
Abstract
Chronic cerebral hypoperfusion (CCH) is a common consequence of various cerebral vascular disorders and hemodynamic and blood changes. Recent studies have revealed an important role of CCH in neurodegeneration and dementia, including vascular dementia and Alzheimer’s disease (AD). This article reviews the recent advances in understanding CCH-induced neurodegeneration and AD-related brain pathology and cognitive impairment. We discuss the causes and assessment of CCH, the possible mechanisms by which CCH promotes Alzheimer-like pathology and neurodegeneration, and animal models of CCH. It appears that CCH promotes neurodegeneration and AD through multiple mechanisms, including induction of oxidative stress, Aβ accumulation and aggravation, tau hyperphosphorylation, synaptic dysfunction, neuronal loss, white matter lesion, and neuroinflammation. Better understanding of the mechanisms of CCH will help develop therapeutic strategies for preventing and treating neurodegeneration, including sporadic AD and vascular dementia, caused by CCH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Senile dementia is a progressive loss of cognitive ability, including memory, attention, language, and problem-solving, in the elderly and is a serious medical, social, and economic burden in modern society because of the growing aged population. Senile dementia mainly includes Alzheimer’s disease (AD), vascular dementia (VaD), and mixed dementia. As the most common form of dementia in older adults, AD is characterized by chronic and progressive neurodegeneration leading to progressive cognitive impairment and eventually to the death of patients. AD in most cases is sporadic, probably caused by multiple factors, and is characterized histopathologically by the presence of both intraneuronal neurofibrillary tangles (NFTs) and extracellular senile plaques together with neurodegeneration in the brain (Braak and Braak 1991). VaD is the second most common form of dementia (Battistin and Cagnin 2010) and is caused by problems in the supply of blood to the brain. Mixed dementia is diagnosed when patients have evidence of both AD and cerebrovascular disorder, either clinically or based on neuroimaging evidence of ischemic lesions. In fact, AD and VaD often coexist in older patients with dementia. It is estimated that as many as 40 % of AD patients actually have mixed dementia (Battistin and Cagnin 2010).
Chronic cerebral hypoperfusion (CCH) (Pristera et al. 2013) is one of the major mechanisms of cerebral vascular disorders and can result from hypertension, diabetes, generalized atherosclerosis, smoking, and heart diseases. These factors can affect the cerebral vascular system and eventually cause decreased blood supply to the brain (Meyer et al. 2000; Roman 2002; Valerio Romanini et al. 2013). Individuals with CCH usually have cognitive deficits to various degrees (Ruitenberg et al. 2005). The important role of CCH in dementia has emerged to the forefront of neurology research (Pluta et al. 2012; Akinyemi et al. 2013; Kelleher and Soiza 2013; Pluta et al. 2013a, b; Roh and Lee 2014). Individuals with moderate or severe intracranial arterial stenosis have a faster decline in cognition and function relative to those without such stenosis (Zhu et al. 2014). Studies in the last decade have suggested that CCH might promote neurodegeneration through neuronal energy failure, production of reactive oxygen species, and pro-inflammatory cytokines through activated microglial cells that, in turn, damage the neurons and contribute to white matter lesions (Kitagawa et al. 2005; Farkas et al. 2007; Adibhatla and Hatcher 2008; Urabe 2012; Bang et al. 2013). This article attempts to review the recent advances focusing on CCH-induced neurodegeneration and AD-related brain pathology and cognitive impairment.
Causes and Assessment of CCH
CCH has a variety of causes and plays an important role in the development of VaD, AD, and subcortical arteriosclerotic encephalopathy. Three main causes lead to CCH: (i) vascular structural lesions resulting from artery stenosis or occlusion caused by atherosclerosis, arteriovenous malformation, takayasu arteritis, moyamoya disease, and cerebral arteriovenous fistula; (ii) cerebral hemodynamic changes, including chronic blood loss, prolonged hypotension, and reduced cardiac output due to heart failure; and (iii) changes in blood components resulting from any reasons that lead to an increase of blood viscosity, such as hyperlipidemia, polycythemia, and hyperhomocysteinemia. The major risk factors of CCH are hypertension, hyperlipidemia, smoking, obesity, age, hyperhomocysteinemia, and obstructive sleep apnea-hypopnea syndrome (Sarti et al. 2002).
Modern development of medical techniques has made the assessment of CCH very practical. In the clinic, cerebral hypoperfusion is usually assessed by transcranial Doppler ultrasonography, computerized tomography angiography, magnetic resonance angiography, computerized tomography perfusion imaging, perfusion-weighted imaging, or Xenon-CT (computerized tomography). For animal studies, CCH can be assessed by laser-Doppler flowmetry (Kitagawa et al. 2005) or by molecular markers such as hypoxia inducible factor-1 (HIF-1), TP53-induced glycolysis, and apoptosis regulator (TIGAR) or glucose transporters (GLUTs) 1 and 3 (Watanabe et al. 2009; Kimata et al. 2010; Chan et al. 2011; Iwabuchi and Kawahara 2011; Yan et al. 2011; Yuan et al. 2011; Hoshino et al. 2012; Wang et al. 2012a, b; Zhao et al. 2014).
Possible Mechanisms from CCH to Alzheimer-Like Pathology and Neurodegeneration
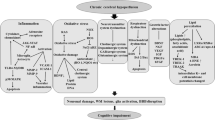
AD and neurodegeneration can be caused by multiple etiological factors. Except for familial, early-onset AD that is caused by mutations of genes encoding presenilin or amyloid β (Aβ) precursor protein (APP), over 95 % AD cases are of sporadic nature. Sporadic AD is likely caused by multiple etiological factors through several pathogenic mechanisms (Iqbal et al. 2010). The major risk factor for sporadic AD is aging, and the most consistently identified AD susceptibility factor is the ε4 allele of apolipoprotein E (Poirier et al. 1993; Strittmatter et al. 1993). Although the amyloid cascade hypothesis continues to exert an important influence in the AD field, recent studies suggest that CCH also promotes Alzheimer-like brain pathology and neurodegeneration through several molecular mechanisms (Fig. 1), as discussed below.

Possible mechanisms by which CCH promotes/causes Alzheimer-like pathology and neurodegeneration. CCH can initiate several pathways that result in oxidative stress, Aβ overproduction and aggregation, tau hyperphosphorylation, synaptic dysfunction, white matter lesion, and neuroinflammation. These neuropathological changes are all seen in AD brain. They can interact and exacerbate each other and eventually promote neuronal cell death and neurodegeneration, resulting in clinical phenotype of memory loss and dementia
CCH-Induced Oxidative Stress
Oxidative stress reflects an imbalance between the systemic manifestation of reactive oxygen species and the biological system’s ability to readily detoxify the reactive intermediates or to repair the resulting damage. Disturbances in the normal redox state of cells can cause toxic effects through the production of peroxides and free radicals that damage almost all components of the cell, including proteins, lipids, and DNA. In humans, oxidative stress is thought to be involved in the development of many diseases including AD (Singh et al. 1995; James et al. 2004; Halliwell 2007; Valko et al. 2007; de Diego-Otero et al. 2009; Dean et al. 2011). CCH can cause mitochondrial dysfunction (Orsucci et al. 2013) and protein synthesis inhibition, which in turn may disturb the balance of antioxidases and reactive oxygen species and produce oxidative damage. At the same time, oxidative injury to vascular endothelial cells, glia, and neurons could further impair vascular function and neurovascular coupling, an orchestrated intercellular communication between neurons, astrocytes and microvessels, which results in a rapid and restricted increase in cerebral blood flow in order to maintain normal brain function in a timely and local manner. Impairment of vascular function and neurovascular coupling may result in a vicious cycle of further reduction of cerebral perfusion (Lyons and Pahwa 2013).
Numerous studies have provided evidence that CCH leads to oxidative stress and have described the mechanism by which oxidative damage results in cognitive impairment. Liu et al. described an alternative CCH rat model by two-stage, three-vessel occlusion and found impaired spatial learning and memory and increased levels of malondialdehyde, the end products of lipid peroxidation (Liu et al. 2012). Xi et al. established a CCH rat model through permanent bilateral common carotid artery occlusion (BCCAO) and found central cholinergic dysfunction and increased oxidative damage that is correlated with spatial learning and memory impairments and working memory dysfunction (Xi et al. 2014). In the BCCAO model, reduction of pyruvate dehydrogenase level and increase of oxidative stress occur in the hippocampus, suggesting that mitochondrial bioenergetic deficits might affect memory directly (Du et al. 2013). Apoptosis signal-regulating kinase 1 (ASK1) appears to be critical to CCH-induced oxidative stress, because the white matter lesions resulting from bilateral common carotid artery stenosis (BCAS) cause oxidative stress-associated cognitive decline in wild-type mice, but not in ASK1-deficient mice (Toyama et al. 2014). ASK1, also known as mitogen-activated protein kinase kinase kinase 5, is a member of the mitogen-activated protein kinase (MAPK) pathway and activates c-Jun N-terminal kinase (JNK) and p38 MAPK in a Raf-independent manner in response to an array of stresses, including oxidative stress, endoplasmic reticulum stress, and calcium influx. ASK1 is also involved in neurodegenerative diseases (Hattori et al. 2009). The oxidative stress-ASK1-p38 cascade appears to play an important role in the pathogenesis of cognitive impairment caused by CCH (Toyama et al. 2014).
CCH-Induced Accumulation and Aggravation of Aβ
Aβ, a peptide of 36–43 amino acids, is the main component of the amyloid plaques found in AD brain. Aβ is derived from APP through proteolytic cleavages by β- and γ-secretases. Aβ pathological deposition occurs both in the brain parenchyma and in the vascular structure in AD brain and in the brains of transgenic animal models with APP mutations (Games et al. 1995; Hsiao et al. 1996; Sturchler-Pierrat et al. 1997; Bornemann and Staufenbiel 2000). It has been reported that CCH accelerates Aβ deposition. BCAS-induced CCH can increase Aβ fibrillization and induce Aβ deposition in the intracellular compartment and, therefore, may accelerate the pathological changes of AD in APPSwe/Ind-Tg mice one month after BCAS (Kitaguchi et al. 2009). CCH induced by permanent unilateral common carotid artery occlusion (UCCAO) causes spatial learning impairments that correlate with the number of cortical Aβ plaques in young APPSwe/PS1 mice (Pimentel-Coelho et al. 2013). In a mouse model of cerebral amyloid angiopathy [C57BL/6-Tg(Thy1-APPSwDutIowa)], BCAS increased Aβ deposition 12 weeks after BCAS surgery (Okamoto et al. 2012). CCH may cause Aβ deposition in aged wild-type animals too. Time-dependent accumulation of oligomeric Aβ in the hippocampus, especially in the axonal terminals of aged rats, occurs after CCH induced by BCCAO (Wang et al. 2010). It is worth noting that the amyloid deposits as seen nine months after transient middle cerebral artery occlusion cannot be stained with Congo red or Thioflavine S (van Groen et al. 2005), which are routinely used to detect the β-pleated sheet conformation that is typical of mature Aβ plaques in AD. Thus, acute cerebral ischemia might lead to Aβ deposition that is somewhat different from the mature Aβ plaques seen in AD brain.
Lots of evidences have showed that CCH and other hypoxia conditions up-regulate β and γ secretase-mediated APP processing (Sun et al. 2006; Li et al. 2009; Zhiyou et al. 2009; Koike et al. 2010; Pluta et al. 2013a, b). A possible mechanism by which CCH up-regulates APP processing and leads to Aβ accumulation could be that CCH induces HIF-1 expression, which then binds to the promoter of β-secretase and consequently increases its expression (Zhang et al. 2007). The Aβ deposition in small arteries caused by CCH could further induce cerebrovascular lesion (Thomas et al. 1996) and worsen cerebral hypoperfusion and finally lead to a vicious circle and irreversible damages.
CCH-Induced Hyperphosphorylation of Tau
The microtubule-associated protein tau becomes abnormally hyperphosphorylated in the brains of individuals with AD and several other neurodegenerative disorders collectively called tauopathies (Grundke-Iqbal et al. 1986). It has been demonstrated that abnormal hyperphosphorylation of tau is crucial to neurodegeneration in AD and probably also in other tauopathies (Gong and Iqbal 2008; Iqbal et al. 2013). Tau hyperphosphorylation can be promoted by several factors. One of these factors could be CCH-induced decrease of brain glucose metabolism because the latter leads to down-regulation of tau O-GlcNAcylation that in turn results in tau hyperphosphorylation (Liu et al. 2004; Li et al. 2006; Liu et al. 2009). In a mouse model of CCH induced by UCCAO, we recently found decreased levels of O-GlcNAcylation, increased levels of tau phosphorylation at several AD-relevant sites, selective neurodegeneration in the brain, and significant short-term memory deficits and mild long-term spatial memory impairment (Zhao et al. 2014).
Several protein kinases, such as glycogen synthase kinase-3β (GSK-3β), extracellular signal-regulated kinases (ERK1/2), cyclin dependent kinases 5 (CDK5), cAMP-dependent protein kinase (PKA), calcium/calmodulin-dependent protein kinase II (CaMK-II), and JNKs, have been implicated in hyperphosphorylation of tau in AD (Gong et al. 2010). Tau phosphorylation is also regulated by protein phosphatase 2A (PP2A) (Gong et al. 2000), which accounts for over 70 % of total tau phosphatase activity in the mammalian brain (Liu et al. 2005). Increased tau hyperphosphorylation with concurrent activation of GSK-3β, CDK5, and CaMK-II, as well as inhibition of PP2A is observed in a rat model of CCH, which shows spatial learning/memory deficits (Yao et al. 2012). It appears that CCH can induce abnormal hyperphosphorylation of tau through several pathways.
Tau hyperphosphorylation and Aβ overproduction appear to be very sensitive to cerebral hypoperfusion. Koike et al. have reported that a single, mild, cerebral hypoperfusion has profound and long lasting effects on tau hyperphosphorylation and Aβ overproduction in 3xTg-AD mice (Koike et al. 2010). It is thus reasonable to speculate that repeated transient cerebral ischemia could contribute to the development of AD.
CCH-Induced Synaptic Dysfunction
Synapses are the functional unit of information transfer between neurons and comprise presynaptic membrane, synaptic cleft, and postsynaptic membrane. Synaptic integrity is essential for normal brain functions, including learning and memory. The severity of clinical symptoms correlates highly with synaptic loss in the brain in AD (Scheff et al. 2011). Synaptic integrity can be studied by using electron microscopy and also indirectly through electrophysiology and studies of various synaptic proteins, such as synapsin, synaptophysin, synaptobrevin, synaptotagmin, synaptoporin, and postsynaptic density protein 95 (PSD95). Altered levels of these synaptic proteins have been seen in the brains of both individuals with AD (Mukaetova-Ladinska et al. 2000) and mouse models of AD (Chen et al. 2012, 2013).
Decreased levels of PSD95 and synaptophysin are found in rat brains, especially in the axonal terminals of the hippocampus, 5 weeks after BCCAO (Wang et al. 2010). These rats also show alterations of synaptic ultrastructure in the CA1 area of the hippocampus, as evaluated by electron microscopy. Thus, these synaptic alterations might be the molecular basis of the memory deficits observed in these animals. In a recent study, we also found increased pre-synaptic protein synapsin and post-synaptic protein PSD95, as well as decreased pre-synaptic protein synaptophysin, in the cerebral cortex and the hippocampus of CCH mice 2.5 months after UCCAO (Zhao et al. 2014). These studies confirmed the important role of synaptic integrity in CCH-induced cognitive deficits. The CCH-induced dysfunction of neural plasticity can be observed directly by determination of long-term potentiation (LTP) in CCH animal models. LTP has been found to be inhibited in the hippocampal CA1 region of rat models of CCH for 3 and 6–7 months (Sekhon et al. 1997; Hai et al. 2009).
CCH-Induced Neuronal Loss
Synaptic and neuronal loss in AD correlates directly to the severity of dementia symptoms (Mukaetova-Ladinska et al. 2000; Scheff et al. 2011) and is also seen after chronic cerebral ischemia (Wang et al. 2010; Zhao et al. 2014). Animal studies have shown apoptotic morphology and DNA strand breaks in hippocampal pyramidal neurons 27 weeks after BCCAO, and the working memory impairment correlates strongly to the number of apoptotic neurons in the CA1 region, suggesting that apoptotic loss of pyramidal neurons may underlie memory impairment associated with CCH (Bennett et al. 1998). Hippocampal atrophy with pyknotic and apoptotic cells is also seen in the brain 8 months after BCAS (Nishio et al. 2010). The neuronal loss in the CA1 subfield, together with the decrease of central acetylcholine levels in the cortex, striatum, and hypothalamus, appears 4 months, but not 1 month, after permanent BCCAO, suggesting that the neuronal loss caused by CCH might result from a long period of failure of neuronal excitation transmission (Ni et al. 1995). Significant numbers of degenerative neurons are detected with Fluoro-Jade staining in the ipsilateral hippocampus and cerebral cortex, especially in the granule cells of the crest of the dentate gyrus, of the CCH mice 2.5 months after UCCAO (Zhao et al. 2014). This study also suggests that the dentate gyrus is the most vulnerable area in the brain for CCH-induced neurodegeneration. Interestingly, CCH-induced neurodegeneration is associated with tau hyperphosphorylation (Zhao et al. 2014), suggesting that the CCH-induced neuronal degeneration might be caused by or associated with abnormal hyperphosphorylation of tau.
CCH-Induced White Matter Lesion and Glial Activation
White matter damage and glial cell activation are seen in both human brains with CCH and in the brains of animal models of CCH (Fernando et al. 2006; Scherr et al. 2012; Thiebaut de Schotten et al. 2014). The degree of ischemic damage correlates positively to the degree of white matter lesions (Shibata et al. 2004; Kitaguchi et al. 2009). White matter damage with increased levels of pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and IL-6, and decreased level of anti-inflammatory cytokines, such as IL-4 and IL-10, is seen in the corpus callosum of the mouse brains 30 days after UCCAO (Yoshizaki et al. 2008). In rat models, CCH appears to cause more severe white matter damage in the corpus callosum than in the striatum (Wakita et al. 1994). This finding is consistent with the more glial activation in the corpus callosum under CCH (Yoshizaki et al. 2008). The activation of glial cells is closely associated with white matter damage (Wakita et al. 1994; Shibata et al. 2004; Nakaji et al. 2006) and may worsen white matter lesions (Wakita et al. 1995).
Two possible mechanisms have been considered for white matter lesion induced by CCH. First, chronic cerebral ischemia results in oxidative stress and increases reactive oxygen species, which cause white matter damage. A large number of inflammatory glial cells are activated immediately after white matter damage occurs (Wakita et al. 1994; Petito et al. 1998). Second, CCH causes blood–brain barrier damage that facilitates the entry of inflammatory cells into the brain parenchyma and causes the generation of inflammatory immune response and the release of a large number of serine proteases, matrix metalloproteinase-2, elastase, collagenase, IL-1β, and tumor necrosis factor-α, which in turn lead to white matter damage (Farkas et al. 2005; Crawford et al. 2008).
Animal Models of CCH for the Studies of Neurodegeneration and AD
Several animal models of CCH have been produced by restricting cerebral blood flow in rodents in order to investigate the roles and mechanisms of CCH in cognitive impairment and to evaluate the therapeutic efficacy of potential drugs. The most commonly used model is a rat model with permanent BCCAO/2-vessel occlusion (BCCAO/2-VO) (Pappas et al. 1996; Ji et al. 2010; Shonesy et al. 2012). Because of the bridging blood supply from posterior communicating arteries, an approximately 50 % decrease of frontal cerebral blood supply can be achieved using this procedure (Tanaka et al. 1996). Numerous studies have reported spatial memory deficits in this CCH model (Wang et al. 2010; Shu et al. 2013). Progressive spatial memory deficits as tested by using Morris water maze, decreased synaptic density and alterations of synaptic ultrastructure in the CA1 area of hippocampus, decreased levels of PSD-95 and synaptophysin, and time-dependent accumulation of oligomeric Aβ in the hippocampus are found 30 days after the occlusion surgery (Wang et al. 2010). Deficits of both short-term non-spatial working memory and long-term spatial memory are observed eight weeks after BCCAO (Shu et al. 2013). Neuroinflammation with microglial and astroglial activation and white matter lesions also occur six weeks after BCCAO (Choi et al. 2011).
UCCAO is a procedure used to produce CCH in mice. A 35–55 % decrease of cerebrocortical perfusion was reported in the ipsilateral hemisphere in mice 28 days after UCCAO (Kitagawa et al. 2005). After UCCAO for 2.5 months, these mice develop significant short-term memory deficits and mild long-term spatial memory impairment, as well as decreased level of protein O-GlcNAcylation, increased level of tau phosphorylation, dysregulated synaptic proteins and insulin signaling, and selective neurodegeneration in the brain (Zhao et al. 2014). UCCAO of the transgenic mouse models with APP mutations exacerbates cognition deficits (Yoshizaki et al. 2008; Lee et al. 2011; Pimentel-Coelho et al. 2013).
BCAS is also used for producing CCH in mice. This approach is theoretically better than the two approaches above, but practically, it is more difficult to achieve the same level of cerebral hypoperfusion due to the more challenging technique of BCAS. Mice after BCAS develop learning and memory impairment (Nishio et al. 2010). Proliferation of activated microglia and astroglia is observed in the white matter after 3 days, and white matter lesions occurred after 14 days of BCAS (Shibata et al. 2004). Impaired reference and working memory, as well as hippocampal atrophy with pyknotic and apoptotic cells, is found 8 months after BCAS (Nishio et al. 2010). BCAS induces more severe cognitive impairment in the APPSw/Ind-Tg mice (Shibata et al. 2004; Nakaji et al. 2006; Shibata et al. 2007; Kitaguchi et al. 2009; Nishio et al. 2010). Rarefied white matter, proliferated astroglia, and Aβ1-40 immunoreactivity appear in some axons in the white matter of APP-Tg mice soon after BCAS, whereas Aβ1-42 accumulates later in the scattered cortical neurons and their axons (Kitaguchi et al. 2009). BCAS also exacerbates Aβ aggregation, neuronal loss, and learning impairment in APP-Tg mice (Yamada et al. 2011).
The above approaches are aimed to reduce cerebral perfusion. However, some of the alterations in these animals might be partially caused by other factors associated with or as the consequence of the procedures. Some artery occlusion paradigms are confounded by parallel damage to pyramidal and cholinergic neurons (Volpe et al. 1988; Mizobuchi 1989; Sugai 1989; Volpe et al. 1992; Ni et al. 1997), making it challenging to know whether it is hypoperfusion or the consequent neuronal damage that causes the cognitive dysfunction.
Concluding Remarks
CCH caused by vascular structural lesions, cerebral hemodynamic changes, or increased blood viscosity is common in the elderly and often contributes to memory impairment, neurodegeneration, and sporadic AD (Fig. 1). Because CCH rarely exists alone and is usually accompanied by other brain pathologies, it is challenging to dissect the exact role of CCH in neurodegeneration and sporadic AD. As discussed in this article, many studies have shown an active and even causative role of CCH in Alzheimer-like brain pathology and neurodegeneration. On the other hand, there is evidence showing cerebrovascular lesions in AD brain and transgenic mouse models of AD (Gold et al. 2007; Tang et al. 2009; Austin et al. 2011), which in turn could lead to CCH. It is highly likely that both scenarios are true and may be co-existing. CCH could be a major contributing or even a causative factor for AD in some patients and the consequence of AD in others.
Future research should focus on dissecting the major molecular mechanisms by which CCH causes or promotes cognitive impairment and neurodegeneration. Better-characterized animal models and detailed studies of the time-dependent changes during CCH in the brain will shed new light on the roles and mechanisms of CCH in cognitive impairment and neurodegeneration. Creating CCH in animal models of other conditions of neurodegeneration and memory loss, such as AD mouse models, will help elucidate the contributions of CCH to those disorders. These studies will help identify potential therapeutic targets to prevent and treat cognitive impairment in individuals with CCH.
Abbreviations
- AD:
-
Alzheimer’s disease
- APP:
-
Amyloid β precursor protein
- ASK1:
-
Apoptosis signal-regulating kinase 1
- BCAS:
-
Bilateral common carotid artery stenosis
- BCCAO:
-
Bilateral common carotid artery occlusion
- CaMK-II:
-
Calcium/calmodulin-dependent protein kinase II
- CCH:
-
Chronic cerebral hypoperfusion
- CDK5:
-
Cyclin dependent kinases 5
- ERK1/2:
-
Extracellular signal-regulated kinases
- GLUTs:
-
Glucose transporters
- GSK-3β:
-
Glycogen synthase kinase-3β
- HIF-1:
-
Hypoxia inducible factor-1
- IL-1β:
-
Interleukin-1β
- JNK:
-
c-Jun N-terminal kinase
- LTP:
-
Long-term potentiation
- MAPK:
-
Mitogen-activated protein kinase
- NFTs:
-
Neurofibrillary tangles
- PP2A:
-
Protein phosphatase 2A
- PSD95:
-
Postsynaptic density protein 95
- TIGAR:
-
TP53-induced glycolysis and apoptosis regulator
- UCCAO:
-
Unilateral common carotid artery occlusion
- VaD:
-
Vascular dementia
References
Adibhatla RM, Hatcher JF (2008) Phospholipase A(2), reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB Rep 41:560–567
Akinyemi RO, Mukaetova-Ladinska EB, Attems J, Ihara M, Kalaria RN (2013) Vascular risk factors and neurodegeneration in ageing related dementias: Alzheimer’s disease and vascular dementia. Curr Alzheimer Res 10:642–653
Austin BP, Nair VA, Meier TB, Xu G, Rowley HA, Carlsson CM, Johnson SC, Prabhakaran V (2011) Effects of hypoperfusion in Alzheimer’s disease. J Alzheimers Dis 26(Suppl 3):123–133
Bang J, Jeon WK, Lee IS, Han JS, Kim BY (2013) Biphasic functional regulation in hippocampus of rat with chronic cerebral hypoperfusion induced by permanent occlusion of bilateral common carotid artery. PLoS One 8:e70093
Battistin L, Cagnin A (2010) Vascular cognitive disorder. A biological and clinical overview. Neurochem Res 35:1933–1938
Bennett SA, Tenniswood M, Chen JH, Davidson CM, Keyes MT, Fortin T, Pappas BA (1998) Chronic cerebral hypoperfusion elicits neuronal apoptosis and behavioral impairment. NeuroReport 9:161–166
Bornemann KD, Staufenbiel M (2000) Transgenic mouse models of Alzheimer’s disease. Ann N Y Acad Sci 908:260–266
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Chan JY, Tsai CY, Wu CH, Li FC, Dai KY, Sun EY, Chan SH, Chang AY (2011) Sumoylation of hypoxia-inducible factor-1alpha ameliorates failure of brain stem cardiovascular regulation in experimental brain death. PLoS One 6:e17375
Chen Y, Tian Z, Liang Z, Sun S, Dai CL, Lee MH, LaFerla FM, Grundke-Iqbal I, Iqbal K, Liu F, Gong CX (2012) Brain gene expression of a sporadic (icv-STZ Mouse) and a familial mouse model (3xTg-AD mouse) of Alzheimer’s disease. PLoS One 7:e51432
Chen Y, Liang Z, Blanchard J, Dai CL, Sun S, Lee MH, Grundke-Iqbal I, Iqbal K, Liu F, Gong CX (2013) A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol Neurobiol 47:711–725
Choi BR, Lee SR, Han JS, Woo SK, Kim KM, Choi DH, Kwon KJ, Han SH, Shin CY, Lee J, Chung CS, Kim HY (2011) Synergistic memory impairment through the interaction of chronic cerebral hypoperfusion and amlyloid toxicity in a rat model. Stroke 42:2595–2604
Crawford GL, Hart GW, Whiteheart SW (2008) Murine platelets are not regulated by O-linked beta-N-acetylglucosamine. Arch Biochem Biophys 474:220–224
de Diego-Otero Y, Romero-Zerbo Y, el Bekay R, Decara J, Sanchez L, Rodriguez-de Fonseca F, del Arco-Herrera I (2009) Alpha-tocopherol protects against oxidative stress in the fragile X knockout mouse: an experimental therapeutic approach for the Fmr1 deficiency. Neuropsychopharmacology 34:1011–1026
Dean OM, van den Buuse M, Berk M, Copolov DL, Mavros C, Bush AI (2011) N-acetyl cysteine restores brain glutathione loss in combined 2-cyclohexene-1-one and d-amphetamine-treated rats: relevance to schizophrenia and bipolar disorder. Neurosci Lett 499:149–153
Du J, Ma M, Zhao Q, Fang L, Chang J, Wang Y, Fei R, Song X (2013) Mitochondrial bioenergetic deficits in the hippocampi of rats with chronic ischemia-induced vascular dementia. Neuroscience 231:345–352
Farkas E, Timmer NM, Domoki F, Mihaly A, Luiten PG, Bari F (2005) Post-ischemic administration of diazoxide attenuates long-term microglial activation in the rat brain after permanent carotid artery occlusion. Neurosci Lett 387:168–172
Farkas E, Luiten PG, Bari F (2007) Permanent, bilateral common carotid artery occlusion in the rat: a model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res Rev 54:162–180
Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F, Wharton SB, Shaw PJ, O’Brien JT, Ince PG (2006) White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37:1391–1398
Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F et al (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373:523–527
Gold G, Giannakopoulos P, Herrmann FR, Bouras C, Kovari E (2007) Identification of Alzheimer and vascular lesion thresholds for mixed dementia. Brain 130:2830–2836
Gong CX, Iqbal K (2008) Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem 15:2321–2328
Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K (2000) Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem 275:5535–5544
Gong CX, Grundke-Iqbal I, Iqbal K (2010) Targeting tau protein in Alzheimer’s disease. Drugs Aging 27:351–365
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917
Hai J, Wan JF, Lin Q, Wang F, Zhang L, Li H, Chen YY, Lu Y (2009) Cognitive dysfunction induced by chronic cerebral hypoperfusion in a rat model associated with arteriovenous malformations. Brain Res 1301:80–88
Halliwell B (2007) Oxidative stress and cancer: have we moved forward? Biochem J 401:1–11
Hattori K, Naguro I, Runchel C, Ichijo H (2009) The roles of ASK family proteins in stress responses and diseases. Cell Commun Signal 7:9
Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, Ikeda K, Ueyama T, Okigaki M, Matsubara H (2012) p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol 52:175–184
Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102
Iqbal K, Wang X, Blanchard J, Liu F, Gong CX, Grundke-Iqbal I (2010) Alzheimer’s disease neurofibrillary degeneration: pivotal and multifactorial. Biochem Soc Trans 38:962–966
Iqbal K, Gong CX, Liu F (2013) Hyperphosphorylation-induced tau oligomers. Front Neurol 4:112
Iwabuchi S, Kawahara K (2011) Inducible astrocytic glucose transporter-3 contributes to the enhanced storage of intracellular glycogen during reperfusion after ischemia. Neurochem Int 59:319–325
James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA (2004) Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr 80:1611–1617
Ji HJ, Hu JF, Wang YH, Chen XY, Zhou R, Chen NH (2010) Osthole improves chronic cerebral hypoperfusion induced cognitive deficits and neuronal damage in hippocampus. Eur J Pharmacol 636:96–101
Kelleher RJ, Soiza RL (2013) Evidence of endothelial dysfunction in the development of Alzheimer’s disease: is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis 3:197–226
Kimata M, Matoba S, Iwai-Kanai E, Nakamura H, Hoshino A, Nakaoka M, Katamura M, Okawa Y, Mita Y, Okigaki M, Ikeda K, Tatsumi T, Matsubara H (2010) p53 and TIGAR regulate cardiac myocyte energy homeostasis under hypoxic stress. Am J Physiol Heart Circ Physiol 299:H1908–H1916
Kitagawa K, Yagita Y, Sasaki T, Sugiura S, Omura-Matsuoka E, Mabuchi T, Matsushita K, Hori M (2005) Chronic mild reduction of cerebral perfusion pressure induces ischemic tolerance in focal cerebral ischemia. Stroke 36:2270–2274
Kitaguchi H, Tomimoto H, Ihara M, Shibata M, Uemura K, Kalaria RN, Kihara T, Asada-Utsugi M, Kinoshita A, Takahashi R (2009) Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res 1294:202–210
Koike MA, Green KN, Blurton-Jones M, Laferla FM (2010) Oligemic hypoperfusion differentially affects tau and amyloid-{beta}. Am J Pathol 177:300–310
Lee JS, Im DS, An YS, Hong JM, Gwag BJ, Joo IS (2011) Chronic cerebral hypoperfusion in a mouse model of Alzheimer’s disease: an additional contributing factor of cognitive impairment. Neurosci Lett 489:84–88
Li X, Lu F, Wang JZ, Gong CX (2006) Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci 23:2078–2086
Li L, Zhang X, Yang D, Luo G, Chen S, Le W (2009) Hypoxia increases Abeta generation by altering beta- and gamma-cleavage of APP. Neurobiol Aging 30:1091–1098
Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX (2004) O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci USA 101:10804–10809
Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22:1942–1950
Liu F, Shi J, Tanimukai H, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX (2009) Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 132:1820–1832
Liu H, Zhang J, Yang Y, Zhang L, Zeng X (2012) Decreased cerebral perfusion and oxidative stress result in acute and delayed cognitive impairment. Curr Neurovasc Res 9:152–158
Lyons K, Pahwa R (2013) Statement of Retraction: “Mohamad Goldust, Mahnaz Talebi, Jafar Majidi, Mohammad Amin Rezazadeh Saatlou, and Elham Rezaee. Evaluation of antiphospholipid antibodies in youths suffering from cerebral ischemia.”. Int J Neurosci 123:597
Meyer JS, Rauch G, Rauch RA, Haque A (2000) Risk factors for cerebral hypoperfusion, mild cognitive impairment, and dementia. Neurobiol Aging 21:161–169
Mizobuchi H (1989) Changes in muscarinic cholinergic receptor and choline acetyltransferase in experimental ischemic brain. Nihon Geka Hokan 58:93–106
Mukaetova-Ladinska EB, Garcia-Siera F, Hurt J, Gertz HJ, Xuereb JH, Hills R, Brayne C, Huppert FA, Paykel ES, McGee M, Jakes R, Honer WG, Harrington CR, Wischik CM (2000) Staging of cytoskeletal and beta-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am J Pathol 157:623–636
Nakaji K, Ihara M, Takahashi C, Itohara S, Noda M, Takahashi R, Tomimoto H (2006) Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke 37:2816–2823
Ni JW, Matsumoto K, Li HB, Murakami Y, Watanabe H (1995) Neuronal damage and decrease of central acetylcholine level following permanent occlusion of bilateral common carotid arteries in rat. Brain Res 673:290–296
Ni B, Stephenson D, Wu X, Smalstig EB, Clemens J, Paul SM (1997) Selective loss of neuronal Na + -dependent phosphate cotransporter mRNA in CA1 pyramidal neuron following global ischemia. Brain Res Mol Brain Res 48:132–139
Nishio K, Ihara M, Yamasaki N, Kalaria RN, Maki T, Fujita Y, Ito H, Oishi N, Fukuyama H, Miyakawa T, Takahashi R, Tomimoto H (2010) A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke 41:1278–1284
Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, Kitamura A, Washida K, Yamada M, Ito H, Tomimoto H, Takahashi R, Ihara M (2012) Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 123:381–394
Orsucci D, Mancuso M, Ienco EC, Simoncini C, Siciliano G, Bonuccelli U (2013) Vascular factors and mitochondrial dysfunction: a central role in the pathogenesis of Alzheimer’s disease. Curr Neurovasc Res 10:76–80
Pappas BA, de la Torre JC, Davidson CM, Keyes MT, Fortin T (1996) Chronic reduction of cerebral blood flow in the adult rat: late-emerging CA1 cell loss and memory dysfunction. Brain Res 708:50–58
Petito CK, Olarte JP, Roberts B, Nowak TS Jr, Pulsinelli WA (1998) Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol 57:231–238
Pimentel-Coelho PM, Michaud JP, Rivest S (2013) Effects of mild chronic cerebral hypoperfusion and early amyloid pathology on spatial learning and the cellular innate immune response in mice. Neurobiol Aging 34:679–693
Pluta R, Kocki J, Maciejewski R, Ulamek-Koziol M, Jablonski M, Bogucka-Kocka A, Czuczwar SJ (2012) Ischemia signalling to Alzheimer-related genes. Folia Neuropathol 50:322–329
Pluta R, Furmaga-Jablonska W, Maciejewski R, Ulamek-Koziol M, Jablonski M (2013a) Brain ischemia activates beta- and gamma-secretase cleavage of amyloid precursor protein: significance in sporadic Alzheimer’s disease. Mol Neurobiol 47:425–434
Pluta R, Jablonski M, Ulamek-Koziol M, Kocki J, Brzozowska J, Januszewski S, Furmaga-Jablonska W, Bogucka-Kocka A, Maciejewski R, Czuczwar SJ (2013b) Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol Neurobiol 48:500–515
Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S (1993) Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 342:697–699
Pristera A, Saraulli D, Farioli-Vecchioli S, Strimpakos G, Costanzi M, di Certo MG, Cannas S, Ciotti MT, Tirone F, Mattei E, Cestari V, Canu N (2013) Impact of N-tau on adult hippocampal neurogenesis, anxiety, and memory. Neurobiol Aging 34:2551–2563
Roh JH, Lee JH (2014) Recent updates on subcortical ischemic vascular dementia. J Stroke 16:18–26
Roman GC (2002) Vascular dementia revisited: diagnosis, pathogenesis, treatment, and prevention. Med Clin North Am 86:477–499
Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM (2005) Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol 57:789–794
Sarti C, Pantoni L, Bartolini L, Inzitari D (2002) Cognitive impairment and chronic cerebral hypoperfusion: what can be learned from experimental models. J Neurol Sci 203–204:263–266
Scheff SW, Price DA, Schmitt FA, Scheff MA, Mufson EJ (2011) Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 24:547–557
Scherr M, Trinka E, Mc Coy M, Krenn Y, Staffen W, Kirschner M, Bergmann HJ, Mutzenbach JS (2012) Cerebral hypoperfusion during carotid artery stenosis can lead to cognitive deficits that may be independent of white matter lesion load. Curr Neurovasc Res 9:193–199
Sekhon LH, Spence I, Morgan MK, Weber NC (1997) Chronic cerebral hypoperfusion inhibits calcium-induced long-term potentiation in rats. Stroke 28:1043–1047
Shibata M, Ohtani R, Ihara M, Tomimoto H (2004) White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 35:2598–2603
Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R, Ihara M, Takahashi R, Tomimoto H (2007) Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 38:2826–2832
Shonesy BC, Thiruchelvam K, Parameshwaran K, Rahman EA, Karuppagounder SS, Huggins KW, Pinkert CA, Amin R, Dhanasekaran M, Suppiramaniam V (2012) Central insulin resistance and synaptic dysfunction in intracerebroventricular-streptozotocin injected rodents. Neurobiol Aging 33:430-e5
Shu Y, Zhang H, Kang T, Zhang JJ, Yang Y, Liu H, Zhang L (2013) PI3 K/Akt signal pathway involved in the cognitive impairment caused by chronic cerebral hypoperfusion in rats. PLoS One 8:e81901
Singh N, Dhalla AK, Seneviratne C, Singal PK (1995) Oxidative stress and heart failure. Mol Cell Biochem 147:77–81
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981
Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94:13287–13292
Sugai K (1989) The effects of ischemia and postischemic reperfusion on the brain cholinergic system of the rat. Masui 38:350–356
Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W (2006) Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci USA 103:18727–18732
Tanaka K, Ogawa N, Asanuma M, Kondo Y, Nomura M (1996) Relationship between cholinergic dysfunction and discrimination learning disabilities in Wistar rats following chronic cerebral hypoperfusion. Brain Res 729:55–65
Tang KF, Cai L, Zhou JN (2009) Observation of the density and size of cells in hippocampus and vascular lesion in thalamus of GFAP-apoE transgenic mice. Neurosci Bull 25:167–178
Thiebaut de Schotten M, Tomaiuolo F, Aiello M, Merola S, Silvetti M, Lecce F, Bartolomeo P, Doricchi F (2014) Damage to white matter pathways in subacute and chronic spatial neglect: a group study and 2 single-case studies with complete virtual “in vivo” tractography dissection. Cereb Cortex 24:691–706
Thomas T, Thomas G, McLendon C, Sutton T, Mullan M (1996) beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 380:168–171
Toyama K, Koibuchi N, Uekawa K, Hasegawa Y, Kataoka K, Katayama T, Sueta D, Ma MJ, Nakagawa T, Yasuda O, Tomimoto H, Ichijo H, Ogawa H, Kim-Mitsuyama S (2014) Apoptosis signal-regulating kinase 1 is a novel target molecule for cognitive impairment induced by chronic cerebral hypoperfusion. Arterioscler Thromb Vasc Biol 34:616–625
Urabe T (2012) Molecular mechanism and new protective strategy for ischemic white matter damages. Rinsho Shinkeigaku 52:908–910
Valerio Romanini C, Dias Fiuza Ferreira E, Correia Bacarin C, Verussa MH, de Weffort Oliveira RM, Milani H (2013) Neurohistological and behavioral changes following the four-vessel occlusion/internal carotid artery model of chronic cerebral hypoperfusion: comparison between normotensive and spontaneously hypertensive rats. Behav Brain Res 252:214–221
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39:44–84
van Groen T, Puurunen K, Maki HM, Sivenius J, Jolkkonen J (2005) Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 36:1551–1556
Volpe BT, Waczek B, Davis HP (1988) Modified T-maze training demonstrates dissociated memory loss in rats with ischemic hippocampal injury. Behav Brain Res 27:259–268
Volpe BT, Davis HP, Towle A, Dunlap WP (1992) Loss of hippocampal CA1 pyramidal neurons correlates with memory impairment in rats with ischemic or neurotoxin lesions. Behav Neurosci 106:457–464
Wakita H, Tomimoto H, Akiguchi I, Kimura J (1994) Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: an immunohistochemical study. Acta Neuropathol 87:484–492
Wakita H, Tomimoto H, Akiguchi I, Kimura J (1995) Protective effect of cyclosporin A on white matter changes in the rat brain after chronic cerebral hypoperfusion. Stroke 26:1415–1422
Wang X, Xing A, Xu C, Cai Q, Liu H, Li L (2010) Cerebrovascular hypoperfusion induces spatial memory impairment, synaptic changes, and amyloid-beta oligomerization in rats. J Alzheimers Dis 21:813–822
Wang F, Wang Y, Geng X, Asmaro K, Peng C, Sullivan JM, Ding JY, Ji X, Ding Y (2012a) Neuroprotective effect of acute ethanol administration in a rat with transient cerebral ischemia. Stroke 43:205–210
Wang Z, Tsai LK, Munasinghe J, Leng Y, Fessler EB, Chibane F, Leeds P, Chuang DM (2012b) Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke 43:2430–2436
Watanabe M, Masaoka N, Nakajima Y, Nagaishi M, Yamamoto T (2009) Changes of expression of glucose transporters in the fetal lamb brain after MCI-186 administration to the maternal circulation with 10-min persistent umbilical cord occlusion. J Matern Fetal Neonatal Med 22:829–836
Xi Y, Wang M, Zhang W, Bai M, Du Y, Zhang Z, Li Z, Miao J (2014) Neuronal damage, central cholinergic dysfunction and oxidative damage correlate with cognitive deficits in rats with chronic cerebral hypoperfusion. Neurobiol Learn Mem 109:7–19
Yamada M, Ihara M, Okamoto Y, Maki T, Washida K, Kitamura A, Hase Y, Ito H, Takao K, Miyakawa T, Kalaria RN, Tomimoto H, Takahashi R (2011) The influence of chronic cerebral hypoperfusion on cognitive function and amyloid beta metabolism in APP overexpressing mice. PLoS One 6:e16567
Yan J, Zhou B, Taheri S, Shi H (2011) Differential effects of HIF-1 inhibition by YC-1 on the overall outcome and blood-brain barrier damage in a rat model of ischemic stroke. PLoS One 6:e27798
Yao ZH, Zhang JJ, Xie XF (2012) Enriched environment prevents cognitive impairment and tau hyperphosphorylation after chronic cerebral hypoperfusion. Curr Neurovasc Res 9:176–184
Yoshizaki K, Adachi K, Kataoka S, Watanabe A, Tabira T, Takahashi K, Wakita H (2008) Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp Neurol 210:585–591
Yuan LB, Dong HL, Zhang HP, Zhao RN, Gong G, Chen XM, Zhang LN, Xiong L (2011) Neuroprotective effect of orexin-A is mediated by an increase of hypoxia-inducible factor-1 activity in rat. Anesthesiology 114:340–354
Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW (2007) Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem 282:10873–10880
Zhao Y, Gu JH, Dai CL, Liu Q, Iqbal K, Liu F, Gong CX (2014) Chronic cerebral hypoperfusion causes decrease of O-GlcNAcylation, hyperphosphorylation of tau and behavioral deficits in mice. Front Aging Neurosci 6:10
Zhiyou C, Yong Y, Shanquan S, Jun Z, Liangguo H, Ling Y, Jieying L (2009) Upregulation of BACE1 and beta-amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer’s disease. Neurochem Res 34:1226–1235
Zhu J, Wang Y, Li J, Deng J, Zhou H (2014) Intracranial artery stenosis and progression from mild cognitive impairment to Alzheimer disease. Neurology 82:842–849
Acknowledgments
Studies performed in the authors’ laboratory were supported in part by the New York State Office for People with Developmental Disabilities. We thank Ms. M. Marlow of the New York State Institute for Basic Research in Developmental Disabilities for her editorial assistance.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhao, Y., Gong, CX. From Chronic Cerebral Hypoperfusion to Alzheimer-Like Brain Pathology and Neurodegeneration. Cell Mol Neurobiol 35, 101–110 (2015). https://doi.org/10.1007/s10571-014-0127-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-014-0127-9