
Abstract
We describe a novel method for the production of lightweight fibrous structures of densities as low as 8.8 kg m\(^{-3}\). The method is based on the use of liquid foam as a carrier medium for dispersed Kraft fibres. Different to the process of foam forming, where the quick removal of the foam results in the formation of thin fibrous sheets, our samples are allowed to slowly drain and dry until all foam has disappeared. This procedure results in bulk samples whose height (up to 25 mm) and density are controlled by initial fibre concentration and liquid fraction of the foam. Above a minimum density, the compression modulus of elasticity of the samples increases linearly with density. Furthermore, we show compressive strength of the structures being controlled via the initial liquid fraction of the foam, making this an important process parameter for the fabrication of such structures.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Non-woven fibrous structures made from natural materials find many applications, including thermal insulation (Poehler et al. 2017), acoustic dampening, and visually pleasing wall panels (Härkäsalmi et al. 2017). Here we describe a novel technique for producing bulk materials from northern bleached softwood Kraft fibres. The technique involves the initial dispersion of the fibres in a liquid foam which is then slowly dried. Height, density and compressive strength of the samples can be tuned via the fibre concentration and the initial liquid fraction \(\phi _i\) of the foam, defined as the fraction of liquid contained in the foam to the total volume of the foam. Figure 1 shows an example of the low density fibrous structure, together with the pulp sheet of Kraft fibres from which it was made. The density of the sheet is 450 kg m\(^{-3}\), while that of the structure on the right is 25 kg m\(^{-3}\). Both the sheet and the foam-formed structure in the figure contain the same mass of fibres.
The use of foam for the formation of pulp sheets dates back to the early 1960s (Radvan 1964). Surfactant is added to an aqueous solution of fibres, which is then agitated to produce a foam dispersion containing fibres. The bubbles in the foam act as spacer particles between fibres, preventing their flocculation. If the foam is quickly removed via suction, this results in sheets of fibres with a greater uniformity than may be achieved in water-based papermaking, in particular when long fibres are used (Al-Qararah et al. 2012; Lehmonen et al. 2013). If the foam is allowed to drain freely it is possible to create three dimensional fibrous materials (Alimadadi and Uesaka 2016).

Low density fibrous structure (right) obtained using the foam-forming technique described in the text. The sample was produced from a thin sheet of compressed Kraft fibres, as shown on the left
An advantage to the foam-forming method is that it allows for a control of porosity via the control of the average bubble size. For sheets it was shown that their density is decreased by using longer fibres which allow for more and/or larger bubbles to aggregate between them, while fibre-fibre contacts are still maintained (Madani et al. 2014). As the foam is removed this results in larger voids, and thus decreased sample density. The initial liquid fraction of the foam, \(\phi _i\), is a further control parameter for varying sheet density (Madani et al. 2014). Also fibre-orientation may be controlled by the use of foam. Compared to water-formed structures, where there is a tendency for the fibres to orientate in the horizontal plane, fibres dispersed in foam were found to orientate through a broad range of angles in a 12 mm thick sample produced by Alimadadi and Uesaka 2016.
In the following we describe a novel, foam based process for the production of lightweight fibrous structures. Allowing the foam to drain freely results in sample heights exceeding that of previously published work, where immediately after the foam has been poured into the drainage vessel a vacuum is applied from below (Alimadadi and Uesaka 2016). The description of our method is followed by demonstrations of how density and compressive strength can be tailored via the choice of fibre concentration and initial liquid fraction of the foam.
Adding foam to the production process of fibrous materials thus allows for a much greater control of their properties.
Sample preparation
The samples described in the following were made from bleached and dried softwood Kraft fibres with average length \(2.0\pm 0.1\) mm and diameter of 35 \(\upmu\)m, and thus an aspect ratio of approximately 60 (Al-Qararah et al. 2013). As a surfactant we used commercial SDS from Sigma-Aldrich with a purity of 98.5%.

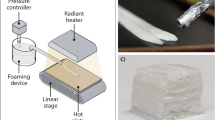
Sketch of the experimental set-up used to create foam-formed fibrous structures. a Steps taken to create a foamed fibre dispersion. After shearing, the dispersion is poured into a container with a wire gauze base. b A non-airtight plastic cover is placed on top. The foam is then left to drain under gravity. As the sample dries out eventually all the bubbles disappear, leaving a three dimensional fibre network
Sample preparation is illustrated in Fig. 2. Disintegration of dry pulp fibre sheets was achieved by soaking them in a specified amount of water for 24 h (Al-Qararah et al. 2013). The samples described below were produced with weight to weight ratios (referred to as fibre concentration) ranging from 1 to 6.25%. Following this, the aqueous fibre suspension is placed into a vessel and sheared with a mixing disk at a constant speed (5000 RPM) for 3 min. The diameter of the mixing disk was 50 mm, with two opposing 25\(^{\circ }\) bends. The design of the disk allows air to be drawn into the suspension during shearing. SDS is then added at a weight per weight (w/w) concentration of 0.66%, in practice 1 g of SDS per 150 ml of water and thus above the critical micelle concentration of 2.3 g/L. The aqueous dispersion is foamed up by shearing once again for 3 min. The use of such a high surfactant concentration is necessary to create foams which are stable for a longer time period than the few seconds that is required for the foam forming of paper sheets (Lee et al. 2014).
The initial volume of the aqueous fibre suspensions ranged from 300 to 600 ml; this resulted in a final volume of fibre containing foam of 1200 ml, corresponding to initial average liquid fractions \(\phi _i\) of 0.25, 0.33, 0.42 and 0.50 for the samples described below.
The fibre-containing foam was then poured into a container (width 125 mm, length 200 mm, depth 50 mm) with a wire mesh (50 \(\upmu\)m hole size) as a base (Fig. 2b) to allow the liquid to drain from the foam.
A rigid plastic sheet was placed on top of the container during this drying process. The plastic sheet did not touch the fibre-foam, nor did it make an airtight seal. However, we found through initial experiments that the sheet plays a crucial role, particularly for samples with lower fibre concentrations, which would collapse completely without its presence. The sheet reduces the evaporation of the liquid and creates a pressure gradient that slows down the drainage; the resulting extension of the drying time may contribute to a better bonding of the fibres, enabling us to reduce the minimum fibre concentrations required for mechanically stable structures.
After approximately 12 h of drying we uncover the plastic sheet and remove the sample, together with the mesh that supports it, from the container. Doing so speeds up the drying time by exposing more of the surface area to air. At this point it is observed that all the foam has decayed from the structure, however there is still a significant volume of liquid contained within. When the samples are dry to the touch we monitor their weight at regular intervals until the drying has effectively ceased (see "Drying process" section).
The dried samples are then carefully removed from the mesh and cut into equal sized pieces using a surgical scalpel (see Fig. 3). The dimensions of each piece are 33 mm by 33 mm, with a height that is dependent on fibre concentration, as discussed in "Variation of sample height with fibre concentration" section.
Figure 1 shows a foam-formed three dimensional fibre structure alongside a fibre sheet. Both samples contain the same type and mass of fibres. However, there is a large difference between the densities of both, the sheet on the left has a density of 450 kg m\(^{-3}\), while the density of the foam-formed structure is 25 kg m\(^{-3}\).

Top view of a foam-formed fibrous structure that has dried but has not yet been cut into samples for further testing. The average height of the structure shown is 25 mm
Drying process
The rate at which the samples dry varies with time; it is also dependent on the concentration of fibres used. Figure 4 shows the ratio of the total sample mass \(m_d\) to the total fibre mass \(m_0\) for one sample from the initial pouring of the fibre laden foam until the sample has fully dried. The data was obtained by placing the fibre-foam dispersion onto a mass balance, the liquid was able to drain from the sample, which was supported by the wire gauze.
Immediately after the fibre-foam dispersion has been poured into the container, the ratio \(m_d / m_0\) decreases very rapidly, as gravitationally induced drainage removes most of the liquid from the foam within the first few minutes (Haffner et al. 2017). This regime can be described by a numerical solution of the foam drainage equation (Verbist et al. 1996) (see inset of Fig. 4).

Drying curve for a fibre-foam dispersion containing 3% fibre concentration showing the ratio of the mass of the dispersion to the mass of the fibres (\(m_d/m_o\)) as a function of time. The accelerated mass loss in the initial 20 min (inset), is due to gravitationally induced foam drainage and can be modelled using the foam drainage equation. Further mass loss is mainly due to evaporation of liquid. After 21 h the sample was removed from the container, doing so increases the rate of evaporation by exposing a larger surface area, as is evident by the sharp decrease in \(m_d/m_0\)
As drainage slows down and eventually stops, further drying is mainly due to evaporation. This is initially accompanied by the rupturing of foam films, but then also due to the evaporation of liquid attached to fibre surfaces and at fibre-fibre contacts. A higher concentration of fibres will allow more water to attach to fibres and is therefore responsible for the slow-down in drying that we observe in this case.
We can define a “drying time” \(T_d\) as the time it takes for the sample to reduce its weight to 0.95 times the weight of the initial fibres, to within ± 5% of the mass of fibres, allowing for any fibre loss that may have occurred during the pouring stage. The typical drying times of about \(T_d \simeq\) 40 h found for our experiments are impractical for any large-scale production of fibrous samples. Several techniques are available for the reduction of \(T_d\), such as placing samples into an oven, impingement-infrared drying, as well as other methods, such as impingement drying under vacuum (Timofeev et al. 2016). Impingement drying is well suited to the fibrous samples due to the porosity of both the structure and the gauze it is formed on. The technique consists of blowing hot air through the sample, which is then recirculated back to a air drier where the moisture is extracted. The benefit of this technique is that it can evenly and rapidly dry the samples, unlike oven drying, which usually dries the outermost part of the samples first (Timofeev et al. 2016). We will address the possible shortening in drying times of our samples in future work.
Sample characterisation
We analysed our samples in terms of homogeneity (local density), sample height as a function of fibre concentration, and compression modulus. Here, fibre concentration is defined by the weight of fibres added to the weight of liquid (before foaming). Each quantity was determined by averaging the measurements from four samples produced under the same conditions. In our experiments fibre concentrations ranged from 1 to 6.3%.
Sample uniformity
Most practical applications of fibrous structures require a uniform sample density. Our structures dry under gravity which may lead to a vertical density variation. We have probed for this by measuring optical absorption in 1 mm thick slices of our samples. To achieve such thin slices we placed the sample in a holder, with a 1 mm protruding edge which is then cut using a thin surgical scalpel.
An LED backlight was placed behind each slice and images were taken using a digital camera (Canon EOS 50D). The images were then converted to greyscale and ImageJ (Schneider et al. 2012) was used to compute an average grey value through the horizontal plane of each sample. The resulting grey value was then plotted as a function of sample height. In order to smooth the profiles we averaged the results over four slices to reduce the contribution any single large void would have on the overall distribution.

The graphs on the left show measurements of optical transmittance for samples obtained with different fibre concentrations (c). (x-axis: sample height in pixels). The straight line fit to the data (dashed line) shows that sample uniformity away from top and bottom increases with fibre concentration. The photographs on the right shows the height variation of the samples with an increase in fibre concentration, see also Fig. 6
The light transmitted through the sample is represented by the greyscale value, i.e. a reduction in transmittance indicates a higher fibre content. The transmission data thus provides a (qualitative) measure of sample uniformity.
Figure 5 shows photographs of the thin slices, as well as the averaged profiles for fibre concentrations of 2.4%, 3.3% and 6.3%, respectively. In all cases both top and bottom of the profiles feature a peak which represents sample edges. The steep curves, rather than an abrupt drop at the top and bottom is due to the edges of the sample being orientated slightly off plane to the plane of the pixels.
The data indicates that an increase in fibre concentration leads to a decrease of the gradient in the sample profiles. The fibre concentration of 6.3% results in a nearly uniform profile away from top and bottom of the sample. The increased density of the samples made from higher fibre concentrations also leads to reduced sample transmittance, i.e. lower greyscale values, as seen in Fig. 5.
Our samples differ greatly from the ones made by (Haffner et al. 2017), also produced via foam forming, but in narrow vertical cylinders of 150 mm in length. In this case drainage of the liquid resulted in a sharp downward gradient of fibre concentration, presumably due to the large volume of liquid drained per cross-sectional area of foam. Also, unlike the experiments carried out here, there was no mesh to act as a stable platform to support the drying fibre structure after the foam had drained.
Variation of sample height with fibre concentration
For all our experiments we kept the initial volume (hence height) of the fibre-foam dispersions constant at 1200 ml. After pouring the fibre-foam into the vessel, the height of the dispersions decreased as drainage and drying took place, asymptoting to a constant value, provided a minimum fibre concentration is exceeded, see section "Stress–strain behaviour under uni-axial compression". This height is reached long before the sample has reached the drying time \(T_d\), defined in section "Drying process". An increase of fibre concentration leads to an increase of up to \(20\%\) in the final height (defined as height at time \(T_d\)), as shown in Fig. 6. This is presumably due to the increased number of fibre-fibre-contacts being formed during the initial decay of the foam.

The final height of the fibrous structures increases with fibre concentration. Each data point is an average over four samples, the dashed line indicates a linear trend. The data shown is for the samples made from fibre-foam dispersions with an initial liquid fraction of \(\phi _i=0.42\), a similar trend was also observed for other values of \(\phi _i\)
Variation of sample density with fibre concentration
The final sample density \(\rho\) is controlled by the fibre concentration c used in its preparation, as shown in Fig. 7a. The increase is linear in c but a minimum concentration \(c_{crit}\sim 0.6\%\) is required for sample stability, see section "Stress–strain behaviour under uni-axial compression". The rate of increase scales roughly linearly with the initial liquid fraction \(\phi _i\) of the foam, see Fig. 7b, highlighting its role as a control parameter in sample production.

a Variation of sample density \(\rho\) as a function of fibre concentration c for samples produced with Kraft fibres. For all values of initial liquid fraction we find \(\rho \propto c\), however, there is a minimum fibre concentration \(c_{crit}\) below which no stable structures can be formed (see section "Stress–strain behaviour under uni-axial compression"). Each data point is an average over four samples. b The constant of proportionality, \(\rho\)/c, scales linearly with the initial liquid fraction of the foam
Stress–strain behaviour under uni-axial compression
We tested the mechanical strength of the dried samples by subjecting them to uni-axial compression. The samples were placed in a rheometer (Anton Paar Physica MCR301) with a plate-plate configuration (Plate diameter 50 mm). All our samples had a contact area of 33 mm by 33 mm. Stress–strain curves were obtained by applying an increasing load to the sample and measuring the resulting deformation up to a maximum strain of 0.6. For each sample density the tests were carried out on four samples and the results were then averaged.

Stress–strain curves for three dimensional fibrous structures for four different sample densities, produced from foams with \(\phi _i=0.25\). Each data point is averaged over four experimental runs carried out with four individual samples. The inset shows the initially linear part of these data sets, where the stress is proportional to strain
Figure 8 shows the stress–strain curves for samples produced with fibre-foam dispersions of \(\phi _i=0.25\). Similar curves were obtained for initial liquid fractions of 0.33, 0.42 and 50. The compression modulus of elasticity \(E_c\) (BS EN826 1996) is given by the slope of the initial linear increase of the stress–strain data, see inset of Fig. 8. Figure 9 shows the compression modulus computed from fits over a strain of 0.15 as a function of sample density for samples produced with four different values of \(\phi _i\). In all cases \(E_c\) increases linearly with sample density. Linear extrapolation of the data leads to a minimum sample density \(\rho _c\), dependent on \(\phi _i\), which is required for mechanical stability, i.e. \(E_c>0\). Above \(\rho _c\) the data is described by

Variation of compressive modulus \(E_c\) with sample density. Linear extrapolation of the data suggests that there is a lower limit to the density of the structures. The corresponding critical densities for the range of initial liquid fractions (\(\phi _i=0.25, 0.33, 0.42 \text{ and }0.50\)) were found to be 3.2 kg m\(^{-3}\), 4.4 kg m\(^{-3}\), 6 kg m\(^{-3}\) and 7.2 kg m\(^{-3}\) respectively

a The minimum sample density \(\rho _c(\phi _i)\), from the fits to the data in Fig. 9 and equation 1, scales linearly with initial liquid fraction. b The rate at which the compressive strength of the structures scales with density is also dependent on the initial liquid fraction
Figure 10a shows that the minimum sample density \(\rho _c\) varies roughly linear with \(\phi _i\). These values are in line with \(\rho _c\)= 5 kg m\(^{-3}\), as reported by Alimadadi and Uesaka (2016). Also the slope \(s(\phi _i)\) (Fig. 10b), varies linearly with the initial liquid fraction for \(\phi _i \ge 0.22\). The minimum fibre concentrations \(c_{crit}\) corresponding to the minimum sample densities \(\rho _{c}(\phi _{i})\), were obtained by inserting the values of \(\rho _{c}(\phi _{i})\) into the fits to the data in Fig. 7a. This results in values of \(c_{crit}\) around 0.6%.
Figures 7 and 9 demonstrate that both the sample density and compressive strength can be controlled by varying the initial liquid fraction of the fibre-foam dispersions. In particular, Fig. 9 shows that samples of a similar density can have a range of compressive strengths which are determined by the initial liquid fraction. Two of our samples highlight this point. The first was produced from a fibre-foam dispersion with an initial liquid fraction of 0.25 and contained a 5% fibre concentration. The second sample was made from a fibre-foam dispersion of an initial liquid fraction of \(\phi _i =0.42\) and a 3% fibre concentration. The structures have similar densities (26 kg m\(^{-3}\) and 25 kg m\(^{-3}\) respectively), since the mass of fibres in each dispersion was almost the same. However, the compression modulus of the samples produced with the higher initial liquid fraction is almost seven times that of the samples produced with a lower initial liquid fraction.
Fibre network and percolation theory
The experimental finding that a finite sample density is required for sample stability, i.e. compression modulus \(E_{c} > 0\) (Fig. 9), allows us to make a connection to percolation theory (Gu et al. 2016). Equation (1) can be re-expressed in terms of a volume fraction of fibres in the sample, \(\phi _v=\rho\)/\(\rho _{f}\) where \(\rho _{f}\) is the fibre density (\(\rho _{f}\) = 1500 kg m\(^{-3}\)) and \(\rho\) is the sample density (Al-Qararah et al. 2016). This results in
where, as before, \(\phi _{i}\) is the initial liquid fraction of the foam used in producing the sample. The value of the constant \(E_{c,0}\) = \(s(\phi _{i})\)/\(\rho _{f}\) is determined by the material properties of the fibres (length, cross-section, density, elastic modulus) as well as by the structure of the network. From Fig. 10a we then obtain that the value of the critical volume fraction \(\phi _{v,c}(\phi _i) = \rho _c(\phi _i)/\rho _f\) varies linearly between 0.002 and 0.005.
Gu et al. (2016) performed computer simulations of fibre networks. For a fibre aspect ratio of approximately 60, as for the fibres in our experiments they report a critical volume fraction \(\phi _{v,c}\) of about 0.005, consistent with our data.
Note however that these authors studied networks formed from randomly distributed fibres. A preliminary analysis of our samples suggest that there is some fibre alignment. We will carry out X-ray scans of our samples to determine how this alignment correlates with the initial liquid fraction of the foam. This should elucidate the origin of the dependence of both \(E_{c,0}\) and \(\phi _{v,c}\) on the initial liquid fraction \(\phi _i\).
Conclusions and Outlook
We have described how a foam forming process can be used to produce lightweight three dimensional fibrous structures, using Kraft fibres. Their height and densities can be controlled by the fibre concentration and the initial liquid fraction of the fibre-foam dispersion. The densities of our fibrous structures ranged from 8.8 to 46 kg m\(^{-3}\). Sample density varies linearly with fibre concentration. From uni-axial compression tests we found a linear increase also of the compression modulus with sample density. There is a lower density limit below which samples are no longer mechanically stable corresponding to the percolation threshold of fibre networks. The initial liquid fraction of the foam is an important control parameter over both sample density and compressive strength. X-ray imaging will be required to relate these sample properties to the structure of the fibre network (isotropy, alignment). The obvious shortcoming in the current production of these fibrous samples is the long time required for their drying. Several methods exists for shortening this (detailed in section "Drying process") and we will address this in future work, together with an analysis of the shape of the drying curves.
The foam forming technique introduced here can be applied to produce a variety of structures from a range of fibre types; we have successfully produced similar structures using fibres from peat and spent grain. This will broaden the range of applications for such non-woven fibrous materials (Härkäsalmi et al. 2017). The compressive strength of the structures can be greatly increased with the addition of a bonding agent (PVA) to the process as found in our preliminary work.
References
Al-Qararah A, Ekman A, Hjelt T, Kiiskinen H, Timonen J, Ketoja J (2016) Porous structure of fibre networks formed by a foaming process: a comparative study of different characterization techniques. J Microsc 264(1):88–101
Al-Qararah A, Hjelt T, Kinnunen K, Beletski N, Ketoja J (2012) Exceptional pore size distribution in foam-formed fibre networks. Nordic Pulp Paper Res J 27(2):226–230
Al-Qararah A, Hjelt T, Koponen A, Harlin A, Ketoja J (2013) Bubble size and air content of wet fibre foams in axial mixing with macro-instabilities. Coll Surf A Physicochem Eng Asp 436(Supplement C):1130–1139
Alimadadi M, Uesaka T (2016) 3d-oriented fiber networks made by foam forming. Cellulose 23(1):661–671
Bs EN 826 (1996) Thermal insulation products for building applications. Determination of compression behaviour, Standard, European Standardization Organisation (ESO)
Gu H, Wang J, Yu C (2016) Three-dimensional modeling of percolation behavior of electrical conductivity in segregated network polymer nanocomposites using monte carlo method. Adv Mater 5(1):1–8
Haffner B, Dunne F, Burke S, Hutzler S (2017) Ageing of fibre-laden aqueous foams. Cellulose 24(1):231–239
Härkäsalmi T, Lehmonen J, Itälä J, Peralta C, Siljander S, Ketoja J (2017) Design-driven integrated development of technical and perceptual qualities in foam-formed cellulose fibre materials. Cellulose 24(11):5053–5068
Lee J, Nikolov A, Wasan D (2014) Foam stability: the importance of film size and the micellar structuring phenomenon. Can J Chem Eng 92(12):2039–2045
Lehmonen J, Jetsu P, Kinnunen K, Hjelt T (2013) Potential of foam-laid forming technology in paper applications. Nordic Pulp Paper Res J 28(3):392–398
Madani A, Zeinoddini S, Varahmi S, Turnbull H, Phillion AB, Olson JA, Martinez DM (2014) Ultra-lightweight paper foams: processing and properties. Cellulose 21(3):2023–2031
Poehler T, Jetsu P, Fougeron A, Barraud V (2017) Use of papermaking pulps in foam-formed thermal insulation materials. Nordic Pulp Paper Res J 32(3):367–374
Radvan B (1964) Basic radfoam process. Br Pat 1329409(0):20
Schneider C, Rasband W, Eliceiri K (2012) Nih image to imagej: 25 years of image analysis. Nat Methods 9(7):671–675
Timofeev O, Jetsu P, Kiiskinen H, Keränen J (2016) Drying of foam-formed mats from virgin pine fibers. Dry Technol 34(10):1210–1218
Verbist G, Weaire D, Kraynik AM (1996) The foam drainage equation. J Phys Condens Matter 8:3715–3731
Acknowledgments
We thank D. Weaire and J. Ketoja for critical comments in laying out the manuscript. We also thank B. Haffner for his assistance on the project. This publication has emanated from research supported in part by a research grant from Science Foundation Ireland (SFI) under Grant Number 13/IA/1926. We also acknowledge the support of cost action MP1305 ‘Flowing matter’ and the European Space Agency ESA, Project microG-Foam, AO-99-075 and Contract 4000115113, ‘Soft Matter Dynamics’. T. Hjelt is supported by the Academy of Finland (Project “Surface interactions and rheology of aqueous cellulose-based foams”).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Burke, S.R., Möbius, M.E., Hjelt, T. et al. Properties of lightweight fibrous structures made by a novel foam forming technique. Cellulose 26, 2529–2539 (2019). https://doi.org/10.1007/s10570-018-2205-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-2205-5