
Abstract
The study of neural dendrite formation is of great significance both in theory and applications. However, the molecular mechanisms of regulation remain unclear. We previously described a novel EST, which has high homology with dentritic cell factors (DCF1), expressed differentially between undifferentiated and differentiated neural stem cells (NSCs). In this study, we cloned, expressed, and silenced the dcf1 gene and offered insight into its function in regulating dendrite formation during the differentiation of NSCs. The results indicated that dcf1 encoded a 42 kD protein and could be successfully expressed both in Escherichia coli and NSCs. In order to silence dcf1 gene, three different kinds of siRNA vectors were constructed and transformed into the NSC line C17.2 and primary NSCs, resulting in down regulation of the dcf1 mRNA. Analysis of immunofluorescence or GFP illuminated that with overexpression of the dcf1 gene, the NSCs were maintained in undifferentiated status. After the dcf1 gene was silenced, cells tended to differentiate into neurons and astrocytes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neural stem cells (NSCs) are multipotent progenitor cells that have self-renewal activity as well as the ability to differentiate into neurons, astrocytes, and oligodendrocytes (Temple 2001). These characteristics have generated great interest in NSCs from the aspects of both basic developmental biology and clinical therapeutic applications to the damaged brain (Gage et al. 1998; Seiler et al. 1998; Arenas et al. 2002) . However, the mechanisms of how NSCs differentiate into different kinds of cells remain unknown. A number of external cues, as well as intrinsic cellular programs, are thought to regulate maintenance or differentiation of NSCs (Edlund et al. 1999). From the aspect of intrinsic gene expression, it is still not very clear how many genes affect this differentiation. In vivo, several transcription factors are expressed during early neural cell determination. Sox genes, such as Sox1 and Sox2, are among the earliest genes ubiquitously expressed in the early neural plate and appear to have a conserved function in neural cell determination (Sasai et al. 2001). Sox8 and Sox10 are jointly required for the maintenance of these vagal neural crest stem cells (Marzena et al. 2005). Other genes coding for transcription factors expressed in the newly formed neural plate are Otx2, Pax3, Pax7, Msx1, and Msx2 (Plouhinec et al. 2005; Ramos et al. 2005; Engleka et al. 2005; Muhr et al. 1999). In the absence of the repressor-type basic helix-loop-helix genes Hes1, Hes3, and Hes5, which are considered to be primary targets of Notch signal pathway (Kageyama et al. 2005), NSCs do not proliferate sufficiently but prematurely differentiate into neurons. Hes genes are essential for the maintenance of NSCs to make cells not only in correct numbers but also in full diversity (Kageyama et al. 2005). Wnt and Pax genes regulate midbrain–hindbrain development (Gerri et al. 2004; Arne et al. 2003; Muroyama 2004). Previously, we reported an EST termed SHD11 using the mRNA differentiation display and reverse Northern blot analysis and showed that this EST only expresses in undifferentiated NSCs (Wen et al. 2002). Analysis of the ORF of dcf1 revealed a high homology of 98% with dendritic cell factor 1 (DCF1) that is a signaling molecule involved in differentiation and migration of the dendritic cell (Wen et al. 2002; Dietz et al. 2000). The regulation of the neural dendrite is important for the formation of functional neural networks. Ohkawa et al. (2007) show that stathmin, a microtubule destabilizing factor, is downregulated at both the expression and activity levels during cerebellar development, and this down-regulation contributes to dendritic arborization. In this study, we successfully cloned and expressed DCF1 and employed C17.2 (Snyder et al. 1992) cells and primary NSCs to identify dcf1 dendrite formation function by RNAi. Further study illustrated that DCF1 interacts with Na+, K+-ATPase beta1 subunit by a yeast-two hybrid. The mechanism of neural dendrite formation by dcf1 regulation is primary disclosed.
Materials and Methods
Construction of siRNA Vector
In order to accomplish the intracellular expression of siRNA, a vector with 21 bp hairpin representing sequences directed against dcf1 gene was constructed. A schematic presentation of the vectors is shown in Fig. 2a. The hairpin cDNA insert was subcloned into the psiRNA vector through two Bpi1 sites. The clones were verified by Nco1 digestion. The integrity of the insert was confirmed by DNA sequencing.
Cell Culture
Mouse NSC line C17.2, which was kindly provided by Dr. Evan Y. Snyder, was cultured as previously described (Snyder et al. 1992) in high-glucose DMEM with 10% fetal bovine serum (Sigma), 5% horse serum (Gibco), 1 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, culture dishes in standard humidified 5% CO2 at 37°C. Cells were maintained in culture by splitting 1:5 into fresh medium. Cells were fed with fresh medium twice weekly and passaged once per week.
Primary NSCs were prepared from the hippocampi of neonatal mice as described previously (Wen et al. 2002). Following trituration, the cell suspensions were plated at a density of 5 × 105 cells/ml in 1:1 F12/DMEM (Gibco) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin, 20 ng/ml EGF (Sigma), 20 μg/ml bFGF (Sigma), and 1% B27 (Gibco).
Ttransfection of NSCs and Resistant Screen
C17.2 cells were plated onto 24 well plates at a density of 1 × 105 cells per well and cultured at 37°C with 5% CO2 overnight for transfection. Total of 5 μg psiRNA-dcf1 DNA per well was transfected into C17.2 using Lipofectamin2000 (from Invitrogen) according to the manufacturer’s protocol. Vector was used to transfect the cell line for further analysis.
The lowest concentration of G418 causing death of C17.2 cells was tested. Selection was begun at 48 h with G418. The transfected cells were cultured in G418-containing medium for 14 days to enrich the cells that were successfully transfected.
RNA Isolation and RT-PCR Analysis
DNA-free RNA was prepared from the transfected C17.2 cells, which were selected by G418 for 2 weeks by using the total RNA extract kit from Sangon Company. RNA was reverse-transcribed by using oligo dT primers. About 1 μl of 0.1 M of forward primer (5′-CTA TGGTACCATGGCGGCGCCAAAGGGGAA-3′) and reverse primer (5′-ACTAGGTAC CGATTTCTGAGTGAGCAAGGT) was used for the PCR reaction. PCR was conducted for 30 cycles with denaturation at 94°C for 60 s, annealing at 56°C for 45 s, and extension at 72°C for 90 s. β-Actin PCR (forward primer, 5′-CAAGAGATGGCCACGGCTGCT-3′; reverse primer, 5′-TCCTTCTGCATCCTGTCGGCA-3′) was used as an internal control. For the amplification of β-actin, PCR was processed by 30 cycles of 94°C for 60 s, 65°C for 60 s, and 72°C for 90 s. The PCR products were run on 1% agarose gels and stained with ethidium bromide.
Real-time Quantitative RT-PCR
In order to quantify the expression levels of the dcf1 gene, real-time quantitative PCR analysis was further performed. First, DNA-free RNA was prepared from the transfected primary NSCs by using the total RNA extract kit (Sangon Company). RNA was reverse-transcribed by using oligo dT primers. When, respectively, silenced by siRNA vectors psiRNA1, psiRNA2, and control psiRNA3, the expression of dcf1 mRNA was tested via real-time PCR. About 1 μl of 0.1 M forward primer (5′-CTATGGTA CCATGGCGGCGCCAAAGGGGAA-3′) and reverse primer (5′-ACTAGGTACCGATT TCTGAGTGAGCAAGGT) was used for the quantitative PCR reaction of dcf1 gene. PCR was conducted for 40 cycles with denaturation at 94°C for 60 s, annealing at 56°C for 45 s, and extension at 72°C for 90 s. Gene expression was quantified relative to β-actin expression. RT-PCR of β-actin was processed by 40 cycles of 94°C for 60 s, 65°C for 60 s, and 72°C for 90 s.
Immunofluorescence
Transfected cells were selected by G418 for 2 weeks and rinsed with PBS, fixed in 4% paraformaldehyde for 30 min at room temperature, and permeabilized by washing with PBS containing 0.2% Triton X-100 thrice for 5 min. The cells were blocked with normal goat serum at room temperature for 30 min and then incubated with primary antibodies (glial fibrillary acidic protein GFAP 1:80 or neuron-specific enloase NSE 1:200, Santa Cruz) at 4°C overnight. After washing with PBS containing 0.1% Triton X-100, cells were incubated with secondary antibodies TRITC or FITC labeled goat anti-rabbit IgG (1:200, Santa Cruz) according to the primary antibodies. Cells were washed with PBS thrice before observation. The results were analyzed using a Nikon TE300 nuorescence microscope. Pictures were taken with a CCD camera (SPOT, Diagnostic Instrument, USA) equipped with a computer-based image acquisition system.
Results
Overexpression of DCF1 Maintains NSCs in Undifferentiated Status
The cDNA encoding the DCF1 protein was subcloned into pEGFP-C1 (Fig. 1a), a mammalian expression vector with a CMV promoter, which could drive the expression of the DCF1 protein in cultured cells. In order to detect the protein, the cDNA of DCF1 protein was tagged with enhanced green fluorescent protein (EGFP) at the N-terminus. This pEGFP-C1-DCF1 construct was transfected into C17.2 cells and the expression of DCF1 protein was observed through a Nikon TE300 fluorescence microscope (Fig. 1c). As the negative control, pEGFP-C1 was transfected into C17.2 cells under the same conditions with pEGFP-C1-DCF1 (Fig. 1b). The data showed that the vector could drive the expression of DCF1 protein in the C17.2 cells (Fig. 1c). Differences between the neurite lengths of cells transfected by pEGFP-C1 and pEGFP-C1-DCF1 were analyzed (Fig. 1d). The data indicated that overexpression of DCF1 led to a draw back of the cell’s neuritis or maintained NSCs in an undifferentiated status (DCF1 protein expression in Escherichia coli was showed in Supplementary Fig. 1).

Over-expression of dcf1 gene maintains NSCs in undifferentiated status. The dcf1 fragment was inserted in pEGFP-C1-DCF1, the positive clones were digested by BamH1 and released a fragment of 972 bp (a); C17.2 cells were transfected by pEGFP-C1 vectors as a control by EGFP fluorescence observation. Long and ramose neurite can be seen (b). Over-expression of the DCF1 gene was observed by transfecting pEGFP-C1-DCF1. NSCs maintained in undifferentiated status (c). Statistical analysis of neurite length showed in (d). It indicated a significantly different
Knockdown of dcf1 Expression in the Cultured NSCs by RNAi
In order to locate the target site of siRNA in the dcf1 genome, we selected two sequences of 19–21 nucleotides within the coding region. Those sequences start with an A, which corresponds to the transcription initiation point of the H1 promoter. As an empirical design, we preferably chose the sites with GC/AT ratios between 30 and 60% (Li et al. 2005). Also we used the selected sequences to search GenBank to confirm their specificity. In matching those criteria, two specific target sequences were determined. The detailed sequences are listed in Table 1. As a negative control, psiRNA-3 is a scrambled sequence. Although siRNA could be chemically synthesized directly, the cDNA vector driven by a promoter is used in most cases (Shi 2003) for convenience and economy reasons. In this study, we adopted the H1 promoter vector (Sui et al. 2002) and constructed a 19–21 bp siRNA through the hairpin, which could be produced by the DNA vector (Fig. 2a). The hairpin cDNAs were generated through annealing of the complementary oligos synthesized, where two BpiI sites were constructed. The alpha peptide (339 bp) was replaced by inserted cDNA (54 bp) in the psiRNA vector through BpiI sites. So, the size of positive vectors is smaller than the negative vectors. The clones were sequence-confirmed (Supplementary Fig. 2a–c). siRNA vectors, named psiRNA-1, psiRNA-2, and psiRNA-3 were transfected into the C17.2 NSC line. After being selected by G418 for 2 weeks, RT-PCR was performed. Both psiRNA-1 and psiRNA-2 could specifically down-regulate the transcription of the dcf1 gene in cultured cells (Fig. 2b, top panel, lanes 1 and 2), while scrambled sequence psiRNA-3 or the untreated sample did not (Fig. 2b, top panel, lanes 3 and 4).

Silence of dcf1 gene. Schematic diagram of RNAi vector construction (a). The dcf1 gene was effectively down-regulated by psiRNA-1 and psiRNA-2 (b, lanes 1 and 2), while the scrambled sequence psiRNA-3 or blank sample had no affect (b, lanes 3 and 4)
The quantification of the DCF protein expression within cells was tested by immunostaining. The results indicated that the expression of DCF1 protein was significantly reduced after dcf1 gene was silenced (Fig. 3d).

Immunostaining analysis of DCF1 protein expression. NSCs transfected with psiRNA-2 appeared a down-regulation of DCF1 protein expression (c) compared control (g). (a–d) RNAi silence. (e–h) Control group. (a, e) Photos in the white light. (b, f) Nuclei were stained blue with 4,6-diaminodino-2-phenylindole (DAPI). (c, g) NSCs (C17.2) were stained with DCF1 antibody after incubation for 4 days (d, h) were merge images
Effects of Down-regualted dcf1 Expression on the Differentiation of the NSC Cell Line 17.2 and Primary NSCs
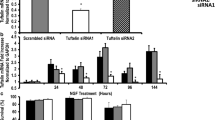
After transfection by siRNA, NSCs differentiated into neurons and astrocytes. The decrease in the number of dcf1 mRNA was correlated with an increase in the number of differentiated C17.2 cells. Analysis of immunostaining of NSE and GFAP indicated that about 10% of cells differentiated into astrocytes (Fig. 4f–i, k) while about 1% C17.2 cells differentiated into neurons (Fig. 4a–d, k). In order to identify the function of dcf1, further study was carried out using primary NSCs. When dcf1 was silenced, primary NSCs were also differentiated into astrocytes (10%) (Fig. 5a) and neurons (1%) (Fig. 5c) in a manner similar to cell line C17.2. Real-time quantitative measurements of dcf1 gene expression revealed that siRNAs could electively reduce the expression of the dcf1 gene (Fig. 5e). Both psiRNA-1 and psiRNA-2 could obviously down-regulate the expression of dcf1 gene, while scrambled sequence psiRNA-3 did not. All these data suggest that siRNA targeting of dcf1 could effectively block or silence the expression of the dcf1 gene in primary NSCs. Statistical analyses showed significant differences (P < 0.001 and P < 0.0001).

Differentiated fate after knockdown of DCF1 in cell line C17.2. After 2 weeks of selection by G418, the transfected cells were immunostained for neuron specific marker NSE with FITC (green, a–e) or astrocyte specific marker GFAP with TRITC (red, f–j). (e, j) The results of transfection by scrambled psiRNA-3 vectors. The average percent of immunoreactive cells were obtained by counting four different fields of view on each well in at least two independent cultures (k)

Differentiated fate after knockdown of dcf1 in primary NSCs. When primary NSCs were transfected by psiRNA-2, they could be differentiated into astrocytes (a) and neurons (c). Photos in white light are showed in (b) and (d), respectively. Real-time PCR was carried out to quantitatively measure dcf1 gene expression. Both psiRNA-1 and psiRNA-2 effectively silence dcf1 (e)
Discussion
In this study, we report the function of the dcf1 gene in regulating NSC differentiation. Our data indicate that the effects of dcf1 on the differentiation of NSCs and dendrite formation when up- and down-regulation. When DCF1 is over-expressed, NSCs maintain undifferentiated status whereas silence of dcf1 initiates NSCs differentiation. This result is consistent with our previous research (Wen et al. 2002).
For many years, scientists have been searching for the mechanisms regulating the differentiation of NSCs. The answer for this question is likely to be complex and involve a variety of contributing factors. Our data suggest on possible aspect of regulating cell differentiation by the control of dcf1. RNAi has been established as a powerful biological strategy for gene silencing (Sui et al. 2002; Thijin et al. 2002; Shi 2003). In this paper, the effectiveness of RNAi techniques was clearly demonstrated. While these RNAi vectors are accepted for use in academic study, more important is their potential as clinical candidates for regulating endogenic cell differentiation. As far as we know, DCF1 is a membrane protein serving as a signaling molecule involved in the differentiation and migration of dendritic cells (Wen et al. 2002; Dietz et al. 2000). The dendritic cell is the most important of the antigen presentation cells in the immune system (Steven et al. 2000). Recently, scientists found that dendritic cells appear in the neural systems of inflammatory mice (Fischer et al. 2000). Because the neural and the immune systems share many of same regulatory factors such as IL-1, IL-6, and TNF-alfa (Fischer et al. 2000; Rita et al. 1999), we speculate the dcf1 gene may serve as a common mechanism of differentiation and migration in both the neural and the immune system.
References
Arenas E et al (2002) Stem cells in the treatment of Parkinson’s disease. Brain Res Bull 57:795–808
Arne C et al (2003) wnt1 and wnt10b function redundantly at the zebrafish midbrain–hindbrain boundary. Dev Biol 254:172–187
Dietz AB, Bulur PA, Knutson GJ, Matasic R, Vuk-Pavlovic S et al (2000) Maturation of human monocyte-derived dendritic cells studied by microarray hybridization. Biochem Biophys Res Commun 275:731–738
Edlund T et al (1999) Progression from extrinsic to intrinsic signaling in cell fate specification: a view from the nervous system. Cell 96:211–224
Engleka KA et al (2005) Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev Biol 280:396–406
Fischer HJ et al (2000) Phenotype and functions of brain dendritic cells emerging during chronic infection of mice with toxoplasma Gondii1. J Immunol 164: 4826–4834
Gage FH et al (1998) Cell therapy. Nature 392(Suppl):18–24
Gerri R et al (2004) Combinatorial Wnt control of zebrafish midbrain–hindbrain boundary formation. Mech Dev 121:437–447
Kageyama R et al (2005) Roles of bHLH genes in neural stem cell differentiation. Exp Cell Res 306:343–348
Li T et al (2005) siRNA targeting the Leader sequence of SARS-CoV inhibits virus replication. Gene Ther 12:751–761
Marzena M et al (2005) Identification of Sox8 as a modifier gene in a mouse model of Hirschsprung disease reveals underlying molecular defect. Dev Biol 277:155–169
Muhr J et al (1999) Convergent inductive signals specify midbrain, hindbrain, and spinal cord identity in gastrula stage chick embryos. Neuron 23:689–702
Muroyama Y (2004) Wnt proteins promote neuronal differentiation in neural stem cell culture. Biochem Biophy Res Commun 313:915–921
Ohkawa N, Fujitani K, Tokunaga E, Furuya S, Inokuchi K (2007) The microtubule destabilizer stathmin mediates the development of dendritic arbors in neuronal cells. J Cell Sci 120(8):1447–1456
Plouhinec JL et al (2005) Comparative analysis of gnathostome Otx gene expression patterns in the developing eye: implications for the functional evolution of the multigene family. Dev Biol 278:560–575
Ramos C et al (2005) msh/Msx gene family in neural development. Trends Genet 21:624–632
Rita M et al (1999) Skewed differentiation of bone marrow CD34+ cells of tumor bearers from dendritic toward monocytic cells, and the redirection of differentiation toward dendritic cells by 1a,25-dihydroxyvitamin D3. Int J Immunopharmacol 21:675–688
Sasai Y et al (2001) Roles of Sox factors in neural determination: conserved signaling in evolution. Int J Dev Biol 45:321–326
Seiler MJ et al (1998) Intact sheets of fetal retina transplanted to restore damaged rat retinas. Invest Ophthalmol Vis Sci 39:2121–2131
Shi Y (2003) Mammalian RNAi for the masses. Trends Genet 19:9–12
Snyder EY et al (1992) Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell 68:33–51
Steven DR et al (2000) The control of T cell responses by dendritic cell subsets. Curr Opin Immunol 12:114–121
Sui G et al (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 99:5515–5520
Temple S (2001) The development of neural stem cells. Nature 414:112–117
Thijn R et al (2002) A system for stable expression of short interfering RNAs in mammalian. Cells Sci 296:550–553
Wen T et al (2002) Discovery of two novel functional genes from differentiation of neural stem cells in the striatum of the fetal rat. Neurosci Lett 329:101–105
Acknowledgments
We thank Dr. Snyder, E.Y. for kindly donating C17.2 cell for this research. This work was supported by grants from the National Basic Research Program of China (No2006CB500702), the National Science Foundation of China (30570590, 30770695), the Shanghai Commission of Science and Technology Basic Research Fund (No.03JC14030), and the Shanghai Commission of Education Science and Technology Innovation Fund (Grant No.08 ZZ41).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10571_2008_9266_MOESM1_ESM.tif
Supplementary Fig. 1 DCF1 protein expression in E. coli. (A) Construction of expression vector containing 972bp DCF1 protein coding sequence. (B) Expression of DCF1 protein arrowed in B 2 (TIF 117 kb)
10571_2008_9266_MOESM2_ESM.tif
Supplementary Fig. 2. Sequencing of psiRNA-1 (A), psiRNA-2 (B), and psiRNA-3 (C). Exactitude with designed sequences is confirmed (TIF 175 kb)
Rights and permissions
About this article
Cite this article
Wang, L., Wang, J., Wu, Y. et al. A Novel Function of dcf1 During the Differentiation of Neural Stem Cells In vitro. Cell Mol Neurobiol 28, 887–894 (2008). https://doi.org/10.1007/s10571-008-9266-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-008-9266-1