granule cells. J Neurochem 92:1228–1242
Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L (2004) Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes 53:3226–3232
Chiueh C, Lee S, Andoh T, Murphy D (2003) Induction of antioxidative and antiapoptotic thioredoxin supports neuroprotective hypothesis of estrogen. Endocrine 21:27–31
Cohen GM (1997) Caspases: the executioners of apoptosis. Biochem J 326(Pt 1):1–16
Colditz GA (1999) Hormones and breast cancer: evidence and implications for consideration of risks and benefits of hormone replacement therapy. J Womens Health 8:347–357
Cortina B, Torregrosa G, Castello-Ruiz M, Burguete MC, Moscardo A, Latorre A, Salom JB, Valles J, Santos MT, Alborch E (2013) Improvement of the circulatory function partially accounts for the neuroprotective action of the phytoestrogen genistein in experimental ischemic stroke. Eur J Pharmacol 708:88–94
Daly E, Vessey MP, Hawkins MM, Carson JL, Gough P, Marsh S (1996) Risk of venous thromboembolism in users of hormone replacement therapy. Lancet 348:977–980
Duelli R, Kuschinsky W (2001) Brain glucose transporters: relationship to local energy demand. News Physiol Sci 16:71–76
Fornazari M, Nascimento IC, Nery AA, da Silva CC, Kowaltowski AJ, Ulrich H (2011) Neuronal differentiation involves a shift from glucose oxidation to fermentation. J Bioenerg Biomembr 43:531–539
Garrido P, Moran J, Alonso A, Gonzalez S, Gonzalez C (2013) 17beta-estradiol activates glucose uptake via GLUT4 translocation and PI3 K/Akt signaling pathway in MCF-7 cells. Endocrinology 154:1979–1989
Ginet V, Puyal J, Clarke PG, Truttmann AC (2009) Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol 175:1962–1974
Gu JH, Ge JB, Li M, Xu HD, Wu F, Qin ZH (2013) Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS ONE 8:e61641
Gulinello M, Lebesgue D, Jover-Mengual T, Zukin RS, Etgen AM (2006) Acute and chronic estradiol treatments reduce memory deficits induced by transient global ischemia in female rats. Horm Behav 49:246–260
Guo F, Wang H, Li L, Zhou H, Wei H, Jin W, Wang Q, Xiong L (2013) A novel domain of amino-Nogo-A protects HT22 cells exposed to oxygen glucose deprivation by inhibiting NADPH oxidase activity. Cell Mol Neurobiol 33:443–452
Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, Di Lorenzo A, Giordano FJ (2012) Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1alpha-dependent function. Proc Natl Acad Sci USA 109:17478–17483
Huttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J, Ramzan R, Vogt S, Grossman LI, Doan JW, Marcus K, Lee I (2012) Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta 1817:598–609
Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK (2001) Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ 12:363–369
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728
Kaplan RC, Tirschwell DL, Longstreth WT Jr, Manolio TA, Heckbert SR, Lefkowitz D, El-Saed A, Psaty BM (2005) Vascular events, mortality, and preventive therapy following ischemic stroke in the elderly. Neurology 65:835–842
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA (1998) Interaction of estrogenic chemicals and phytoestrogen with estrogen receptor beta. Endocrinology 139:4252–4263
Kuroda S, Siesjo BK (1997) Reperfusion damage following focal ischemia: pathophysiology and therapeutic windows. Clin Neurosci 4:199–212
Liang HW, Qiu SF, Shen J, Sun LN, Wang JY, Bruce IC, Xia Q (2008) Genistein attenuates oxidative stress and neuronal damage following transient global cerebral ischemia in rat hippocampus. Neurosci Lett 438:116–120
Lopez-Alonso I, Aguirre A, Gonzalez-Lopez A, Fernandez AF, Amado-Rodriguez L, Astudillo A, Batalla-Solis E, Albaiceta GM (2013) Impairment of autophagy decreases ventilator-induced lung injury by blockade of the NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol 304:L844–L852
Maher F, Simpson IA (1994) The GLUT3 glucose transporter is the predominant isoform in primary cultured neurons: assessment by biosynthetic and photoaffinity labelling. Biochem J 301:379–384
Maher F, Davies-Hill TM, Lysko PG, Henneberry RC, Simpson IA (1991) Expression of two glucose transporters, GLUT1 and GLUT3, in cultured cerebellar neurons: evidence for neuron-specific expression of GLUT3. Mol Cell Neurosci 2:351–360
Malagelada C, Xifro X, Minano A, Sabria J, Rodriguez-Alvarez J (2005) Contribution of caspase-mediated apoptosis to the cell death caused by oxygen–glucose deprivation in cortical cell cultures. Neurobiol Dis 20:27–37
Marino G, Ugalde AP, Salvador-Montoliu N, Varela I, Quiros PM, Cadinanos J, van der Pluijm I, Freije JM, Lopez-Otin C (2008) Premature aging in mice activates a systemic metabolic response involving autophagy induction. Hum Mol Genet 17:2196–2211
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399:271–275
McEwen BS, Reagan LP (2004) Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol 490:13–24
Mehta SL, Kumari S, Mendelev N, Li PA (2012) Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci 13:79
Moran J, Garrido P, Alonso A, Cabello E, Gonzalez C (2013) 17beta-Estradiol and genistein acute treatments improve some cerebral cortex homeostasis aspects deteriorated by aging in female rats. Exp Gerontol 48:414–421
Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM (1998) Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem 273:33533–33539
Pelligrino DA, Santizo R, Baughman VL, Wang Q (1998) Cerebral vasodilating capacity during forebrain ischemia: effects of chronic estrogen depletion and repletion and the role of neuronal nitric oxide synthase. NeuroReport 9:3285–3291
Qian Y, Guan T, Huang M, Cao L, Li Y, Cheng H, Jin H, Yu D (2012) Neuroprotection by the soy isoflavone, genistein, via inhibition of mitochondria-dependent apoptosis pathways and reactive oxygen induced-NF-kappaB activation in a cerebral ischemia mouse model. Neurochem Int 60:759–767
Qin AP, Liu CF, Qin YY, Hong LZ, Xu M, Yang L, Liu J, Qin ZH, Zhang HL (2010) Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 6:738–753
Reggiori F, Klionsky DJ (2002) Autophagy in the eukaryotic cell. Eukaryot Cell 1:11–21
Ryan HE, Lo J, Johnson RS (1998) HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 17:3005–3015
Sandau KB, Faus HG, Brune B (2000) Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI 3 K pathway. Biochem Biophys Res Commun 278:263–267
Schreihofer DA, Redmond L (2009) Soy phytoestrogen are neuroprotective against stroke-like injury in vitro. Neuroscience 158:602–609
Schurr A, Rigor BM (1998) Brain anaerobic lactate production: a suicide note or a survival kit? Dev Neurosci 20:348–357
Semenza GL (2004) Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 19:176–182
Sharp FR, Bergeron M, Bernaudin M (2001) Hypoxia-inducible factor in brain. Adv Exp Med Biol 502:273–291
Sheldahl LC, Shapiro RA, Bryant DN, Koerner IP, Dorsa DM (2008) Estrogen induces rapid translocation of estrogen receptor beta, but not estrogen receptor alpha, to the neuronal plasma membrane. Neuroscience 153:751–761
Shimada S, Hirabayashi M, Ishige K, Kosuge Y, Kihara T, Ito Y (2010) Activation of dopamine D4 receptors is protective against hypoxia/reoxygenation-induced cell death in HT22 cells. J Pharmacol Sci 114:217–224
Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, Bodor N, Day AL (1997) Estrogen may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg 87:724–730
Singer CA, Rogers KL, Strickland TM, Dorsa DM (1996) Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett 212:13–16
Spinella F, Rosano L, Del Duca M, Di Castro V, Nicotra MR, Natali PG, Bagnato A (2010) Endothelin-1 inhibits prolyl hydroxylase domain 2 to activate hypoxia-inducible factor-1alpha in melanoma cells. PLoS ONE 5:e11241
Sun YY, Lin SH, Lin HC, Hung CC, Wang CY, Lin YC, Hung KS, Lien CC, Kuan CY, Lee YH (2013) Cell type-specific dependency on the PI3 K/Akt signaling pathway for the endogenous Epo and VEGF induction by baicalein in neurons versus astrocytes. PLoS ONE 8:e69019
Suzuki S, Brown CM, Wise PM (2009) Neuroprotective effects of estrogen following ischemic stroke. Front Neuroendocrinol 30:201–211
Tian F, Deguchi K, Yamashita T, Ohta Y, Morimoto N, Shang J, Zhang X, Liu N, Ikeda Y, Matsuura T, Abe K (2010) In vivo imaging of autophagy in a mouse stroke model. Autophagy 6:1107–1114
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76:4350–4354
Towfighi A, Saver JL, Engelhardt R, Ovbiagele B (2007) A midlife stroke surge among women in the United States. Neurology 69:1898–1904
van Wijk SJ, Hageman GJ (2005) Poly(ADP-ribose) polymerase-1 mediated caspase-independent cell death after ischemia/reperfusion. Free Radic Biol Med 39:81–90
Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92:5510–5514
Wang L, Chen M, Yuan L, Xiang Y, Zheng R, Zhu S (2014a) 14,15-EET promotes mitochondrial biogenesis and protects cortical neurons against oxygen/glucose deprivation-induced apoptosis. Biochem Biophys Res Commun 450:604–609
Wang S, Wei H, Cai M, Lu Y, Hou W, Yang Q, Dong H, Xiong L (2014b) Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of ERK activity in ovariectomized mice. Int J Biol Sci 10:457–465
Yu J, Li J, Zhang S, Xu X, Zheng M, Jiang G, Li F (2012) IGF-1 induces hypoxia-inducible factor 1alpha-mediated GLUT3 expression through PI3 K/Akt/mTOR dependent pathways in PC12 cells. Brain Res 1430:18–24
Zhang Z, Yan J, Taheri S, Liu KJ, Shi H (2014) Hypoxia-inducible factor 1 contributes to N-acetylcysteine’s protection in stroke. Free Radic Biol Med 68:8–21
Acknowledgments
This study was funded by European Union FEDER funds, Plan de Ciencia, Tecnología e Innovación del Principado de Asturias, FICYT (GRUPIN 14-069).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Morán, J., Perez-Basterrechea, M., Garrido, P. et al. Effects of Estrogen and Phytoestrogen Treatment on an In Vitro Model of Recurrent Stroke on HT22 Neuronal Cell Line. Cell Mol Neurobiol 37, 405–416 (2017). https://doi.org/10.1007/s10571-016-0372-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-016-0372-1