
Abstract
The extent of neuronal damage/death in some brain regions is highly correlated to duration time of transient ischemia. In the present study, we carried out neuronal degeneration/death and glial changes in the septum 4 days after 5, 10, 15, and 20 min of transient cerebral ischemia using gerbils. To examine neuronal damage, Fluoro-Jade B (F-J B, a marker for neuronal degeneration) histofluorescence staining was used. F-J B positive (+) cells were detected in the septo-hippocampal nucleus (SHN) of the septum only in the 20 min ischemia-group; the mean number of F-J B+ neurons was 14.9 ± 2.5/400 μm2 in a section. Gliosis of astrocytes and microglia was examined using anti-glial fibrillary acidic protein (GFAP) and anti-ionized calcium-binding adapter molecule 1 (Iba-1), respectively. In all the ischemia-groups, GFAP- and Iba-1-immunoreactive astrocytes and microglia, respectively, were increased in number, and apparently tended to be increased in their immunoreactivity. Especially, in the 20 min ischemia-group, the number and immunoreactivity of Iba-immunoreactive microglia was highest and strongest in the ischemic SHN 4 days after ischemia–reperfusion. In brief, our findings showed that neuronal damage/death in the SHN occurred and gliosis was apparently increased in the 20 min ischemia-group at 4 days after ischemia–reperfusion.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Transient global cerebral ischemia occurs when the blood supply to entire brain or a large part of the brain is disrupted, and it results in tissue deprivation of oxygen and glucose and may give permanent brain damage (de Araujo et al. 2012; Ohk et al. 2012; Yu et al. 2012). In humans, selective neuronal death in the brain occurs frequently after cardiocirculatory arrest, which is a worldwide devastating health problem (Horn and Schlote 1992; Petito et al. 1987).
The Mongolian gerbil has been used as a good model of transient cerebral ischemia to investigate the molecular mechanism of selective neuronal death following ischemia–reperfusion injury, because about 90 % of gerbils lacks the communicating vessels between the carotid and vertebral circulation (Levine and Sohn 1969; Levy and Brierley 1974). Thus, the bilateral occlusion of the carotid arteries essentially completely eliminates blood flow to the forebrain; it completely spares the vegetative center of the brainstem. Several regions of the brain are especially sensitive to transient cerebral ischemia. These sensitive regions in the gerbil brain include the cerebral cortex, striatum, and hippocampus (Hwang et al. 2006, 2007; Lin et al. 1990; Ohk et al. 2012).
We recently reported that the pattern of neuronal death was very different according to ischemic duration in the gerbil hippocampus and striatum induced by various durations of transient cerebral ischemia (Ohk et al. 2012; Yu et al. 2012). However, studies regarding neuronal damage in interconnecting specific brain areas related to the hippocampus, which has contacts with various areas, have been limited, although there is a great deal of information on ischemic brain damage in gerbils (Fukuchi et al. 1998; Janac et al. 2006; Selakovic et al. 2011).
The septum is a set of structures in the middle anteroventral cerebrum and forms an integral part of the limbic system, and it is composed of medium-size neurons that are classified into the medial, lateral, and posterior groups. The septal nuclei contribute to the medial forebrain bundle and receive reciprocal connections from the hippocampus, amygdala, hypothalamus, midbrain, habenula, cingulate gyrus, and thalamus (Freund and Antal 1988; Kiss et al. 1990; Meibach and Siegel 1977; Risold and Swanson 1997; Yoshida and Oka 1995).
Some researchers have reported ischemic damage in the septum, however, we have thought that the studies are not sufficient, because their methods were old (Buchan and Pulsinelli 1990; Crain et al. 1988). Therefore, we examined the pattern of neuronal death/damage and gliosis in the septal nuclei induced by various durations of transient cerebral ischemia in gerbils, which serve a good animal model of transient cerebral ischemia (Hu et al. 2007; Lorrio et al. 2009; Salazar-Colocho et al. 2008).
Materials and Methods
Experimental Animals
The progeny of male Mongolian gerbils (Meriones unguiculatus) was obtained from the Experimental Animal Center, Kangwon National University, Chuncheon, South Korea. Gerbils were used at 6 months (B.W., 65–75 g) of age. The animals were housed in a conventional state under adequate temperature (23 °C) and humidity (60 %) control with a 12-h light/12-h dark cycle, and were provided with free access to food and water. The procedures for animal handling and care adhered to guidelines that are in compliance with the current international laws and policies (Guide for the Care and Use of Laboratory Animals, The National Academies Press, 8th Ed, 2011), and they were approved by the Institutional Animal Care and Use Committee (IACUC) at Hallym’s Medical Center. All the experiments were conducted to minimize the number of animals used and the suffering caused by the procedures used in the present study.
Induction of Transient Cerebral Ischemia
The animals were anesthetized with a mixture of 2.5 % isoflurane in 33 % oxygen and 67 % nitrous oxide. Bilateral common carotid arteries were isolated and occluded using non-traumatic aneurysm clips. The complete interruption of blood flow was confirmed by observing the central artery in retinae using an ophthalmoscope. After 5 min (5 min ischemia–reperfusion group), 10 min (10 min ischemia–reperfusion group), 15 min (15 min ischemia–reperfusion group), or 20 min (20 min ischemia–reperfusion group) of occlusion, the aneurysm clips were removed from the common carotid arteries. The body (rectal) temperature under free-regulating or normothermic (37 ± 0.5 °C) conditions was monitored with a rectal temperature probe (TR-100; Fine Science Tools, Foster City, CA) and maintained using a thermometric blanket before, during, and after the surgery until the animals completely recovered from anesthesia. Thereafter, animals were kept in the thermal incubator (Mirae Medical Industry, Seoul, South Korea) to maintain the body temperature of animals until the animals were euthanized. Sham-operated animals (sham-operated group) were subjected to the same surgical procedures except that the common carotid arteries were not occluded.
Tissue Processing for Histology
For histology, sham- and ischemia-operated adult gerbils (n = 10 in each group) at 4 days after reperfusion were sacrificed. The animals were anesthetized with pentobarbital sodium and perfused transcardially with 0.1 M phosphate-buffered saline (PBS, pH 7.4) followed by 4 % paraformaldehyde in 0.1 M phosphate-buffer (PB, pH 7.4). The brains were removed and postfixed in the same fixative for 6 h. The brain tissues were cryoprotected by infiltration with 30 % sucrose overnight. Thereafter, frozen tissues were serially sectioned on a cryostat (Leica, Germany) into 30 μm coronal sections, and they were then collected into six-well plates containing PBS.
Hematoxylin and Eosin (H–E) Staining
The brain tissues were embedded in paraffin and then were sectioned on microtome (Leica) into 5 μm coronal sections. The sections were mounted on slides, and were stained with H–E followed by standard histochemical procedures. After dehydration, the sections were mounted with Canada Balsam (Kanto, Tokyo, Japan).
Cresyl Violet (CV) Staining
To examine the neuronal death in the brain after transient cerebral ischemia, sham- and ischemia-operated animals (n = 10 in each group) were used 4 days after the ischemic surgery for CV staining. The sections were mounted on gelatin-coated microscopy slides. Cresyl violet acetate (Sigma, MO, USA) was dissolved at 1.0 % (w/v) in distilled water, and glacial acetic acid was added to this solution. The sections were stained and dehydrated by immersing in serial ethanol baths, and they were then mounted with Canada balsam (Kanto).
Fluoro-Jade B (F-J B) Histofluorescence
To confirm the neuronal death in the brain after transient forebrain ischemia, sham- and ischemia-operated animals (n = 10 in each group) were used 4 days after the ischemic surgery for F-J B (a high-affinity fluorescent marker for the localization of neuronal degeneration) histofluorescence under the same conditions. F-J B histofluorescence staining procedures were conducted according to the method by Candelario-Jalil et al. (2003). The sections were first immersed in a solution containing 1 % sodium hydroxide in 80 % alcohol, and followed by 70 % alcohol. They were then transferred to a solution of 0.06 % potassium permanganate, and transferred to a 0.0004 % F-J B (Histochem, Jefferson, AR, USA) staining solution. After washing, the sections were placed on a slide warmer (approximately 50 °C), and then examined using an epifluorescent microscope (Carl Zeiss, Germany) with blue (450–490 nm) excitation light and a barrier filter. With this method neurons that undergo degeneration brightly fluoresce in comparison to the background (Schmued and Hopkins 2000).
Immunohistochemistry for NeuN, GFAP, and Iba-1
In order to examine the changes of neurons, astrocytes, and microglia in the septal nuclei after ischemia–reperfusion, we carried out immunohistochemical staining with mouse anti-neuronal nuclei (NeuN; 1:1,000, Chemicon International, Temecula, CA) for neurons, rabbit anti-glial fibrillary acidic protein (GFAP, 1:800, Chemicon, Temecular, CA) for astrocytes, rabbit anti-ionized calcium-binding adapter molecule 1 (Iba-1, 1:500, Wako, Japan) for microglia, and biotinylated goat anti-mouse or -rabbit IgG (Vector, Burlingame, CA) for secondary antibody. In brief, the sections were sequentially treated with 0.3 % hydrogen peroxide (H2O2) in PBS for 30 min and 10 % normal goat serum in 0.05 M PBS for 30 min. The sections were next incubated with anti-NeuN, anti-GFAP, or Iba-1 overnight at 4 °C. Thereafter the tissues were exposed to biotinylated goat anti-mouse or -rabbit IgG (Vector, Burlingame, CA) and streptavidin peroxidase complex (1:200, Vector). And they were visualized by staining with 3,3′-diaminobenzidine tetrahydrochloride in 0.1 M Tris–HCl buffer (pH 7.2) and mounted on gelatin-coated slides. After dehydration, the sections were mounted with Canada balsam (Kanto).
Cell Counts
All measurements were performed to insure objectivity in blind conditions, by two observers for each experiment, carrying out the measures of experimental samples under the same conditions. The studied tissue sections were selected with 120 μm interval according to anatomical landmarks corresponding to AP from +1.1 to −0.1 mm of gerbil brain atlas, and cell counts were obtained by averaging the counts from 25 sections taken from each animal and the total cell numbers from each animal per group. To evaluate the neuronal degeneration after ischemia–reperfusion, the measurement of degenerated neuronal numbers was performed using an image analyzing system equipped with a computer-based digital camera (software: Optimas 6.5, CyberMetrics, Scottsdale, AZ) and the number of F-J B positive neurons was calibrated as the number of neurons in 400 μm2. According to the previous method (Sugawara et al. 2002), the densities of all GFAP- and Iba-1-immunoreactive structures were evaluated on the basis of optical density (OD), which was obtained after the transformation of the mean gray level using the formula: OD = log (256/mean gray level). The OD of background was taken from areas adjacent to the measured area. After the background density was subtracted, a ratio of the OD of image file was calibrated as % (relative optical density, ROD) using Adobe Photoshop version 8.0 and then analyzed using NIH Image 1.59 software.
Statistical Analysis
Data are expressed as the mean ± SEM. The data were evaluated by a Tukey test for post hoc multiple comparisons following one-way ANOVA. Statistical significance was considered at P < 0.05.
Results
H–E Positive Cells
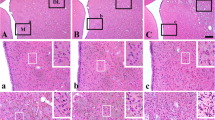
In the sham-group, neurons (cell body diameter >10 μm) in the septum were well stained with H–E (Fig. 1A, a). In all the ischemia–reperfusion groups, the markedly reduction of H–E positive neurons was not detected in the ischemic septum 4 days after ischemia–reperfusion compared to that in the sham-group (Fig. 1B–E, b–e).

H–E staining in the septum of the sham-group (A, a) and ischemia-groups (5 min (B, b), 10 min (C, c), 15 min (D, d), and 20 min (E, e)) 4 days after ischemia–reperfusion. In all the ischemia-groups, H–E staining is not apparently changed compared to that in the sham-group. However, H–E positive small cells tend to be increased with duration time of ischemia in the septo-hippocampal nucleus (SHN) of the septum. a–e High magnification framed in A–E. Scale bar 200 μm (A–E), 50 μm (a–e)
On the other hand, glia-like cells (cell body diameter <10 μm) were identified by their relatively smaller size and lack of stained cytoplasm in the H–E stained sections. In the 10 min ischemia-group, these small H–E stained cells showed a tendency to be increased in the septo-hippocampal nucleus (SHN) 4 days after ischemia–reperfusion compared to those in the sham-group (Fig. 1a–e).
CV Positive Cells
In the sham-group, neurons in the SHN were well stained with CV (Fig. 2a). In all the ischemia-groups, a markedly reduction of CV positive neurons was not detected in the SHN 4 days after ischemia–reperfusion compared to those in the sham-group (Fig. 2b–e).

CV staining (a–e) and immunohistochemistry for NeuN (f–J) in the SHN of the sham-group (a, f) and ischemia-groups (5 min (b, g), 10 min (c, h), 15 min (d, i), and 20 min (e, j)) 4 days after ischemia–reperfusion. In all the ischemia-groups, numbers of CV positive neurons are not apparently changed in the ischemic SHN; however, NeuN-immunoreactive neurons tend to be slightly decreased in number. Scale bar 50 μm
NeuN Positive Cells
In the sham-group, neurons in the SHN were well immunostained with NeuN (Fig. 2f). In the 5, 10, and 15 min ischemia-groups, a distinctive change in the distribution of NeuN positive neurons was not found 4 days after ischemia–reperfusion (Fig. 2g–h). However, in the 20 min ischemia-group, we found that NeuN positive neurons were somewhat decreased in the SHN 4 days after ischemia–reperfusion compared to those in the sham-group (Fig. 2i).
F-J B Positive Cells
Neuronal degeneration in the SHN following ischemia–reperfusion was examined using F-J B histofluorescence staining. In the sham-groups, 5, 10, and 15 min ischemia-groups, F-J B positive neurons were not observed in the SHN 4 days after ischemia–reperfusion (Fig. 3a–d, f). However, in the 20 min ischemia-group, some neurons in the SHN were positive to F-J B (Fig. 3e) 4 days after ischemia–reperfusion: the mean number of F-J B positive neurons was about 14.9 ± 2.5/400 μm2 in a section (Fig. 3f).

F-J B staining of the SHN of the sham-group (a) and ischemia-groups (5 min (b), 10 min (c), 15 min (d), and 20 min (e)) 4 days after ischemia–reperfusion. No F-J B positive neurons are observed in the SHN of the sham-group and 5–15 min ischemia-group. However, in the 20 min ischemia-group, many F-J B positive cells (arrows) are observed in the SHN. Scale bar 50 μm. f Mean number of F-J B positive neurons/40 mm2 in a section of the SHN 4 days after ischemia–reperfusion (n = 10 per each group; *P < 0.05, significantly different from the sham-group; # P < 0.05, significantly different from the preceding-group). The bars indicate the mean ± SEM
GFAP-Immunoreactive Astrocytes
In the SHN of the sham-group, GFAP-immunoreactive astrocytes showed a rest form with a small cell body and thin processes (Fig. 4a). In the 5 min ischemia-group, GFAP-immunoreactive astrocytes 4 days after ischemia–reperfusion were distinctively increased in number and showed stronger immunoreactivity than that in the sham-group (Figs. 4b, 5a). In the 10 min ischemia-group, GFAP immunoreactivity was more increased in the SHN 4 days after ischemia–reperfusion, and GFAP-immunoreactivity in the 15 min ischemia-group was more increased; however, in the 20 min ischemia-group, GFAP-immunoreactivity was similar to that in the 15 min ischemia-group (Figs. 4c–e, 5a).

Immunohistochemistry for GFAP (a–e) and Iba-1 (f–j) in the SHN of the sham-group (a) and ischemia-groups (5 min (b), 10 min (c), 15 min (d), and 20 min (e)) 4 days after ischemia–reperfusion. In the sham-group, typical GFAP-immunoreactive astrocytes (arrows) and Iba-1-immunoreactive microglia (arrows) are easily detected. In all the ischemia-groups, GFAP and Iba-1 immunoreactivity tends to be increased according to the duration of ischemia. Scale bar 50 μm

Relative optical density (ROD) as % of GFAP (a) and Iba-1 (b) immunoreactive structures/400 μm2 of the SHN 4 days after ischemia–reperfusion (n = 10 per each group; + P < 0.05, significantly different from the sham-group; # P < 0.05, significantly different from the preceding-group). The bars indicate the mean ± SEM
Iba-1-Immunoreactive Microglia
Iba-1-immunoreactive microglia showed a typical rest form in the SHN of the sham-group (Fig. 4f), and Iba-1-immunoreactivity was slightly increased in the SHN of the 5 min ischemia-group 4 days after ischemia–reperfusion (Figs. 4g, 5b). Iba-1 immunoreactivity and numbers of microglia in the ischemic SHN were gradually increased according to the duration of ischemia 4 days after ischemia–reperfusion; Iba-1 immunoreactivity was strongest in the 20 min ischemia-group 4 days after ischemia–reperfusion (Figs. 4h–j, 5b).
Discussion
The septum (septal area) is an important component of the limbic system that is involved in the regulation of cognitive and behavioral functions (Winson 1978). The septum is extensively inter-connected with the hippocampus (Raisman 1966). For example, the fibers of CA1, CA2, CA3, and entorhinal regions project to the lateral septal nucleus, and reciprocal fibers connect the medial septal nucleus with the dentate gyrus, CA1, CA2, subiculum, and entorhinal cortex (DeFrance et al. 1973; Swanson and Cowan 1979). The septum includes diverse neuronal populations with heterogeneous morphology, electrophysiological properties, and connectivity (Brashear et al. 1986; Manns et al. 2000; Panula et al. 1984).
In the hippocampus, the vulnerability differs from each hippocampal subregion. The hippocampal CA1 region is the most susceptible to transient cerebral ischemia, and neuronal death occurs at 4–5 days post-ischemia, which is called “delayed neuronal death (DND)” due to that it occurs very slowly (Kirino 1982). We demonstrated that degenerating CA1 pyramidal neurons were found at 4 days post-ischemia in the 5, 10, 15, and 20 min ischemia–reperfusion group; most of the CA1 pyramidal neurons of all the groups were almost completely degenerated in the CA1 region induce by at least 5 min of transient cerebral ischemia (Yu et al. 2012). In addition, Selakovic et al. (2011) and we (2012) reported that some of neurons in the stratum pyramidale of the CA1 region were survived after a longer duration of ischemia–reperfusion (15 min). On the other hand, a few studies regarding neuronal damage in the ischemic CA2 region have been reported. No definite CA2 cell injury was found after brief ischemia–reperfusion (5 min); however, with a longer ischemia–reperfusion (20–30 min), CA2 pyramidal neurons showed reactive changes in gerbils (Kirino and Sano 1984). We recently also found that many CA2 pyramidal neurons were positive to F-J B after a longer duration of ischemia–reperfusion (10–20 min) at 4 days after ischemia–reperfusion (Yu et al. 2012). In addition, we reported that an abundance of degenerating neurons were found in the striatum of the 20 min ischemia-group (Ohk et al. 2012). Therefore, our present finding in the SHN is fully supported by previous reports that showed that CA2 pyramidal and striatal neurons showed reactive changes with a longer ischemia–reperfusion (20 min) (Ohk et al. 2012; Yu et al. 2012).
An important feature of cerebral ischemic damage is vulnerability of specific neuronal populations. In the central nervous system, certain brain areas are selectively damaged even after a brief ischemic insult, and this topographical heterogeneity is known as “selective vulnerability of the brain”. The Mongolian gerbil has been used as a good animal model to investigate mechanisms of selective neuronal death following transient global cerebral ischemia (Kirino and Sano 1984). While numerous studies regarding transient ischemia have emphasized neuronal damage/death in the hippocampus, some brain regions such as the cerebral cortex, striatum, thalamus, and septal nuclei are also susceptible to ischemic damage. In addition, the extent of neuronal death in ischemic regions is highly correlated to the duration of ischemia. We recently reported that the pattern of neuronal death was very different in the hippocampus, striatum and somatosensory cortex according to various durations of transient cerebral ischemia in gerbils (Ohk et al. 2012; Yu et al. 2012). In the present study, we found that H–E staining showed that small cells were increased in the SHN only in the 20 min-ischemia group, and that F-J B positive cells were detected in the SHN, not in other nuclei, of the 20 min ischemia-group alone 4 days after ischemia–reperfusion. Little is known about mechanisms underlying neuronal damage in SHN after cerebral ischemia. It has been reported that the rapid restoration of blood flow increases the level of tissue oxygenation and accounts for a second burst of reactive oxygen species generation that leads to reperfusion injury (Rodrigo et al. 2013).
In general, H–E staining is a very important staining for general cell morphology. It is known that degenerating neurons tend to be hyperchromic with H–E stain (Stensaas et al. 1972) and damaged cells show various features including a shrunken cell body with pyknosis and chromatolysis (Bartus et al. 1995). However, this staining is insufficient to discriminate neuronal degeneration, because argyrophilic dark neurons, which are damaged by insults, would ultimately result in dying or recovering neurons (Gallyas et al. 1992). On the other hand, it is important to count neuronal, not glial, loss in a damaged brain, because NeuN immunohistochemistry shows an apparent neuronal loss. In addition, F-J B has a good affinity for entirely degenerating neurons (cell bodies, dendrites, axons, and axon terminals), and it is a useful marker for study on neuronal degeneration after ischemic injury (Schmued and Hopkins 2000).
In the present study, small H–E stained cells showed a tendency to be increased in the SHN at 4 days after ischemia–reperfusion compared to those in the sham-group. These small H–E stained cells were counted by their relatively smaller size and lack of stained cytoplasm (cell body diameter <10 μm). The small H–E stained cells may be glial cells. Like many other neurodegenerative disorders, reactive gliosis associated with cerebral ischemia involves both astrocytes and microglia (Kriz 2006). Their glial responses can vary according to the severity and extent of brain injury (Gehrmann et al. 1995; Guo et al. 2007; Kumar and Evans 1997; McRae et al. 1995). It is well known that, in the hippocampus, neuronal damage/death is accompanied by glial activation that releases a variety of cytotoxic agents that lead to neuronal injury (Giulian and Vaca 1993).
To verify changes in reactive gliosis in the SHN after various durations of transient ischemia, we conducted immunohistochemistry of GFAP for astrocytes. Our result showed that reactive astrogliosis in the gerbil SHN 4 days after ischemia–reperfusion; the strongest astrogliosis was shown in the 15 and 20 min ischemia-groups, although we could not exactly explain why strong astrogliosis in the 15 min ischemia-group was similar to that in the 20 min ischemia-group. It is well known that increased expression of GFAP, which shows a main constituent of intermediate filaments in astrocytes, is a hallmark of reactive astrogliosis (Petito et al. 1990). Many researchers have reported that change in GFAP immunoreactivity in the ischemic CA1 region was associated with neuronal death (Ordy et al. 1993; Petito and Halaby 1993; Steward et al. 1992; Stoll et al. 1998). Moreover, GFAP-up-regulated astrocytes were able to uptake harmful substances and could produce neurotrophic factors under pathological conditions (Kraig et al. 1991; Lascola and Kraig 1997; Matsushima et al. 1998). Therefore, our finding suggests that the marked increase of GFAP immunoreactivity in the SHN after ischemia–reperfusion may be associated with an uptake of harmful substances by cerebral ischemic injury.
Changes in morphology and function of microglia are closely correlated with the development of delayed neuronal death in cerebral ischemia (Hailer et al. 1996; Hwang et al. 2006; Schwartz et al. 2006). Microglia can contribute to the elimination of deleterious debris, promotion of tissue repair, and return to tissue homeostasis, and they may involve in neuroprotection (Hashimoto et al. 2005; Laurenzi et al. 2001; Lu et al. 2005). To verify the change in microglia, conducted immunohistochemistry of Iba-1 for microglia in the SHN with longer time of ischemia–reperfusion. In the 20 min ischemia-group, microglia were largest in size and showed the highest immunoreactivity in the SHN 4 days after ischemia–reperfusion. It seems that Iba-1-immunoreactive microglia may be closely related to neuronal degeneration detected by F-J B staining after cerebral ischemic damage, although we could not exactly explain why the activation of microglia was increased in the ischemia–reperfusion group. Our present finding could be supported by papers that showed that microglial activation after cerebral ischemic damage led to neuronal death/damage through excitotoxic mechanisms (Barron 1995; Kreutzberg 1996).
In conclusion, we found that neuronal degeneration/death (F-J B-positive cells) occurred in the SHN of the septum only in the 20 min ischemia–reperfusion group 4 days after ischemia–reperfusion, and that activations of astrocytes and microglia in the ischemic SHN were also highest in the 20 min ischemia-group. These results indicate that the degree of neuronal damage/death and gliosis in an ischemic brain area must be distinctively different according to the duration time of transient cerebral ischemia.
References
Barron KD (1995) The microglial cell. A historical review. J Neurol Sci 134(Suppl):57–68
Bartus RT, Dean RL, Cavanaugh K, Eveleth D, Carriero DL, Lynch G (1995) Time-related neuronal changes following middle cerebral artery occlusion: implications for therapeutic intervention and the role of calpain. J Cereb Blood Flow Metab 15(6):969–979
Brashear HR, Zaborszky L, Heimer L (1986) Distribution of GABAergic and cholinergic neurons in the rat diagonal band. Neuroscience 17(2):439–451
Buchan AM, Pulsinelli WA (1990) Septo-hippocampal deafferentation protects CA1 neurons against ischemic injury. Brain Res 512(1):7–14
Candelario-Jalil E, Alvarez D, Merino N, Leon OS (2003) Delayed treatment with nimesulide reduces measures of oxidative stress following global ischemic brain injury in gerbils. Neurosci Res 47(2):245–253
Crain BJ, Westerkam WD, Harrison AH, Nadler JV (1988) Selective neuronal death after transient forebrain ischemia in the Mongolian gerbil: a silver impregnation study. Neuroscience 27(2):387–402
de Araujo FL, Bertolino G, Goncalves RB, Marini Lde C, Coimbra NC, de Araujo JE (2012) Neuropathology and behavioral impairments after three types of global ischemia surgery in Meriones unguiculatus: evidence in motor cortex, hippocampal CA1 region and the neostriatum. J Neurol Sci 312(1–2):73–78
DeFrance JF, Kitai ST, Shimono T (1973) Electrophysiological analysis of the hippocampal-septal projections. I. Response and topographical characteristics. Exp Brain Res 17(5):447–462
Freund TF, Antal M (1988) GABA-containing neurons in the septum control inhibitory interneurons in the hippocampus. Nature 336(6195):170–173
Fukuchi T, Katayama Y, Kamiya T, McKee A, Kashiwagi F, Terashi A (1998) The effect of duration of cerebral ischemia on brain pyruvate dehydrogenase activity, energy metabolites, and blood flow during reperfusion in gerbil brain. Brain Res 792(1):59–65
Gallyas F, Zoltay G, Dames W (1992) Formation of “dark” (argyrophilic) neurons of various origin proceeds with a common mechanism of biophysical nature (a novel hypothesis). Acta Neuropathol 83(5):504–509
Gehrmann J, Banati RB, Wiessner C, Hossmann KA, Kreutzberg GW (1995) Reactive microglia in cerebral ischaemia: an early mediator of tissue damage? Neuropathol Appl Neurobiol 21(4):277–289
Giulian D, Vaca K (1993) Inflammatory glia mediate delayed neuronal damage after ischemia in the central nervous system. Stroke 24(12 Suppl):I84–I90
Guo X, Nakamura K, Kohyama K, Harada C, Behanna HA, Watterson DM, Matsumoto Y, Harada T (2007) Inhibition of glial cell activation ameliorates the severity of experimental autoimmune encephalomyelitis. Neurosci Res 59(4):457–466
Hailer NP, Jarhult JD, Nitsch R (1996) Resting microglial cells in vitro: analysis of morphology and adhesion molecule expression in organotypic hippocampal slice cultures. Glia 18(4):319–331
Hashimoto M, Nitta A, Fukumitsu H, Nomoto H, Shen L, Furukawa S (2005) Involvement of glial cell line-derived neurotrophic factor in activation processes of rodent macrophages. J Neurosci Res 79(4):476–487
Horn M, Schlote W (1992) Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol 85(1):79–87
Hu Z, Zeng L, Xie L, Lu W, Zhang J, Li T, Wang X (2007) Morphological alteration of Golgi apparatus and subcellular compartmentalization of TGF-beta1 in Golgi apparatus in gerbils following transient forebrain ischemia. Neurochem Res 32(11):1927–1931
Hwang IK, Yoo KY, Kim DW, Choi SY, Kang TC, Kim YS, Won MH (2006) Ionized calcium-binding adapter molecule 1 immunoreactive cells change in the gerbil hippocampal CA1 region after ischemia/reperfusion. Neurochem Res 31(7):957–965
Hwang IK, Yoo KY, Kim DO, Lee BH, Kwon YG, Won MH (2007) Time course of changes in pyridoxal 5′-phosphate (vitamin B6 active form) and its neuroprotection in experimental ischemic damage. Exp Neurol 206(1):114–125
Janac B, Radenovic L, Selakovic V, Prolic Z (2006) Time course of motor behavior changes in Mongolian gerbils submitted to different durations of cerebral ischemia. Behav Brain Res 175(2):362–373
Kirino T (1982) Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239(1):57–69
Kirino T, Sano K (1984) Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol 62(3):201–208
Kiss J, Patel AJ, Freund TF (1990) Distribution of septohippocampal neurons containing parvalbumin or choline acetyltransferase in the rat brain. J Comp Neurol 298(3):362–372
Kraig RP, Dong LM, Thisted R, Jaeger CB (1991) Spreading depression increases immunohistochemical staining of glial fibrillary acidic protein. J Neurosci 11(7):2187–2198
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19(8):312–318
Kriz J (2006) Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol 18(1–2):145–157
Kumar K, Evans AT (1997) Effect of hypothermia on microglial reaction in ischemic brain. NeuroReport 8(4):947–950
Lascola C, Kraig RP (1997) Astroglial acid-base dynamics in hyperglycemic and normoglycemic global ischemia. Neurosci Biobehav Rev 21(2):143–150
Laurenzi MA, Arcuri C, Rossi R, Marconi P, Bocchini V (2001) Effects of microenvironment on morphology and function of the microglial cell line BV-2. Neurochem Res 26(11):1209–1216
Levine S, Sohn D (1969) Cerebral ischemia in infant and adult gerbils. Relation to incomplete circle of Willis. Arch Pathol 87(3):315–317
Levy DE, Brierley JB (1974) Communications between vertebro-basilar and carotid arterial circulations in the gerbil. Exp Neurol 45(3):503–508
Lin CS, Polsky K, Nadler JV, Crain BJ (1990) Selective neocortical and thalamic cell death in the gerbil after transient ischemia. Neuroscience 35(2):289–299
Lorrio S, Negredo P, Roda JM, Garcia AG, Lopez MG (2009) Effects of memantine and galantamine given separately or in association, on memory and hippocampal neuronal loss after transient global cerebral ischemia in gerbils. Brain Res 1254:128–137
Lu YZ, Lin CH, Cheng FC, Hsueh CM (2005) Molecular mechanisms responsible for microglia-derived protection of Sprague-Dawley rat brain cells during in vitro ischemia. Neurosci Lett 373(2):159–164
Manns ID, Alonso A, Jones BE (2000) Discharge profiles of juxtacellularly labeled and immunohistochemically identified GABAergic basal forebrain neurons recorded in association with the electroencephalogram in anesthetized rats. J Neurosci 20(24):9252–9263
Matsushima K, Schmidt-Kastner R, Hogan MJ, Hakim AM (1998) Cortical spreading depression activates trophic factor expression in neurons and astrocytes and protects against subsequent focal brain ischemia. Brain Res 807(1–2):47–60
McRae A, Gilland E, Bona E, Hagberg H (1995) Microglia activation after neonatal hypoxic-ischemia. Brain Res Dev Brain Res 84(2):245–252
Meibach RC, Siegel A (1977) Efferent connections of the septal area in the rat: an analysis utilizing retrograde and anterograde transport methods. Brain Res 119(1):1–20
Ohk TG, Yoo KY, Park SM, Shin BN, Kim IH, Park JH, Ahn HC, Lee YJ, Kim MJ, Kim TY, Won MH, Cho JH (2012) Neuronal damage using fluoro-jade B histofluorescence and gliosis in the striatum after various durations of transient cerebral ischemia in gerbils. Neurochem Res 37(4):826–834
Ordy JM, Wengenack TM, Bialobok P, Coleman PD, Rodier P, Baggs RB, Dunlap WP, Kates B (1993) Selective vulnerability and early progression of hippocampal CA1 pyramidal cell degeneration and GFAP-positive astrocyte reactivity in the rat four-vessel occlusion model of transient global ischemia. Exp Neurol 119(1):128–139
Panula P, Revuelta AV, Cheney DL, Wu JY, Costa E (1984) An immunohistochemical study on the location of GABAergic neurons in rat septum. J Comp Neurol 222(1):69–80
Petito CK, Halaby IA (1993) Relationship between ischemia and ischemic neuronal necrosis to astrocyte expression of glial fibrillary acidic protein. Int J Dev Neurosci 11(2):239–247
Petito CK, Feldmann E, Pulsinelli WA, Plum F (1987) Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 37(8):1281–1286
Petito CK, Morgello S, Felix JC, Lesser ML (1990) The two patterns of reactive astrocytosis in postischemic rat brain. J Cereb Blood Flow Metab 10(6):850–859
Raisman G (1966) The connexions of the septum. Brain 89(2):317–348
Risold PY, Swanson LW (1997) Connections of the rat lateral septal complex. Brain Res Brain Res Rev 24(2–3):115–195
Rodrigo R, Fernandez-Gajardo R, Gutierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W (2013) Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets [Epub ahead of print]
Salazar-Colocho P, Del Rio J, Frechilla D (2008) Neuroprotective effects of serotonin 5-HT 1A receptor activation against ischemic cell damage in gerbil hippocampus: involvement of NMDA receptor NR1 subunit and BDNF. Brain Res 1199:159–166
Schmued LC, Hopkins KJ (2000) Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874(2):123–130
Schwartz M, Butovsky O, Bruck W, Hanisch UK (2006) Microglial phenotype: is the commitment reversible? Trends Neurosci 29(2):68–74
Selakovic V, Korenic A, Radenovic L (2011) Spatial and temporal patterns of oxidative stress in the brain of gerbils submitted to different duration of global cerebral ischemia. Int J Dev Neurosci 29(6):645–654
Stensaas SS, Edwards CQ, Stensaas LJ (1972) An experimental study of hyperchromic nerve cells in the cerebral cortex. Exp Neurol 36(3):472–487
Steward O, Torre ER, Tomasulo R, Lothman E (1992) Seizures and the regulation of astroglial gene expression. Epilepsy Res Suppl 7:197–209
Stoll G, Jander S, Schroeter M (1998) Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 56(2):149–171
Sugawara T, Lewen A, Noshita N, Gasche Y, Chan PH (2002) Effects of global ischemia duration on neuronal, astroglial, oligodendroglial, and microglial reactions in the vulnerable hippocampal CA1 subregion in rats. J Neurotrauma 19(1):85–98
Swanson LW, Cowan WM (1979) The connections of the septal region in the rat. J Comp Neurol 186(4):621–655
Winson J (1978) Loss of hippocampal theta rhythm results in spatial memory deficit in the rat. Science 201(4351):160–163
Yoshida K, Oka H (1995) Topographical projections from the medial septum-diagonal band complex to the hippocampus: a retrograde tracing study with multiple fluorescent dyes in rats. Neurosci Res 21(3):199–209
Yu DK, Yoo KY, Shin BN, Kim IH, Park JH, Lee CH, Choi JH, Cho YJ, Kang IJ, Kim YM, Won MH (2012) Neuronal damage in hippocampal subregions induced by various durations of transient cerebral ischemia in gerbils using Fluoro-Jade B histofluorescence. Brain Res 1437:50–57
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A1A2001404), and by 2012 Kangwon National University Hospital Grant.
Conflict of interest
The authors have declared that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Chan Woo Park and Jae-Chul Lee contributed equally to this article.
Rights and permissions
About this article
Cite this article
Park, C.W., Lee, JC., Ahn, J.H. et al. Neuronal Damage Using Fluoro-Jade B Histofluorescence and Gliosis in the Gerbil Septum Submitted to Various Durations of Cerebral Ischemia. Cell Mol Neurobiol 33, 991–1001 (2013). https://doi.org/10.1007/s10571-013-9967-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-013-9967-y