
Abstract
Cellulose II aerogels are light-weight, open pores materials with high specific surface area. They are made in the same way as bio-aerogels based on other polysaccharides, via dissolution-(gelation)-solvent exchange-drying with supercritical CO2. Gelation step is often omitted as cellulose allows keeping 3D shape during solvent exchange (which leads to cellulose coagulation) and drying. Drying in supercritical conditions preserves the porosity of “wet” (coagulated) cellulose. There are numerous ways to vary cellulose II aerogel morphology and properties by changing processing conditions and cellulose type. Together with chemical and physical modifications of cellulose and possibility of making hybrid and composite materials (organic–inorganic and organic–organic), it opens up a huge variety of aerogel properties and applications. On one hand, they are similar to those of classical aerogels, i.e. can be used for absorption and adsorption, as catalysts and catalysts support and in electro-chemistry when pyrolysed. On the other hand, because the preparation of cellulose aerogels may not involve any toxic compounds, they can be used in life science applications such as pharma, bio-medical, food and cosmetics. The review makes an overview of results reported in literature on the structure and properties of cellulose II aerogels and their applications. The reader may be surprised finding more questions than answers and clear trends. The review shows that several fundamental questions still remain to be answered and applications to be explored.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
This review is devoted to cellulose II based aerogels and the term “aerogel” will be first defined as literature provides different approaches. According to IUPAC Gold Book, aerogel is a “Gel comprised of a microporous solid in which the dispersed phase is a gas” with examples such as “Microporous silica, microporous glass and zeolites” (IUPAC. Compendium of Chemical Terminology 2014). This definition is very restrictive as it includes only microporous materials, i.e. with pore sizes below 2 nm, and thus excludes, for example, classical silica aerogels which have pores of some tens of nanometers. Aerogel scientists now agree that aerogels are open pores solid networks with high porosity (at least 90%), high specific surface area (“although no official convention really exists” (Pierre 2011) and are nanostructured (mainly mesoporous with small macropores). These structural properties make aerogels very attractive for various applications such as acoustic and thermal insulation (some aerogels are superinsulating materials, i.e. with thermal conductivity lower than that of air in ambient conditions), catalysts and catalyst supports, for adsorption and absorption, particle detectors (Cerenkov counters), electrochemical when pyrolysed and as matrices for drug delivery.
The first aerogels were synthesized via sol–gel chemistry and reported by Kistler; solvent was removed from the gel by drying in super-critical conditions (Kistler 1931). In this case capillary pressure, which develops during drying and is responsible for pores’ collapse, is theoretically zero as no liquid–vapor interface (no meniscus) is formed in super-critical state.
Since that time silica aerogels, with density around 0.1 g/cm3 and specific surface area around 800–1000 m2/g and higher, became the most studied reference aerogel materials. Their major industrialized application is thermal insulation materials due to ultra-low thermal conductivity, around 0.012–0.014 W/m K against 0.025 W/m K for air. It should be noted that very similar properties have been obtained for hydrophobised (silylated) silica dried at ambient pressure and slightly elevated temperature (around 130–150 °C). However, silica gels break during drying in the course of so-called “spring-back” effect, i.e. re-opening of the pores during the last stage of drying due to the repulsion of the grafted groups and certain elasticity of the solid network which recovers its shape after contraction. Ambient-pressure dried silica-based “xerogels” with structure and properties equivalent to supercritically dried aerogels is a unique example of ambient-dried lightweight thermal superinsulating mesoporous materials.
Next generations of aerogels developed in the 1970s–1980s of the last century were based on metal oxides (titanium, zirconium, aluminum) and their “mixtures” with silica (Teichner 1986) and on synthetic polymers [resorcinol–formaldehyde (Pekala 1989), polyurethane (Biesmans et al. 1998), polyimide (Meador et al. 2015), etc.] and their hybrids with silica (Maleki et al. 2014). Polymer aerogels showed improved mechanical properties, as compared to silica ones, some possessed very low thermal conductivity and interesting electro-chemical properties when pyrolysed. For more information on silica and synthetic polymer aerogels the reader is advised to consult Aerogels Handbook (Aegerter et al. 2011).
A new generation of aerogels appeared at the beginning of the twenty first century: they are biomass based, mainly polysaccharide-based, and are thus called bio-aerogels. Their synthesis is inspired by that of classical aerogels, from polymer dissolution to solution gelation (in some case this step can be omitted which is one of the specificities of polysaccharide aerogels) followed by solvent exchange and drying with supercritical carbon dioxide. Compared to silica aerogels which are extremely fragile, bio-aerogels do not break under compression, with plastic deformation up to 80% strain before pore wall collapse (Sescousse et al. 2011a; Rudaz et al. 2014; Pircher et al. 2016). Bio-aerogels are of low density, 0.05–0.2 g/cm3, and rather high specific surface area, from 200 to 600 m2/g. It seems that the latter strongly depends on the type of polysaccharide but why and how is an open question.
The preparation of bio-aerogels does not involve any toxic components. This makes bio-aerogels “human-friendly” and thus very attractive in life-science applications such as matrices for controlled release and scaffolds (García-González et al. 2011; Veronovski et al. 2014). Bio-aerogels also possess properties similar to synthetic polymer and inorganic aerogels: some are with thermal superinsulating properties (Rudaz et al. 2014; Groult and Budtova 2018a) (but cellulose aerogels are not as it will be shown in “Overview on cellulose II aerogels structure and properties” section), some can be used as matrices for catalysis (Chtchigrovsky et al. 2009), in electrochemical applications when pyrolyzed (Budarin et al. 2006; Guilminot et al. 2008) and for adsorption and/or separation (Quignard et al. 2008).
The number of publications on polysaccharide-based aerogels strongly increased the past 10 years. However, not always the term “aerogel” is used for mesoporous material with high specific surface area: for example, in the first publication on starch aerogels in 1995 they were called “microcellular foams” (Glenn and Irving 1995). Cellulose aerogels obtained in 1993 from viscose were simply called “porous cellulose” (Ookuna et al. 1993), which was also the case of a recent publication on cellulose aerogel made from cellulose/ionic liquid solutions (Voon et al. 2016), and also “nanoporous cellulose” (Cai et al. 2009). Cellulose aerogels obtained from cellulose dissolved in direct solvents are sometimes called “aerocellulose” (Gavillon and Budtova 2008) and this term is extended to “aeropolysaccharides” (Rein and Cohen 2011). In our days, the term “aerogel” is sometimes overused as far as porous, but not necessarily mesoporous materials, are called “aerogels”. This is often the case when a polysaccharide “system” (solution or gel or suspension) is freeze-dried leading to ultra-light but highly macroporous materials, thus with low specific surface area. The latter should be named “foams”, as suggested for nanocellulose based low-density materials (Lavoine and Bergstrom 2017). Two excellent recent reviews on nanocellulose gels, aerogels and foams summarise their physical and chemical properties, functionalization routes and potential applications (Lavoine and Bergstrom 2017; De France et al. 2017). A chapter on cellulose I and cellulose II various porous materials makes an overview of the influence of processing conditions on materials’ properties and potential applications (Liebner et al. 2016).
The goal of this review is to focus on cellulose aerogels obtained via dissolution route only, i.e. cellulose II based aerogels and their composites. Only dry lightweight cellulose II with certain mesoporosity, i.e. specific surface area higher than around 100 m2/g, will be considered. This is usually the case when drying is performed with supercritical CO2. Few exceptional cases when other types of dryings, lyophilisation or ambient pressure/low vacuum drying, lead to the elevated specific surface area, will also be briefly presented.
The preparation pathways, structure, properties and potential applications will be analysed and discussed together with some problems and challenges. Despite a certain number of publications on cellulose II aerogels there are still more open questions than clear trends. While the topic “cellulose II aerogels” may look narrow, it contains several fundamental questions, such as the understanding of structure formation during cellulose coagulation. Thanks to drying with supercritical CO2, which keeps reasonably intact the morphology of “wet” cellulose, the latter can be “seen” and analysed. The understanding of the correlations between structure formation, aerogel morphology and properties is the key in the successful development of cellulose II aerogels’ applications which are now mainly at the level of trials and errors.
The review is structured as follows. First, the general pathways in the preparation of cellulose II aerogels are presented, together with characterization methods. Then, “case studies” provide more details on cellulose II aerogels made from different solvents; their main properties are summarized in Table S1 of the Supporting Information. The next section compares structure and properties of aerogels made via different pathways. Finally, potential applications are presented and discussed.
Preparation pathways, mechanisms of structure formation and characterization of bio-aerogels
In this section, the general principles of bio-aerogel preparation are presented, the majority being applicable to cellulose II case. The main differences with other polymer and inorganic aerogel synthesis pathways are discussed. The mechanisms of aerogel structure formation are suggested. The methods for bio-aerogel shaping, drying and characterization are presented.
Overall approach in making bio-aerogels and mechanisms of structure formation
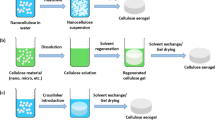
Synthesis pathways for bio-aerogels are schematically presented in Fig. 1. For simplicity we will call “cryogels” those that are obtained via freeze-drying and “xerogels” via ambient pressure or low vacuum drying; they are shown for having a complete overview of options and will be discussed in “Shaping, kinetics of solvent exchange and drying” section. An illustration of samples of cellulose aerogel precursor (or “wet” network with water in the pores, often called “cellulose hydrogel”) together with cellulose cryo-, aero- and xerogel made from the same solution, is presented in Fig. 2.

Schematic presentation of bio-aerogels synthesis pathways

Example of “wet” cellulose aerogel precursor and aero-, cryo- and xerogel obtained from 7 wt% cellulose/1-ethyl-3-methylimidazolium acetate/dimethyl sulfoxide solutions. For more details see (Buchtova and Budtova 2016). Reprinted by permission from: [Springer] [Cellulose] [Buchtova N, Budtova T (2016) Cellulose aero-, cryo- and xerogels: towards understanding of morphology control. Cellulose 23:2585–2595], [2016]
Contrary to inorganic and synthetic polymer aerogels, the starting matter in bio-aerogels is not a solution of monomers or a colloidal suspension, but a solution of “ready” polymers, here, polysaccharides. No polymerization step is involved unless composite or hybrid aerogels are made involving a second component (organic or inorganic) polymerized inside polysaccharide network. For cellulose II aerogels it is the case, for example, of cellulose/silica interpenetrated aerogel network.
As follows from the name “aerogel”, it is made by replacing the solvent in a gel by air. If willing to remove the solvent and preserve mesoporosity, drying with supercritical CO2 is recommended. Because in most of the cases the solvent of polysaccharide, often aqueous, is immiscible with CO2 (except when aerogels are based on cellulose esters soluble in acetone), the solvent should be replaced by a liquid which is miscible with both, solvent and CO2. Acetone and alcohols are often used for this purpose, all being non-solvents for the majority of natural polysaccharides, including cellulose. As it will be demonstrated in the following, gelation step is not a pre-requisite in the case of aerogels based on polysaccharides, and this is one of the significant differences between bio-aerogels and other organic or inorganic aerogels. It is thus possible to make aerogels when the state of the matter before solvent exchange is either solution or gel, as shown in Fig. 1. In both cases coagulated polysaccharide “wet” network is formed (with non-solvent in the pores), but the mechanisms of structure formation are different.
When the state of the matter before solvent exchange is solution, non-solvent induced phase separation occurs. This process is very similar to the formation of membranes via phase inversion also known as “immersion precipitation”, but drying with supercritical CO2 leads to highly porous open-pore network with thin pore walls. Here another specificity of polysaccharides is manifesting: despite certain volume shrinkage, the macromolecules do not totally collapse under solvent → non-solvent exchange even if they are not gelled. Above polymer overlap concentration a 3D network is formed. Chain rigidity and formation of polysaccharide networks stabilized by hydrogen bonds are probably the reasons of polymer “resistance” to coagulant. To avoid packing of polymer chains into dense domains, solvent → non-solvent exchange is usually performed in a gradual way, by slowly increasing the fraction of non-solvent. The kinetics of phase separation probably plays a certain role in structure formation.
When the state of the matter before solvent exchange is gel (for example, case of alginate or pectin cross-linked with polyvalent metal ions or aged cellulose/(7–9)%NaOH-water), the structure of future aerogel network is already pre-formed. Solvent → non-solvent exchange and drying with supercritical CO2 do not seem to strongly affect gel morphology. The examples of different aerogel morphologies obtained from gelled and non-gelled pectin solutions are shown by Groult and Budtova 2018b. For example, aerogels from non-gelled pectin solutions are denser (0.1–0.15 g/cm3) and with higher specific surface area (400–600 m2/g) as compared to their gelled counterparts (density 0.05–0.1 g/cm3 and specific surface area 250–500 m2/g) (Groult and Budtova 2018b).
Contrary to most of polysaccharide-based aerogels, the pathway to make cellulose II aerogels has been, till now, via non-solvent induced phase separation, i.e. without solution physical or chemical gelation. This is probably due to the traditions developed in processing of cellulose from solutions: spinning fibers and casting films are made by direct coagulation or regeneration of cellulose in a non-solvent (usually water). Another reason is that except cellulose/(7–9)%NaOH/water solutions that are spontaneously gelling with time and temperature increase, gelling cellulose solutions is not as easy as gelling other polysaccharides such as alginate, pectin or carrageenan which need just a change of solution pH or addition of metal ions, or of aqueous starch pastes which are gelling during retrogradation.
Shaping, kinetics of solvent exchange and drying
Shaping
Drying with supercritical CO2 preserves the shape of aerogel precursor, i.e. of “wet” polysaccharide network with non-solvent in the pores (Fig. 1). Shaping of bio-aerogels is thus fully governed by shaping of polysaccharide solution before solvent exchange, either via gelation or phase separation route. Both approaches are well known and depend on the type of polysaccharide used and processing conditions such as polymer concentration and molecular weight, solution viscosity, potentially surface and/or interfacial tension (for example, in the case of making beads), temperature, pH and presence of ions or co-solutes. It is thus possible to make bio-aerogels in the shape of monoliths of different forms, beads, fibers and films. This opens a lot of prospects in using 3D printing technique for making bio-aerogels of various and complex shapes which can be very attractive for bio-medical applications such as scaffolds and wound dressings.
Till now the majority of bio-aerogels are made in the form of monoliths and beads (particles); to form fibers and films is possible but is a bit challenging from the point of view of aerogel mechanical properties. Making monoliths is easy and this is what is done in most of laboratory trials: monolithic bio-aerogel takes the shape of the container in which solution was gelled or coagulated. Monoliths allow easy determination of density and testing mechanical properties (usually uniaxial compression of cylindrical samples). In some cases bio-aerogel disks are made to study the release of active substances.
Two main ways of making bio-aerogel beads have been used till now: by dropping a solution in a gelation or coagulation bath and using emulsion technique (Ganesan et al. 2018). As well as “wet” polysaccharide gel particles, bio-aerogels in the form of beads can find applications in various fields such as food, cosmetics, medical, pharma, sorption and separation. Particle size may vary from few microns (usually in the case of emulsion technique) to few millimeters (dropping) and depends on the shaping method used and solution parameters. As compared to monoliths, the whole process efficiency is strongly increased in the case of beads because each processing step (solvent exchange, drying) is diffusion controlled.
Bio-aerogel beads were made by dropping solution either in a very simple way, i.e. using a syringe or pipette (Quignard et al. 2008; Veronovski et al. 2014) or by breaking solution jet (prilling, as shown by De Cicco et al. 2016). For “easy-gelling” polysaccharides, their solutions are dropped in a bath in which a droplet would gel. This is the case when pectin or κ-carrageenan or alginate solution is dropped in a bath containing polyvalent metal salt which induces quick formation of a gelled layer on the droplet surface, stabilizing droplet shape.
Emulsion technique can also be applied to the same “easy gelling” polysaccharides. The classical approach is to disperse aqueous polymer solution in oil phase containing a surfactant. The system is emulsified and polysaccharide droplet is gelled due to an external input (addition of metal salts in the case of pectin or κ-carrageenan or alginate solution (Quignard et al. 2008; Veronovski et al. 2014), or temperature decrease for starch solutions (García-González et al. 2012).
All said above can be partly applied to cellulose aerogels keeping in mind that cellulose solutions are not “easy-gelling”, except the case of cellulose–(7–9)%NaOH/water. The shape of cellulose II aerogel precursor is thus usually stabilized during solvent → non-solvent exchange. Some examples of cellulose aerogels in the shape of monoliths, beads and fibers are shown in Fig. 3. Monoliths are obtained either from gelled solutions (here, from cellulose/8%NaOH/water) or from direct solvent → non-solvent exchange when cellulose solvents are ionic liquids (Fig. 3) or alkali/water (NaOH or LiOH, with urea and/or ZnO added). A special case, different from other bio-aerogels, is when the shape is given during solution solidification (not to be confused with gelation) due to temperature decrease down to room conditions. This happens when cellulose solvents are N-methylmorpholine-N-oxide monohydrate (NMMO), zinc chloride hydrate (ZnCl2·6H2O) and calcium thiocyanate (Ca(SCN)2·6H2O). Sometimes these solutions are called “melts” as they have to be prepared and processed at elevated temperatures; they are of rather high viscosity and thus resemble polymer melts. Some ionic liquids, such as 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]), and their cellulose solutions are also solid at room temperature. Cellulose aerogels in the form of fibers were made by extruding hot cellulose/calcium thiocyanate solution into ethanol (Fig. 3) (Karadagli et al. 2015).

Examples of bio-aerogel monoliths, fibers and beads made from: (1) gelled cellulose/8%NaOH/water. Reprinted with permission from (Gavillon R, Budtova T (2008) Aerocellulose: new highly porous cellulose prepared from cellulose–NaOH aqueous solutions. Biomacromolecules 9:269–277). Copyright (2007) American Chemical Society. (2) Gelled cellulose/organosolv lignin/8%NaOH/water. Reprinted by permission from [Springer], [Cellulose], [Sescousse R, Smacchia A, Budtova T (2010) Influence of lignin on cellulose-NaOH-water mixtures properties and on Aerocellulose morphology. Cellulose 17:1137–1146], [2010]. (3) Extruded hot cellulose/calcium thiocyanate fibers. Reprinted from Karadagli I, Schulz B, Schestakow M, Milow B, Gries T, Ratke L (2015) Production of porous cellulose aerogel fibers by an extrusion process. J Supercrit Fluids 106:105–114, Copyright 2015, with permission from Elsevier. (4) Beads made with JetCutting technique from 2% (4a, b, c) and 3% (4d) cellulose/5-diazabicyclo[4.3.0]non-5-enium propionate solution. Reproduced from Druel L, Niemeyer P, Milow B, Budtova T (2018) Rheology of cellulose-[DBNH][CO2Et] solutions and shaping into aerogel beads. Green Chem 20:3993–4002, with permission from The Royal Society of Chemistry. (5) Particles of various shapes made by syringe-dropping of non-gelled cellulose/8%NaOH/water solutions. Reprinted by permission from: [Springer] [J Mater Sci] [Sescousse R, Gavillon R, Budtova T (2011b) Wet and dry highly porous cellulose beads from cellulose–NaOH–water solutions: influence of the preparation conditions on beads shape and encapsulation of inorganic particles. J Mater Sci 46:759–765], [2010]
Cellulose in the shape of beads is known since long time for using in various applications (immobilization, purification, separation and filtration purposes). In most cases cellulose beads are either never dried, with water in the pores, or, if dried, it is done at ambient pressure which results in a non-porous material. The techniques used to make beads, when cellulose is dissolved either in a direct solvent or via derivatization/regeneration route, are by dropping solution with a syringe (Sescousse et al. 2011b; Trygg et al. 2013, 2014; Mohamed et al. 2015; Voon et al. 2016), with atomizers (De Oliveira and Glasser 1996; Rosenberg et al. 2007) and using emulsion method (Luo and Zhang 2010; Lin et al. 2009a; Zhang et al. 2018). Various ways of production of cellulose beads are summarized in a recent review (Gericke et al. 2013).
Only few publications report on cellulose aerogel beads, and the majority is made with syringe-dropping method from cellulose dissolved in alkali solvents. Using 7%NaOH/12%urea/water solvent, beads were produced via dropping in aqueous non-solvent, and their size and shape were varied by modifying coagulation conditions (bath temperature, from 5 to 50 °C, and concentration of HNO3, from 0.5 to 10 M): particles’ volume varied from 8 to 20 mm3, and circularity was mainly influenced by bath temperature with more deformed particles obtained at lower temperature (Trygg et al. 2013). ZnO of different concentration (from 0 to 2%) was added to the same solvent and beads were formed by dropping in 2 M HCl; their diameter was from 2 to 2.5 mm which increased with the increase of ZnO concentration (Mohamed et al. 2015). Authors suggest that higher ZnO concentration better preserves beads from shrinking. 8%NaOH/water without additives was also used to make aerogel beads via dropping method (Sescousse et al. 2011b). It was shown that by varying solution viscosity, distance between the syringe tip and coagulation bath and bath temperature, different shapes, from very flat plates to spheres, can be obtained (Fig. 3).
Ionic liquids, being powerful cellulose solvents, were also used for making cellulose aerogel beads. Contrary to NaOH/water based solvents, ionic liquids allow dissolution of cellulose in a large range of concentrations and molecular weights. Solution viscosity can additionally be varied by so-called co-solvents such as dimethyl sulfoxide (DMSO) or dimethyl formamide (DMF). Voon et al. (2016) report on making cellulose aerogel beads from cellulose/1-allyl-3-methylimidozoium chloride ([Amim][Cl]) solution by dropping it into water with a syringe. Particles’ diameter was from 0.4 to 2.2 mm and, as expected, the size increased with the increase of needle nozzle diameter. Surprisingly, specific surface area decreased, from 500 to 100 m2/g, with the increase of particle size. An opposite influence of cellulose aerogel geometrical dimensions was reported by Karadagli et al. 2015, where Ca(SCN)2·6H2O was used to make aerogels in the shape of monoliths and fibers. While the density of aerogels did not depend on sample shape and size, specific surface area was lower in fibers as compared to monoliths.
Recently, jet-cutting technology, that can be easily scaled up, was used to make cellulose aerogel beads (Druel et al. 2018). Contrary to “water jet-cutter machine” which is cutting the material, it is the jet of liquid (here, polymer solution) which is cut with high speed rotating wires. Liquid spheres are formed in the air due to surface tension; they are then collected into a bath. This method is developed by GeniaLab (Germany) and used to make “easy-gelling” polysaccharide gel beads. Cellulose beads from cellulose dissolved in ionic liquid 5-diazabicyclo[4.3.0]non-5-enium propionate ([DBNH][CO2Et]) were made with this technology and collected in water, ethanol and isopropanol baths. Cellulose aerogel beads were with mean diameter from 0.5 to 1.8 mm (Fig. 3), density around 0.04–0.07 cm3/g and specific surface area around 240–300 m2/g. They had the same density and specific surface area as the majority of their monolithic counterparts obtained from ionic liquids and other solvents. The rheological properties of “cut” solutions were demonstrated to be crucial for making cellulose aerogel beads with JetCutting method (Druel et al. 2018).
Kinetics of solvent exchange
Whatever the mechanisms of structure formation, gelation or phase separation, and the method of shaping into a “wet” network, the next processing steps are the same for all bio-aerogels: replacing solvent by a fluid miscible with CO2 and drying (see Fig. 1). Solvent in cellulose solutions and gels is usually washed out by water or ethanol or acetone, rarely by isopropanol. If water is used, it is then replaced by ethanol or acetone that are miscible with CO2. All exchanges are diffusion controlled processes and are thus rather slow. Time needed for cellulose solvent to diffuse out and non-solvent to diffuse in depends on cellulose concentration, sample shape and bath temperature (Fig. 4). Higher is bath temperature and lower cellulose concentration, higher is diffusion coefficient, as expected. Roughly, diffusion coefficient is proportional to sample thickness in power 2; to wash out cellulose solvent from a thick monolithic sample takes several days. In order to calculate solvent diffusion coefficient, size changes due to “wet” network shrinkage during solvent exchange should also be taken into account (Sescousse and Budtova 2009).

Diffusion of NaOH from 5 wt%cellulose/7.6% NaOH/water gels into water bath (t is time, l is sample half-thickness) at (1) 25, (2) 50 and (3) 80 °C. The lines are shown to guide the eye. Reprinted with permission from Gavillon R, Budtova T (2007) Kinetics of cellulose regeneration from cellulose-NaOH-water gels and comparison with cellulose-N-methylmorpholine-N-oxide-water solutions. Biomacromolecules 8:424–432. Copyright 2007 American Chemical Society
For the systems used to make cellulose aerogels, the kinetics of solvent → non-solvent exchange (or of cellulose coagulation) was studied for cellulose/NMMO solutions (solid solutions) (Laity et al. 2002; Biganska and Navard 2005), cellulose/8%NaOH/water solutions and gels (Gavillon and Budtova 2007; Sescousse and Budtova 2009) and cellulose/imidazolium ionic liquid solutions (Sescousse et al. 2011a; Hedlund et al. 2017). In all cases cellulose solvent was replaced by water. Overall, it was shown that the process is governed by Fick diffusion. When the release of NaOH from cellulose solution and from gel of the same cellulose concentration was compared, it turned out that diffusion is faster from a gel (Sescousse and Budtova 2009). The reason is that the structure in cellulose gels is rather heterogeneous (they are opaque due to micro-phase separation), with pores being much larger than the size of the diffusing solvent molecule. Local cellulose concentration in “gel pores” is thus lower as compared to a homogeneous solution, making diffusion from the gel faster.
The interactions between cellulose solvent and non-solvent may influence the kinetics of solvent exchange and should also be taken into account. This is the case of cellulose/ionic liquid solutions when placed in water. For example, it was shown that 1-ethyl-3-methylimidazolium acetate ([Emim][OAc]) and water are interacting, with reaction being exothermal and mixture temperature exceeding room temperature by several tens of °C (Hall et al. 2012). Viscosity and diffusion coefficients (measured by NMR) in [Emim][OAc]/water mixtures are several hundred per cent higher than those predicted by the mixing rule (Hall et al. 2012). This can change the overall duration of solvent exchange and, potentially, the morphology of the corresponding aerogels.
Drying
The final step in making aerogels is drying (Fig. 1). While network morphology is stabilized either during gelation or non-solvent induced phase separation, drying is critical to keep the morphology as much intact as possible. The main goal is to avoid pores’ collapse due to capillary pressure. If willing to keep mesoporosity and avoid pores’ chemical treatment to increase the contact angle, drying should be done when the liquid in the pores is in supercritical state in which no meniscus is formed (Fig. 5).

Phase diagram with various ways of drying. Courtesy of C. Rudaz (Rudaz 2013)
Fluid in the supercritical state has diffusivity comparable to that of gases, density in-between gas and liquid and high solvation power. Being discovered in the first half of the nineteenth century, supercritical fluids are now used in various applications such as separation and extraction, in polymer processing due to plasticizing effect and for foaming, in chemical and biochemical reactions, “cleaning” in microelectronics and also for drying when making aerogels and samples for scanning electron microscopy (Knez et al. 2014). Using supercritical fluids involves high-pressure technology (see critical point pressure in Table 1) which has some drawbacks; however, low viscosity, high diffusivity and solvation properties can counterbalance high-pressure disadvantage. For aerogels, CO2 is the easiest solution to be used for drying as it has mild critical point temperature and pressure (Table 1), is chemically inert, non-flammable, non-toxic and cheap. As far as bio-aerogels are concerned, obviously neither water nor acetone or ethanol can be used because of their high critical point temperature; water in supercritical state has, in addition, oxidizing properties.
Other ways of making 3D porous polysaccharide-based materials are also possible, but most of drying methods do not lead to a mesoporous matter, i.e. with high specific surface area. Figure 1 shows the options of drying via lyophilisation (or freeze-drying) and via ambient pressure or low vacuum drying. The terms “cryogel” and “xerogel” are used here for simplicity: strictly speaking, “cryogels” correspond to a matter that is gelling under freezing or storage in the frozen state or under thawing (Lozinsky et al. 2003). This is the case of some polysaccharides such as agarose (Lozinsky et al. 2008). However, the term “cryogels” is often used when water is sublimated from a frozen aqueous system which is also known as ice-templating. However, if no special precautions are taken to decrease the growth of ice crystals, “bio-cryogels” are usually open-pores networks with very low density, very large pores of the size of microns up to several hundreds of microns, rather thick and often non-porous walls and low specific surface area. In the case of cellulose II, water is frozen and sublimated from so-called cellulose “hydrogel” (3D network of coagulated cellulose with water in the pores) (Buchtova and Budtova 2016), and in the case of cellulose I water is sublimated from nanocellulose suspension.
To tune the morphology of “bio-cryogels” the control of the kinetics of ice crystal growth is crucial. This can be done either by spray-freeze-drying which allows fast freezing in sub-micron size pores, or by using mixed solvents (Guizard et al. 2014). Spray-freeze-drying was applied to make “cellulose aerogels” (using the terminology of authors) (Cai et al. 2014; Jiménez-Saelices et al. 2017) from nanofibrillated cellulose resulting in material with specific surface area 80–100 m2/g (Jiménez-Saelices et al. 2017) and 390 m2/g (Cai et al. 2014). It is supposed that this method is not easy to apply for making cellulose II “cryogels” as far as the network is already formed during cellulose coagulation in water and spraying, even if done on mechanically weak wet precursors, will lead only to the macroscopic breakage of the sample. As for using mixed solvents, the most popular way to make “bio-cryogels” with certain mesoporosity is freeze-drying from tert-butanol(TBA)/water (Borisova et al. 2015): for example, pectin “cryogels” of density from 0.044 to 0.144 g/cm3 and specific surface area from 128 to 280 m2/g were made via freeze-drying from TBA/water of various compositions. The lowest density was obtained for samples freeze dried from pure water and the highest when TBA/water was at the composition corresponding to the first eutectic point of this mixture. Freeze-drying from TBA resulted in high specific area of cellulose II, 260–330 m2/g (Hwang et al. 2018). The same mixed solvent was used to make nanofibrillated cellulose “nanopaper” (terminology of authors) with specific surface area from 45 to 117 m2/g (Sehaqui et al. 2011) and esterified nanocellulose “aerogel” (terminology of authors) with specific surface area from 100 m2/g to 180 m2/g (Fumagalli et al. 2013, 2015). Other solvents used for freeze-dried, such as 1,1,2,2,3,3,4-heptafluorocyclopentane, also result in rather high specific surface area 190–210 m2/g (Wang et al. 2012).
The term “xerogel” strictly means “a dry gel”, but it is traditionally employed for meso- and microporous systems, with porosity up to 50%, dried at ambient pressure or low vacuum. An example of xerogels is silylated silica gel dried at ambient pressure and around 130–150 °C; it has the internal structure similar to silica aerogels dried with supercritical CO2. Such silica xerogels are with high specific surface area (500–1000 m2/g) and low density (around 0.1–0.2 g/cm3) and are sometimes called “ambient pressure dried aerogels”.
Very few works report on low density cellulose “xerogels” and most of them are with rather low specific surface are; capillary pressure developing during evaporative drying coupled with hydrogen bonding between polysaccharide chains usually lead to network collapse resulting in a non-porous material. One way to decrease pore closing during drying is to use fluids with surface tension lower than that of water (0.073 N/m): ethanol (0.022 N/m), acetone (0.0237 N/m), hexane (0.0184 N/m), methanol (0.0226 N/m) or pentane (0.0158 N/m). This approach was applied to obtain open-pores cellulose sheets: water was replaced first by methanol, then acetone and finally pentane, and then samples were dried overnight by evaporation under forced convection of argon (Svensson et al. 2013). The specific surface area varied from 75 to 130 m2/g. Solvent exchange resulted in high specific surface area of cellulose II as compared to conventional freeze-drying, 150–190 m2/g versus 70–100 m2/g, respectively (Jin et al. 2004).
Another way is to perform cellulose surface hydrophobisation which can be applied to cellulose pulp (Tejado et al. 2014; Köhnke et al. 2010) and nanofibrilated cellulose (Sehaqui et al. 2014). Hydrophobic cellulose nanopaper was with density 0.4–0.6 g/cm3 and specific surface area 40–60 m2/g (Sehaqui et al. 2014). Highly porous nanocellulose foams were obtained via high-pressure homogenisation technique, cellulose caboxymethylation and drying at 60 °C in an oven without convection; pore size was between 300 and 600 μm and density around 0.03 g/cm3 (Cervin et al. 2013). With such size of pores these foams cannot have high specific surface area. Inspired by the approach used for making low density and high specific surface area silica xerogels, trityl cellulose was synthesised via homogeneous reaction and then xerogels were prepared via dissolution-solvent exchange-ambient drying route (Pour et al. 2015). Low density (0.1 and 0.2 g/cm3) hydrophobic xerogels showing contact angle with water 140° were obtained when the degree of substitution was 0.72. Specific surface area was not high, from 13 to 27 m2/g. While bulky trityl groups on cellulose chain prevent, to a certain extent, the formation of intra- and inter-molecular hydrogen bonds during drying and thus lead to xerogels of low density, still chains aggregation occurs during solvent exchange and drying which may explain the absence of mesoporosity.
The analysis and examples of various drying ways presented above show that if having the goal to obtain light-weight and mesoporous cellulose II materials, drying with supercritical CO2 is, till now, the most successful option.
Characterisation of bio-aerogels
The methods used to characterize bio-aerogels are the same as for classical aerogels. However, some features, specific for bio-aerogels, should be taken into account in order not to obtain artefacts. One is high sensitivity of native polysaccharides to humidity and thus capability to adsorb water vapours. For example, the weight of bio-aerogel may increase in room conditions by 10–20 wt% in the case of cellulose aerogels to several tens of wt% for aerogels based on water-soluble polysaccharides. Higher humidity leads to even higher weight increase. As a result, characteristics such as density and thermal conductivity of aged bio-aerogels should increase. An example of three to five fold increase of thermal conductivity with relative humidity increase from 0 to 60% was demonstrated for cellulose II cryogels (Shi et al. 2013a); no data is reported on cellulose II aerogels. Subsequent drying should lead to pores’ irreversible closing which is known for cellulose as “hornification”. This, in turn, may lead to aerogel shrinkage, change of density, morphology and decrease of specific surface area. Bio-aerogel mechanical properties should also depend on aging time. Till now, there is no quantitative analysis of bio-aerogel aging except some simple kinetics of mass and volume uptake by cellulose aerogels as a function of relative humidity (Demilecamps 2015a). Samples’ storage and characterisation should, ideally, be performed in controlled temperature and humidity environment and sample “age” (time from drying to analysis) reported.
Bulk density ρbulk is the first obvious parameter to report for 3D porous materials; it is usually determined by measuring sample mass and dimensions. Powder densitometer, such as Geopyc from Micromeritics with DryFlo powder, is a useful option for samples with geometrically complex shapes (Rudaz et al. 2014). Powder densitometer measures sample volume by using different chamber volumes and tapping forces. Because bio-aerogels are deformable and compressible, the conditions should be very carefully selected in order to avoid volume decrease during measurement. Skeletal density ρskeletal of polysaccharides is known to be 1.5–1.7 g/cm3.
Scanning electron microscopy (SEM) is a very useful tool to visualize aerogel morphology, however SEM cannot be used to quantify it. Specific surface area SBET and pore size distribution are the main parameters characterizing aerogel texture. As for classical aerogels, specific surface area of bio-aerogels is determined using nitrogen adsorption technique and Brunauer–Emmett–Teller (BET) theory. It should be noted that standard methods for measuring pore volume and size distribution using Barrett–Joyner–Halenda (BJH) approach (via nitrogen adsorption) or mercury porosimetry cannot be applied to the majority of bio-aerogels. Bio-aerogels possess macro- and mesopores, and are often with large macropores (several hundreds of nanometers up to several microns). BJH method mainly considers mesopores and small macropores (below 200 nm), which takes in account only 10–20% of the total pore volume in bio-aerogels (Robitzer et al. 2011; Rudaz et al. 2014; Jiménez-Saelices et al. 2017; Groult and Budtova 2018a). For example, mesopore volume in bio-aerogels is usually around 0.5–2.5 cm3/g while total pore volume Vpores calculated from bulk ρbulk and skeletal densities ρsk (Eq. 1) can reach several tens of cm3/g due to macroporosity (Robitzer et al. 2011; Rudaz et al. 2014; Groult and Budtova 2018a):
Pore size distributions in bio-aerogels are clearly not limited to mesopores region. It may also be possible that bio-aerogel is compressed at higher nitrogen pressure. If not keeping in mind the limitations of BJH method applied to bio-aerogels, the values provided by equipment with inserted programs may lead to a wrong understanding of bio-aerogel morphology. When mercury porosimetry is used, bio-aerogels are often compressed without mercury penetration in the pores, and thus the “value” given by the machine is an artefact (Rudaz 2013; Rudaz et al. 2014). Imaging, such as SEM or 3D tomography, provide only qualitative ways to estimate pore sizes: in the former, no automatic image analysis is available yet to analyse complex bio-aerogel morphology and the latter does not allow the analysis of mesopores.
Thermoporosimetry was suggested to determine pore size distribution; this method was applied to cellulose II aerogels (Pircher et al. 2015, 2016). The approach is based on the measurement of the experimental shift of the melting point of an interstitial liquid caused by its confinement in small pores (Gibbs–Thomson equation). Cellulose aerogels were soaked in o-xylene and crystallization temperatures were recorded using differential scanning calorimeter. Till now, there are only two examples of using thermoporosimetry for the characterization of pore size distribution in bio-aerogels. It provides a reasonable correlation with cellulose aerogel morphology seen by SEM and shows a significant difference with pore sizes predicted by BJH method.
Cellulose II aerogels: case studies
For making cellulose II aerogels, two main ways of cellulose dissolution should be considered, either via cellulose derivatization followed by regeneration or in direct solvents. In the latter case no “regeneration” per se occurs, and thus the process of cellulose “recovery” from solution will be called “coagulation” (or precipitation).
When dissolved in direct solvents, cellulose solutions can be “liquid” at room temperature, gelled or solidified. In the next sections cellulose II aerogels will be discussed from the point of view of the solvent used to dissolve cellulose; a special attention will be paid on the state of the matter before solvent → non-solvent exchange. The mechanisms of cellulose dissolution in a particular solvent and solution properties will not be discussed as far as this would make the article infinite; the reader is advised to address an excellent review of Liebert (2010) and other review articles devoted to cellulose dissolved in a specific solvent (for example, Fink et al. 2001 for cellulose/NMMO, Budtova and Navard 2016 for cellulose/NaOH, Pinkert et al. 2009 and Mäki-Arvelaa et al. 2010 for cellulose-ionic liquids). Table S1 of the Supporting Information summarises the properties of cellulose II aerogels divided by the type of solvent, with the chronological order of publications within each solvent family. Some special cases of porous cellulose with high specific surface area obtained via freeze-drying are also presented at the end of this table.
Aerogels from cellulose dissolved via derivatization
Because the research on aerogels and on cellulose was not intersecting in the past except being just briefly mentioned by Kistler (1931), it seems there is only one publication reporting on cellulose aerogels obtained from viscose process (Ookuna et al. 1993). Aerogel beads of the diameter of several hundreds of microns were produced and specific surface area varied from 15 to 400 m2/g (Table S1). These materials, called “porous cellulose”, were suggested to be used as ion-exchangers (Ookuna et al. 1993). Another example which can be placed in the category of dissolution via derivatization is cellulose carbamate: it was synthesized by kneading cellulose in the excess of urea at 130 °C and dissolving in NaOH/water (Pinnow et al. 2008). Monoliths and beads were made, cellulose regenerated, followed by drying in supercritical CO2; some samples were pyrolysed. Neat cellulose aerogels density varied from 0.06 to 0.22 g/cm3 and specific surface area from 360 to 430 m2/g; pyrolysed counterparts’ density and specific surface area were higher, 0.21–0.27 g/cm3 and 490–660 m2/g, respectively (Table S1).
Surprisingly, no other examples of cellulose aerogels synthesized via derivatization route have been reported. Viscose process is known to be not very eco-friendly and complicated to be done on laboratory scale; however, other ways of making cellulose aerogels via derivatization-regeneration route could be interesting to test. One example is making cellulose aerogels by saponification of cellulose acetate gels. The synthesis of cellulose acetate and cellulose acetate butyrate gels and aerogels via chemical cross-linking with isocyanates had already been described (Tan et al. 2001; Fischer et al. 2006), thus cellulose regeneration before drying could, theoretically, be possible. Cellulose acetate butyrate aerogels were reported to possess high impact strength for this type of porous materials, 0.85 Nm (density 0.15 g/cm3, specific surface area 389 m2/g) versus ten times lower value for resorcinol–formaldehyde aerogel of the same density, 0.08 Nm (density 0.15 g/cm3, specific surface area 526 m2/g) (Tan et al. 2001). The synthesis of many other cellulose esters and ethers is well known but was never used to obtain regenerated cellulose aerogels.
Aerogels from cellulose dissolved in direct aqueous solvents
Despite the difficulties in cellulose dissolution, many direct solvents are known (Liebert 2010). Some, but not many, were used to dissolve cellulose for making aerogels. The classical examples are aqueous alkali-based solvents, NaOH and LiOH, which turned out to be the most popular in making cellulose II aerogels. The great majority of work was performed using additives, such as urea, thiourea or ZnO, which improve cellulose dissolution and delay solution gelation. In these cases the state of the matter before solvent → non-solvent exchange was solution.
4.6%LiOH/15%urea/water was used to fabricate cellulose aerogels mainly as a “support” matrix (Table S1): of metal nanoparticles (Cai et al. 2009; Cui et al. 2018), to make interpenetrated cellulose/poly(methyl methacrylate/butyl methacrylate) and cellulose/poly(methyl methacrylate/butyl acrylate) networks (Shi et al. 2015) and composite aerogels with silica (Cai et al. 2012; Liu et al. 2013). In the latter case the specific surface area of composite aerogels was 270–340 m2/g, similar to that of neat cellulose counterpart (320 m2/g). Cai et al. (2008) performed a systematic study of the influence of cellulose concentration, coagulation bath temperature and cellulose solvent, LiOH/urea versus NaOH/urea, on cellulose aerogel properties. It seems that if keeping all processing parameters the same (origin and concentration of cellulose, coagulation bath type and temperature), there is no influence of solvent type on aerogel properties (density around 0.26 g/cm3 and specific surface area 364–381 m2/g) (Cai et al. 2008). Overall, except the increase in density with the increase of polymer concentration, which is expected, other trends are not very clear most probably because of “too many” processing conditions which are not always easy to consider.
(7–9)%NaOH/water was used as cellulose solvent in two ways, either as is (Gavillon and Budtova 2008; Sescousse and Budtova 2009; Sescousse et al. 2010, 2011a, b; Demilecamps et al. 2016), or with additives: urea (Cai et al. 2008; Trygg et al. 2013), thiourea (Chin et al. 2014) or urea/ZnO (Mohamed et al. 2015) (Table S1). It is well known that cellulose/NaOH based solutions are gelling with time and temperature increase (Roy et al. 2003) causing problems for processing (fiber spinning and film casting), and thus additives are used to delay gelation. However, gelation property can be useful for making aerogels of various and easily controlled shapes as far as sample shape remains the same during all processing steps (only volume decreases). Gelation was used, for example, for making cylindrical and disk carbon aerogels for electro-chemical applications (Rooke et al. 2012). It is also known that in NaOH-based solvents it is not possible to dissolve cellulose of high DP and at concentrations above 7–8 wt% (Egal et al. 2007). To make a self-standing aerogel precursor, polymer concentration should be at least two–three times above the overlap concentration which is around 1 wt% for microcrystalline cellulose in this solvent. These constraints on the minimal and maximal cellulose concentrations make the processing interval in NaOH-based solvents rather narrow, decreasing the possibility of varying aerogel structure and properties.
When NaOH/water solvent was used without additives, solutions gelled. Gelation occurs due to cellulose–cellulose preferential interactions via hydrogen bonding resulting in packing of cellulose chains and formation of cellulose-rich domains; gels become opaque indicating entities that are scattering visible light. This heterogeneous morphology with rather large pores and thick pore walls might be the reason of lower specific surface area of aerogels made from gelled solutions, around 200–250 m2/g (Gavillon and Budtova 2008; Sescousse et al. 2010; Demilecamps et al. 2014), as compared to their non-gelled counterparts of similar density but with surface area of 300–400 m2/g when made from NaOH/water solvent with additives (Cai et al. 2008; Trygg et al. 2013; Mohamed et al. 2015) or from LiOH/urea/water (see Table S1). Similar trend was reported for pectin aerogels: specific surface area for aerogels based on non-gelled solutions was more than twice higher than that of their gelled counterparts (Groult and Budtova 2018b).
As well as urea, ZnO also delays gelation, but its low solubility (around 0.5–0.7 wt% at pH 14 which is pH of 8 wt%NaOH/water) and presence of non-dissolved particles if above the solubility limit should be taken into account (Liu et al. 2011). Mohamed et al. (2015) studied the influence of ZnO concentration on the properties of cellulose aerogels. A non-monotonous behaviour of bulk density and specific surface area as a function ZnO concentration was found. The authors speculate that the increase of specific surface area with the increase of ZnO concentration is correlated with the increase of the number of zincate molecules which are swelling cellulose and thus creating small pores (Mohamed et al. 2015). After the maximum solubility of ZnO is reached (around 0.5 wt% ZnO, according to the authors), the presence of undissolved ZnO leads to the decrease of the amount of zincate, which in turn decreases specific surface area. Bulk density of aerogels shows a maximum at 0.4 wt% ZnO (Mohamed et al. 2015).
Aerogels from cellulose dissolved in direct non-aqueous solvents
Non-aqueous cellulose solvents used to make aerogels are NMMO, ionic liquids and molten salt hydrates such as zinc chloride and calcium thiocyanate.
Aerogels from cellulose/NMMO solutions
Lenzing, Austria, was the first to report on cellulose aerogels using NMMO (Firgo et al. 2004; Innerlohinger et al. 2006a, b). The work was performed within EC 6th framework program, “AeroCell” project, which boosted the research on cellulose aerogels and, probably, on bio-aerogels in general. Within AeroCell project aerogels were also made from cellulose dissolved in 8%NaOH/water (Center for Materials Forming, MINES ParisTech, France), cellulose carbamate dissolved in NaOH/water (Fraunhofer IAP, Germany) and cellulose acetate dissolved in acetone and chemically cross-linked (Centre for processes, renewable energies and energy systems, MINES ParisTech, France). For aerogels based on cellulose dissolved in NMMO, bleached, unbleached and cotton linter pulps were used (Innerlohinger et al. 2006a, b). Samples of various shapes were prepared either by solidifying cellulose/NMMO solution in moulds of different forms or by dropping hot solution in water. Because of large amount of different starting parameters (cellulose DP and concentration, type of pulp, way of structure formation (from solid or liquid solution), type of non-solvent) it was difficult to build correlations except few evident ones such as the increase in aerogel density with the increase of cellulose concentration, as already mentioned for aerogels made from cellulose/alkali solutions. Interestingly, specific surface area of aerogels made via dropping of hot solutions in water bath was the highest (300–350 m2/g) as compared to aerogels prepared from solidified solutions (below 250 m2/g) (Innerlohinger et al. 2006a, b). For cellulose/NMMO solutions it is known that it is free solvent which is crystallising at room temperature leading to “pre-forming” of the morphology of future aerogel, as in the case of cellulose solution gelation. This confirms the hypothesis that aerogels with higher mesoporosity are formed via direct non-solvent induced phase separation.
Further work on cellulose aerogels from NMMO solutions was continued in the group of Falk Liebner (BOKU, Austria) (Liebner et al. 2008, 2009, 2012; Pircher et al. 2016). The majority of the initial solutions were of 3 wt% cellulose (cotton linters, various pulps) resulting in aerogels of density 0.05–0.06 g/cm3 and specific surface area 200–300 m2/g (Table S1); pulp type and cellulose molecular weight (from 80 to 665 kg/mol) did not seem to influence either density or specific surface area (Liebner et al. 2009). It was reported that solvent exchange directly with ethanol (NMMO → ethanol), as compared with two-step exchange to water and then to ethanol (NMMO → water → ethanol), leads to lower aerogel density, 0.06 versus 0.09 g/cm3, respectively (Liebner et al. 2008).
Aerogels from cellulose/ionic liquid solutions
Since ionic liquids became in the focus of cellulose research as the medium for cellulose derivatization and processing at the beginning of the twenty first century, they were also used to make cellulose aerogels. Ionic liquids allow cellulose easy dissolution in a wide range of molecular weights and concentrations, and also the dissolution of lignocellulose and even wood. This opens many ways to perform systematic experiments in order to test and understand processing-structure-properties relationships in cellulose aerogels, and also make aerogels with desired characteristics. Still the research is at the beginning of the long way and a lot of questions remain. For example, the highest value of specific surface area ever obtained for cellulose aerogels, 539 m2/g, was for aerogel prepared from bleached softwood Kraft pulp dissolved at 1.5 wt% in [Bmim][Cl] (Aaltonen and Jauhiainen 2009). Other high surface area values for aerogels from cellulose solutions in imidazolium-based ionic liquids are for aerogels from waste paper, 478 m2/g (Voon et al. 2017) and from eucalyptus pulp with maximum specific surface area 350 m2/g (Wang et al. 2013a) (Table S1). The addition to cellulose (from bleached softwood Kraft pulp) of lignin and xylan, or their presence in spruce wood, strongly decreased specific surface area from 539 m2/g to 210–220 m2/g and 122 m2/g, respectively (Aaltonen and Jauhiainen 2009). Other works report aerogels obtained from cellulose/ionic liquids with densities and specific surface areas similar to those from other solvents: 0.05–0.2 g/cm3 and 130–300 m2/g (Table S1) (Tsioptsias et al. 2008; Sescousse et al. 2011a; Pircher et al. 2015, 2016; Demilecamps et al. 2015; Buchtova and Budtova 2016).
Aerogels from molten salt hydrates
Zinc chloride (ZnCl2·6H2O) and two options of calcium thiocyanate, Ca(SCN)2·6H2O and Ca(SCN)2·8H2O/LiCl, were used to dissolve cellulose and make aerogels (Table S1). Rege et al. (2016) report that within the same interval of cellulose concentrations in solution, from 1 to 5 wt%, the density of cellulose aerogels made from zinc chloride solutions are several times higher than that from Ca(SCN)2·6H2O solutions (Table S1). As a consequence of higher density, Young’s moduli of aerogels from zinc chloride route are much higher than those from Ca(SCN)2·6H2O, in the same range of initial cellulose concentrations, 2–10 MPa versus 5–95 MPa, respectively (Table S1). Cellulose/ZnCl2·6H2O solutions were coagulated in isopropanol and cellulose/Ca(SCN)2·6H2O in ethanol (Rege et al. 2016) which may influence aerogel properties. Indeed, in another work the same team reported that aerogels of the same density obtained from cellulose/ZnCl2·6H2O and coagulated in isopropanol possess Young’s modulus almost twice higher than that when coagulated in ethanol (Schestakow et al. 2016a).
Ca(SCN)2·6H2O and Ca(SCN)2·8H2O/LiCl were used to make cellulose aerogels of dual porosity using porogens, either oil or polymethylmethacrylate solid spheres (Pircher et al. 2015; Ganesan et al. 2016). As expected, the presence of large pores remaining after leached out porogens led to density and Young’s modulus decrease as compared to reference (without porogens) aerogels.
Overview on cellulose II aerogels structure and properties
In this section the analysis of general trends of processing-structure-properties correlations for cellulose II aerogel is performed. Because of a huge number of parameters used to prepare cellulose aerogels an adequate comparison is rather challenging. The main parameters, corresponding to each preparation step, are as follows (see Fig. 1): cellulose origin and presence of other components (hemicellulose, lignin), molecular weight and concentration in solution; type of solvent and presence of additive(s) or co-solvent(s); mechanism of structure formation (via gelation or solidification or non-solvent induced phase separation); type of non-solvent, bath temperature, solvent/non-solvent interactions and way of solvent exchange (gradual or not); parameters of supercritical drying (temperature, pressure, pressurization and depressurisation rate) and, finally, samples’ aging. Considering, in addition, that not all publications provide comprehensive information on aerogel preparation, the understanding and prediction of cellulose II aerogel structure and properties is not an easy task.
Volume change during processing and cellulose II aerogel density
Those who are involved in making bio-aerogels noticed that sample volume decreases from the initial solution to final aerogel, and this is also the case for cellulose II aerogels. Volume shrinkage seems to depend on the type of polysaccharide: for example, for 2–2.5 wt% solutions it is 90–95 vol% for κ-carrageenan while it is 40–50 vol% for chitosan and around 20 vol% for calcium alginate (Quignard et al. 2008). This difference was interpreted by different chain flexibility; low shrinkage of calcium alginate was suggested to be due to the formation of “egg-box” structure during calcium-induced gelation. Rather low shrinkage occurs in nanocellulose based aerogels (Lavoine and Bergstrom 2017). Volume decrease during solvent exchange and drying may look a “too simple” phenomenon to be studied, however, it reflects the fundamental property of polymer chain to change its conformation as a function of external conditions, in particular, in the presence of a non-solvent. Here the mechanism of network structure formation, via gelation or non-solvent induced phase separation, plays a very important role [for example, around 75% volume shrinkage for non-cross-linked versus around 35% for calcium cross-linked pectin aerogels made from 3 wt% low-methylated pectin solutions (Groult and Budtova 2018b)]. A comparison with synthetic polymers of different flexibility would be very interesting.
As far as cellulose II aerogels are concerned, volume shrinkage was reported to depend on cellulose concentration in solution and type of non-solvent (Innerlohinger et al. 2006a; Sescousse and Budtova 2009; Schestakow et al. 2016a; Buchtova and Budtova 2016). Shrinkage occurs at both solvent exchange and drying steps. The reason for the first one is clear: from being in solution, macromolecules tend to decrease their volume in a non-solvent. The extent of this decrease may depend on if the polymer is “stabilized” in a network or not (see the case of pectin mentioned above), but this was never systematically studied for cellulose. Volume decrease during drying with supercritical CO2 is, somehow, “against” the theoretical prediction which states that shrinkage should be zero as far as capillary pressure is zero. However, CO2 is cellulose non-solvent with very low polarity and very different solubility parameter: 5–8 MPa0.5 for CO2 in supercritical state (Zhang et al. 2017) versus 39 MPa0.5 for cellulose (Hansen 2007). This and certain pressure needed to reach supercritical conditions (around 8–10 MPa) may together be the reasons of volume decrease during drying.
An example of the dependence of volume shrinkage during solvent exchange and drying on cellulose initial concentration in solution is shown in Fig. 6, aerogels were made from cellulose/[Emim][OAc]/DMSO solutions coagulated in ethanol (Buchtova and Budtova 2016). Here major shrinkage occurred at drying step; total volume is better preserved at higher cellulose concentration: shrinkage is around 70 vol% for 3 wt% cellulose in solution versus around 20 vol% for 11 wt% cellulose. Higher cellulose concentration helps mechanically “resisting” solvent exchange and drying. The same trend was reported by other authors (Schestakow et al. 2016a, b; Innerlohinger et al. 2006a): for example, for aerogels made from cellulose/NMMO solutions coagulated in water shrinkage was from around 80–85 vol% for 0.5 wt% cellulose solutions to around 40–50 vol% for 9 wt% solutions (Innerlohinger et al. 2006a).

Volume shrinkage during solvent exchange and total shrinkage after drying for cellulose aerogels as a function of cellulose concentration. Cellulose was dissolved in [Emim][OAc]/DMSO, data taken from Buchtova and Budtova (2016)
The influence of non-solvent type on cellulose shrinkage was demonstrated in by Schestakow et al. 2016a. The comparison was made for all processing conditions being the same, except non-solvent type. Higher volume loss was reported for cellulose coagulated in acetone (60–70 vol%) followed by ethanol and isopraponol (40–60 vol%) and then by water (30–40 vol%). This was interpreted by different solubility of cellulose solvent, zinc chloride tetrahydrate, in the corresponding non-solvent; the highest was in water and the lowest in acetone. Similar trend, i.e. higher shrinkage of cellulose II aerogels made from cellulose/[DBNH][CO2Et] solution was reported when non-solvent was ethanol as compared to water (density 0.04 vs. 0.05 g/cm3, respectively) (Druel et al. 2018). However, when cellulose solvent was NMMO, higher shrinkage occurred when non-solvent was water as compared to ethanol (density 0.09 vs. 0.06 g/cm3, respectively) (Liebner et al. 2008).
Aerogel bulk density is inversely proportional to sample shrinkage if no volume and/or mass loss occurs. The latter may happen when pulp is used as far as hemicelluloses can be washed out during coagulation and washing in water. Bulk density can be compared to the ideal case of no volume change during the preparation steps, from solution to aerogel. The density of “no-shrinkage case” can be taken “equal” to cellulose concentration, in a very rough approximation, as far as the majority of solutions are rather dilute, below 10 wt%. A summary of cellulose II aerogel density as a function of cellulose concentration for different solvents and non-solvents is presented in Fig. 7.

Density of cellulose II aerogels as a function of cellulose concentration in solution, for different solvents and non-solvents; solid line corresponds to the case of no shrinkage and no mass loss. Experimental data are from the following references: ref A: Schestakow et al. (2016a), ref B: Rege et al. (2016), ref C: Buchtova and Budtova (2016), ref D: Sescousse et al. (2011a), ref E: Hoepfner et al. (2008), ref F: Pircher et al. (2016), ref G: Cai et al. (2008) and ref H: Gavillon and Budtova (2008)
As already mentioned in the previous section, the first and obvious trend is that aerogel bulk density increases with the increase of cellulose concentration in solution. More matter is in a given volume, higher is material density. The second trend is that all experimental densities are higher than that calculated for the case of no volume shrinkage. As mentioned above, whatever experimental conditions are, shrinkage occurs during solvent exchange and drying. Except ZnCl2·4H2O, there is no significant influence of solvent or non-solvent type on cellulose II aerogel density. It should be kept in mind that different research groups use experimental conditions (for example, the way of solvent exchange (gradual or not) and drying parameters) that differ one from another; the exact match of experimental values is thus not expected. Finally, density does not seem to linearly increase with cellulose concentration; the most probable reason is the decrease of shrinkage with the increase of concentration.
Much higher bulk density was reported for cellulose aerogels from ZnCl2·4H2O solvent whatever is non-solvent type (Fig. 7). This solvent was used only in two publications and more work is needed to understand this trend. Was cellulose well dissolved? The values of specific surface area, which could indicate the presence of non-dissolved fibers not participating to mesoporosity, are similar to those reported for aerogels made from other solvents. The argument of bad solubility of ZnCl2·4H2O in non-solvent (acetone, as reported in Schestakow et al. 2016a) cannot work here as far as density is high even when water was used as coagulation bath, in which ZnCl2·4H2O is highly soluble.
Morphology and specific surface area
Before discussing the morphology of cellulose II aerogels, a brief overview of the representative morphologies of some classical aerogels based on silica, synthetic polymers and bio-aerogels is presented. The microstructure of silica aerogels is shown in Fig. 8a, b. The difference between two is that Fig. 8a shows the morphology of a classical silica aerogel, with “pearl-necklace” structure (Leventis et al. 2002; Katti et al. 2006), and Fig. 8b corresponds to the morphology of prepolymerised silica sol (Markevicius et al. 2017). Classical silica aerogels consist of a “pearl-necklace” mesoporous network of particles of around 5–10 nm in diameter, connected by “necks” and formed by dissolution and reprecipitation of silica during aging (Leventis et al. 2002). Thin “necks” are the main reason of extremely fragile mechanical properties of silica aerogels. Prepolymerised tetraethyl orthosilicate (TEOS) (Fig. 8b) show more fibrous-like structure (Markevicius et al. 2017); by varying silica concentration and, as a consequence, aerogel density, it was possible to obtain aerogels with various mechanical behaviour (from ductile compaction to elastic deformation and to brittle fracture) (Wong et al. 2014).

Silica aerogels based on: a classical base-catalysed silica (Reprinted with permission from Katti A, Shimpi N, Roy S, Lu H, Fabrizio EF, Dass A, Capadona LA, Leventis N (2006) Chemical, physical, and mechanical characterization of isocyanate cross-linked amine-modified silica aerogels. Chem Mater 18:85–296. Copyright 2006 American Chemical Society) and b prepolymerised oligomers of TEOS (Reprinted by permission from [Springer] [J Mater Sci] [Markevicius G, Ladj R, Niemeyer P, Budtova T, Rigacci A (2017) Ambient-dried thermal superinsulating monolithic silica-based aerogels with short cellulosic fibers. J Mater Sci 52:2210–2221], [2017])
The morphology of various synthetic polymer aerogels based on resorcinol–formaldehyde, polyimide, polyurea and polyurethane is shown in Fig. 9. While some show bead-like structure (resorcinol–formaldehyde and polyurethane, Fig. 9a, d, respectively), polyimide and polyurea are represented by a fibrous network (Fig. 9b, c, respectively).

SEM images of morphology of aerogels based on: a acid-catalysed resorcinol–formaldehyde (Reprinted with permission from Mulik S, Sotiriou-Leventis C, Leventis N (2007) Time-efficient acid-catalyzed synthesis of resorcinol-formaldehyde aerogels. Chem Mater 19:6138–6144. Copyright 2007 American Chemical Society), b polyimide (Reprinted with permission from Meador MAB, Agnello M, McCorkle L, Vivod SL, Wilmoth N (2016) Moisture-resistant polyimide aerogels containing propylene oxide links in the backbone. ACS Appl Mater Interfaces 8:29073−29079. Copyright 2016 American Chemical Society), c polyurea (Reprinted from Weigold L, Reichenauer G (2014) Correlation between mechanical stiffness and thermal transport along the solid framework of a uniaxially compressed polyurea aerogel. J Non Cryst Solids 406:73–78, Copyright 2014, with permission from Elsevier) and d polyurethane (Reprinted from Diascorn N, Calas S, Sallée H, Achard P, Rigacci A (2015) Polyurethane aerogels synthesis for thermal insulation–textural, thermal and mechanical properties. J Supercrit Fluids 106:76–84, Copyright 2015, with permission from Elsevier)
Bio-arogels made by dissolution-solvent exchange route possess net-like morphology, see examples for pectin, alginate and starch aerogels in Fig. 10. They are all “easy-gelling” polysaccharides. One exception of aerogel made from non-gelled low-methylated pectin solution is shown in Fig. 10a: it is much denser (bulk density 0.12 vs. 0.045 g/cm3 for its cross-linked counterpart, Fig. 10b) and with higher specific surface area (550 vs. 400 m2/g, respectively) (Groult and Budtova 2018b).

Morphology of bio-aerogels based on: a pectin non-gelled solution and b pectin gelled with calcium (Reprinted from Groult S, Budtova T (2018b) Tuning structure and properties of pectin aerogels. Eur Polym J 108:250–261, Copyright 2018, with permission from Elsevier), c corn starch (Reprinted from García-González CA, Uy JJ, Alnaief M, Smirnova I (2012) Preparation of tailor-made starch-based aerogel microspheres by the emulsion-gelation method. Carbohydr Polym 88:1378–1386, Copyright 2012, with permission from Elsevier) and d alginate gelled with calcium (Reprinted from Escudero RR, Robitzer M, Di Renzo F, Quignard F (2009) Alginate aerogels as adsorbents of polar molecules from liquid hydrocarbons: hexanol as probe molecule. Carbohydr Polym 75:52–57, Copyright 2009, with permission from Elsevier)
The morphology of cellulose II aerogels shows, for the majority of cellulose solvents, a net-like texture. This is the case of aerogels made from cellulose/alkali, cellulose/ZnCl2·4H2O, cellulose/TBAF/DMSO, cellulose/calcium thiocyanate and solid cellulose/NMMO. The examples are shown in Figs. 11, 12, 13, 14 and 15. There are some exceptions which correspond to the cases when aerogels were made from hot cellulose/NMMO and from cellulose/[Emim][OAc] solutions (Figs. 12b, c, 13a, 14b). These solutions are liquid before solvent exchange, and it was suggested that when such solution is placed in a non-solvent, network structure is formed due to spinodal decomposition mechanism leading to periodic bead-like morphology with beads of the same size (Sescousse et al. 2011a). This is not that evident for aerogels made from other cellulose/ionic liquids solutions (Fig. 13b, c): when cellulose/[Bmim][Cl] was used (solvent is solid in room conditions but authors specified that solutions were not solidified before being placed in non-solvent) (Aaltonen and Jauhiainen 2009), beads, if formed, are of much smaller size as compared to cellulose/[Emim][OAc] or cellulose/NMMO case, and aerogels from cellulose/1-hexyl-3-methyl-1H-imidazolium chloride ([Hmim][Cl]) solutions do not show bead-like morphology (Wang et al. 2013a). Most of cellulose/alkali solutions were not gelled before solvent exchange but aerogels do not show bead-like morphology either. The difference in the morphology of aerogels from gelled and not cellulose/NaOH/water solutions was demonstrated (Demilecamps et al. 2014): gelled solutions resulted in net-like aerogel structure and bead-like morphology was recorded when cellulose was mixed with sodium silicate, both dissolved in 8%NaOH/water. Sodium silicate was inducing cellulose coagulation by competing with common solvent. It should be noted that in NaOH/water based solvents cellulose is not dissolved on the molecular level, aggregates are formed (Lu et al. 2011). Overall, the state of the matter (solution or gel), the kinetics of phase separation and the interactions between cellulose and non-solvent have to be taken into account when interpreting the morphology of cellulose II aerogels.

Morphology of cellulose II aerogels from cellulose/alkali solutions: a 4 wt% cellulose/LiOH/urea/water, non-solvent ethanol (With permission from Wiley: Cai J, Kimura S, Wada M, Kuga S, Zhang L (2008) Cellulose aerogels from aqueous alkali hydroxide–urea solution. ChemSusChem 1:149–154), b 5 wt% cellulose/NaOH/ZnO, non-solvent 0.3 M HCl (Reprinted by permission from [Springer] [Cellulose] [Demilecamps A, Reichenauer G, Rigacci A, Budtova T (2014) Cellulose–silica composite aerogels from “one-pot” synthesis. Cellulose 21:2625–2636], [2014]) and c 5 wt% cellulose/NaOH/urea, non-solvent 2 M HCl (Republished with permission of Royal Society of Chemistry, from [Mohamed SMK, Ganesan K, Milow B, Ratke L (2015) The effect of zinc oxide (ZnO) addition on the physical and morphological properties of cellulose aerogel beads. RSC Adv 5:90193–90201]; permission conveyed through Copyright Clearance Center, Inc)

Morphology of cellulose II aerogels from solid (a) and molten (b, c) 5 wt% cellulose/NMMO solutions, non-solvent was water. Image b is courtesy of R. Gavillon (Gavillon 2007) and images a and c are reprinted from Sescousse R, Gavillon R, Budtova T (2011a) Aerocellulose from cellulose–ionic liquid solutions: preparation, properties and comparison with cellulose–NaOH and cellulose–NMMO routes. Carbohydr Polym 83:1766–1774, Copyright 2011, with permission from Elsevier

Morphology of cellulose II aerogels from cellulose/ionic liquid solutions: a 5 wt% cellulose/[Emim][OAc]/DMSO, non-solvent ethanol (Reprinted by permission from [Springer] [Cellulose] [Buchtova N, Budtova T (2016) Cellulose aero-, cryo- and xerogels: towards understanding of morphology control. Cellulose 23:2585–2595], [2016]); b 1.5 wt% bleached pulp/[Bmim][Cl], non-solvent ethanol (Reprinted from Aaltonen O, Jauhiainen O (2009) The preparation of lignocellulosic aerogels from ionic liquid solutions. Carbohydr Polym 75:125–129, Copyright 2009, with permission from Elsevier) and c 1.5 wt% cellulose/[Hmim][Cl], non-solvent ethanol (Wang et al. 2013a)

Morphology of cellulose II aerogels from 3 wt% cotton linters (CL) in a TBAF/DMSO, b [Emim][OAc]/DMSO, c NMMO and d 1.5 wt% cotton linters in Ca(SCN)2·8H2O/LiCl, non-solvent was ethanol (Pircher et al. 2016)

Reprinted from Schestakow M, Karadagli I, Ratke L (2016a) Cellulose aerogels prepared from an aqueous zinc chloride salt hydrate melt. Carbohydr Polym 137:642–649, Copyright 2016, with permission from Elsevier
Morphology of cellulose II aerogels from 5 wt% cellulose/ZnCl2·4H2O solutions coagulated in water, ethanol, isopropanol and acetone.
It should be noted that the interactions between cellulose solvent and non-solvent should also be taken into account when investigating aerogel morphology and properties. For example, it was shown that exothermal reaction occurs when mixing [Emim][OAc] and water (Hall et al. 2012). At the moment of mixing, the temperature of [Emim][OAc]/water can increase as much as by 30–40 °C (Hall et al. 2012). It was hypothised that this may create air bubbles in coagulating cellulose/[Emim][OAc] solution leading the “traces” as channels in cellulose aerogel, as shown in Fig. 16. These large “holes” decrease density and increase porosity; potentially they can modify aerogel mechanical properties. Depending on the application, this phenomenon could be an interesting way to vary cellulose II aerogel morphology making hierarchical structure with pores of very different sizes, however, the control of structure formation is not easy.

Cellulose II aerogels prepared from 15% cellulose/[Emim][OAc]/DMSO solutions coagulated in a water and b ethanol. Courtesy of C. Rudaz (Rudaz 2013)
Specific surface area of cellulose II aerogels is shown in Fig. 17 as a function of cellulose concentration which is the obvious parameter to vary when making aerogels, keeping all the others the same. While density is easy to measure and many data are available, much less systematic results are reported for specific surface which requires special and rather expensive equipment. Aging of cellulose aerogels should be considered: it drastically influences mesoporosity with pores closing due to humidity adsorption and not re-opening during degassing because of hornification effect. The type of aerogel morphology, net-like or bead-like (see Figs. 11, 12, 13, 14, 15), does not seem to influence specific surface area (Fig. 17), but here again more systematic experiments are needed to make convincing conclusions.

Specific surface area of cellulose II aerogels as a function of cellulose concentration in solution for cellulose dissolved in different solvents and coagulated in different non-solvents. Ref A: Wang et al. (2013a), ref B: Trygg et al. (2013), ref C: Buchtova and Budtova (2016), ref D: Karadagli et al. (2015), ref E: Hoepfner et al. (2008), ref F: Gavillon and Budtova (2008) and ref G: Cai et al. (2008)
The increase of cellulose concentration leads to three types of trends for specific surface area, all contradicting each other (Fig. 17): surface area (1) increases (case of cellulose dissolved in NaOH/urea/water and in ionic liquids [Emim][OAc] and [Hmim][Cl], all coagulated in ethanol), (2) decreases (cellulose dissolved and gelled in NaOH/water and in Ca(SCN)2·6H2O) and (3) without any clear trend (cellulose dissolved in Ca(SCN)2·6H2O and in LiOH/urea/water). The increase in specific surface area with the increase of aerogel density (which is proportional to cellulose concentration as shown in Fig. 7) was also reported for four cases when cellulose was dissolved in ZnCl2·4H2O and coagulated in non-solvents such as water, ethanol, acetone and isopropanol (Schestakow et al. 2016a, b). The increase of specific surface area with the increase of cellulose concentration was suggested to be the result of pores “division” into smaller ones and not due to the increase of pore walls thickness (Buchtova and Budtova 2016). More careful and systematic experiments are needed to confirm or not this hypothesis.
Mechanical properties of cellulose II aerogels and their composites
The majority of bio-aerogel’s mechanical properties are tested in the uniaxial compression mode which is due to the easiness of the preparation of cylindrical samples. While theoretical approaches interpreting the mechanical response of silica aerogels have been developed (see, for example, Alaoui et al. 2008), the understanding of the mechanical properties of bio-aerogels and of the influence of various parameters (type of polysaccharide, polymer molecular weight, type of solvent and non-solvent, morphology, etc.) still remain to be unveiled.
Under the uniaxial compression cellulose II aerogel does not buckle, it uniformly decreases its height keeping diameter constant within experimental errors (Fig. 18); it was thus deduced that Poisson ratio is zero (Sescousse et al. 2011a; Schestakow et al. 2016a; Rege et al. 2016). Aerogel can be compressed without breakage till 80% strain (after that the experiments are stopped). Similar properties were reported for other bio-aerogels, for example, based on nanocellulose (Plappert et al. 2017) and pectin (Rudaz et al. 2014). Being highly compressed, bio-aerogels do not recover their shape, strong densification occurs.

Images of cellulose II aerogel under the uniaxial compression, courtesy of R. Gavillon (Gavillon 2007)
Compression stress–strain curves of cellulose II aerogels look as classical ones obtained for porous materials such as foams (Gibson and Ashby 1997) and inorganic and synthetic polymer aerogels (Fig. 19). At low strains (up to few per cent strain units), stress is linearly proportional to strain; this region is characterized by compressive modulus which is also often called Young’s modulus. Further increase of strain leads to progressive buckling of cell walls followed by their collapse; it corresponds to stress plateau the beginning of which is characterized by yield stress. Finally, at high strains cell walls touch each other, broken fragments pack and, theoretically, wall material itself is compressed (densification region). This type of compression behaviour was reported for cellulose II aerogels made from various solvents with different non-solvents.

Reprinted from Sescousse R, Gavillon R, Budtova T (2011a) Aerocellulose from cellulose–ionic liquid solutions: preparation, properties and comparison with cellulose–NaOH and cellulose–NMMO routes. Carbohydr Polym 83:1766–1774, Copyright 2011, with permission from Elsevier
Stress–strain curves of aerogels made from cellulose/[Emim][OAc] solution of various cellulose concentrations, non-solvent was water.
To better understand the mechanical properties of materials, loading–unloading tests should be performed and strain recovery should be followed as a function of strain [see, for example, amine-modified silica aerogels (Katti et al. 2006) or polyurea aerogels (Weigold and Reichenauer 2014)]. For classical aerogels in the linear regime the deformation is recovered, and at higher strains a hysteresis occurs with strain recovery becoming lower and lower with strain increase and finally being irreversible (Alaoui et al. 2008). This type of experiments was not performed on cellulose II aerogels. Strain recovery coupled with density and morphology analysis at each compression step would help the understanding of structure-properties relationships in cellulose aerogels. In particular, a comparison of strain recovery of foams based on cellulose I (Martoïa et al. 2016) and cellulose II aerogels would be very interesting.
A usual way to analyse the mechanical properties of aerogels is to plot compressive modulus E as a function of bulk density. For porous materials E is power-law dependent on bulk density (Gibson and Ashby 1997):
For regular open-cell foams the exponent n = 2, for silica aerogels it is usually around 3–4 (Cross et al. 1989; Alaoui et al. 2008; Wong et al. 2014) and for synthetic polymer aerogels it is, in general, around 2–3 [n = 2 was reported for polyurea aerogels (Weigold and Reichenauer 2014), n = 2.7 for resorsinol-formaldehyde (Pekala et al. 1990) but n = 3.7 for polyurethane aerogels (Diascorn et al. 2015)]. The examples of compressive modulus versus bulk density for silica (n = 3.6) and resorcinol–formaldehyde (n = 2.7) aerogels are shown in Fig. 20a, b, respectively.

Compressive modulus as a function of aerogel density for: a polyethoxydisiloxane aerogels (Reprinted from Wong JCH, Kaymak H, Brunner S, Koebel MM (2014) Mechanical properties of monolithic silica aerogels made from polyethoxydisiloxanes. Microporous Mesoporous Mater 183:23–29, Copyright 2014, with permission from Elsevier) and b resorcinol–formaldehyde aerogels and their carbons (Reprinted from Pekala RW, Alviso CT, LeMay JD (1990) Organic aerogels: microstructural dependence of mechanical properties in compression. J Non Cryst Solids 125:67–75, Copyright 1990, with permission from Elsevier)
Uniaxial compression tests have been performed on cellulose I aerogels and foams. For nanofibrillated freeze-dried cellulose, the exponent 2.29 was obtained for TEMPO-oxidised foams and 3.11 for foams from enzymatically pre-treated cellulose (Martoïa et al. 2016). A linear relationship between compressive modulus and aerogel density was reported by Kobayashi et al. 2014 and Plappert et al. 2017. For pectin aerogels n was 2.8 (Rudaz et al. 2014).
The compression modulus of cellulose II aerogels made from various celluloses dissolved in different solvents is demonstrated in Fig. 21; power law trends are shown by dashed lines. In a narrow density interval modulus can be seen as linearly dependent on aerogel density. However, this is only part of the trend, the straight line is a tangent to modulus versus density curve which clearly follows the power law in a wide range of densities (Eq. 2). The exponent for cellulose aerogels made from cellulose/ZnCl2·4H2O solutions and coagulated in isopropanol is n = 2.6, from cellulose/ZnCl2·6H2O solutions and coagulated in water n = 4.6, from cellulose (DP 1175)/NMMO coagulated in water and from cellulose/calcium thiocyanate coagulated in ethanol n = 1.7 and for cellulose (DP 180)/[Emim][OAc] coagulated in water n = 3.4.

Compressive modulus versus cellulose II aerogel density for various cellulose origins dissolved in different solvents and coagulated in different non-solvents. Dashed lines are power-law fits (see more details in the text). Data are taken from ref A: Mi et al. (2016), ref B: Karadagli et al. (2015); ref C: Rege et al. (2016); ref D: Pircher et al. (2016); ref E: Schestakow et al. (2016a); ref F: Sescousse et al. (2011a)
Linear approximation was used by Rege et al. (2016) to analyse modulus versus density of aerogels made from cellulose/Ca(SCN)2·6H2O and cellulose/ZnCl2·4H2O solutions (Fig. 22). Each shows a linear dependence being within a narrow interval of densities. Authors interpret high moduli values of aerogels from cellulose/ZnCl2·4H2O by the fact that “considerable amount of cellulose type I found in ZC-derived cellulose aerogels leads to the formation of a stronger backbone” (Rege et al. 2016). To find cellulose I after dissolution is surprising; SEM images of aerogel morphology from cellulose/ZnCl2·4H2O do not show any undissolved cellulose and the texture is similar to that from cellulose/Ca(SCN)2·6H2O route (Fig. 22). As mentioned in “Aerogels from cellulose dissolved in direct non-aqueous solvents” section and shown in “Volume change during processing and cellulose II aerogel density” section, all aerogels from cellulose/ZnCl2·4H2O route have much higher density than that of aerogels made from cellulose dissolved in any other solvent which results in very high moduli.

Adapted with permission of Royal Society of Chemistry, from [Rege A, Schestakow M, Karadagli I, Ratke L, Itskov M (2016) Micro-mechanical modelling of cellulose aerogels from molten salt hydrates. Soft Matter 12:7079–7088, copyright 2016]; permission conveyed through Copyright Clearance Center, Inc
Compressive modulus versus density for aerogels made from cellulose/Ca(SCN)2·6H2O coagulated in ethanol (“CT route”) and from cellulose/ZnCl2·4H2O coagulated in isopropanol (“ZC route”) together with the corresponding SEM images.
The trend for compressive modulus versus density for all data plotted together (inset in Fig. 21) gives n = 2 but with low correlation coefficient, R2 = 0.77. The exponent n = 2 was not expected because cellulose II aerogels are far from being regular foams, however, the same exponent was obtained for polyurea aerogels (Weigold and Reichenauer 2014) which shows morphology similar to some of cellulose II aerogels (see “Morphology and specific surface area” section and Fig. 9). To put all data for cellulose II aerogels without distinguishing (at least) by cellulose molecular weight is, certainly, a too rough approximation, however, even if keeping data for low-molecular weight cellulose only, the trend remains with the same exponent. On one hand, Sescousse et al. (2011a) showed that compressive modulus of aerogels from cellulose (Solucell, DP 950) dissolved in NMMO is higher than that made from cellulose (microcrystalline, DP 180) dissolved in 8%NaOH/water. On the other hand, modulus is the same for aerogels made from pulps, hardwood of DP 665 and softwood of DP 148 (Liebner et al. 2009); however, a huge decrease in cellulose DP from 665 to 129 was reported after the dissolution in NMMO which may be the reason of comparable moduli values of aerogels.
One of the problems in the understanding of the trends in the properties of bio-aerogels is their sensitivity to moisture, as mentioned in “Characterisation of bio-aerogels” section. While mechanical testing of classical polymers and their composites is usually performed on conditioned samples and at fixed and controlled humidity and temperature according to the norms, unfortunately this is rare for the case for bio-aerogels. The norms should be applied to bio-aerogels for the adequate comparison of data from different publications.
The mechanical properties of cellulose aerogels vary if another component is added resulting in composite aerogel material. To make composite cellulose aerogels, usually a “wet” cellulose precursor is impregnated by a second component which is polymerized inside cellulose network, the whole is then dried with supercritical CO2. The values obtained for such composite aerogels must be analysed with care as far as aerogel density and morphology are modified as compared to neat cellulose counterpart, and the interactions between the components and the morphology of the second network should be considered. For example, a strong increase in the mechanical properties of cellulose/polymethylmethacrylate interpenetrated network aerogels as compared to neat cellulose aerogels was reported (Pircher et al. 2015). When cellulose/silica aerogel composites were prepared, Cai et al. reported a “softening effect” (decrease of the modulus) due to the presence of silica (Cai et al. 2012), while Demilecamps et al. (2015) and Liu et al. (2013) demonstrated a strong increase in compressive modulus of cellulose/silica aerogel composites. The influence of the conditions in which the second component is polymerised on composite aerogel properties should also be considered: for example, cellulose degradation occurred during acid catalysis of alkoxylane, in the view of making interpenetrated cellulose/silica aerogels (Litschauer et al. 2011). The formation of silica aerogel was not confirmed in this case as far as specific surface area of the composite, 220–290 m2/g, was the same as of neat cellulose aerogel, 255 m2/g, while it is known that TEOS-based silica aerogels possess very high specific surface area, around 700–1000 m2/g. The case of silica particles (dispersed in cellulose aerogel matrix), and not silica aerogel, was reported for cellulose mixed with sodium silicate, both dissolved in 8% NaOH/water, however, the mechanical properties of composite aerogels were slightly improved (Demilecamps et al. 2014).
Thermal conductivity of cellulose II aerogels
Thermal conductivity is the most peculiar and exciting property of aerogels. Because of low density and mesoporosity, some classical aerogels (silica, resorcinol–formaldehyde and polyurethane based) are thermal super-insulating materials, i.e. with thermal conductivity below that of air in ambient conditions, 0.013–0.015 versus 0.025 W/m K. In the first approximation, thermal conductivity λ of a porous material is an additive sum of gaseous (λgas) and solid (λsolid) phase conduction and of the radiative heat transfer (λrad):
Solid phase conduction increases with density increase; it is power-law dependent on aerogel density (Lu et al. 1992). To minimize the conduction of the gaseous phase two options are possible: either evacuation of the gas (air), or decrease of pores’ size down to mesoporous region. According to Knudsen effect, when pore size is below the mean free path of air molecule, which is around 70 nm at 25 °C and 1 atm, λgas is lower than that of ambient air. λrad is not significant at room temperatures and optically thick materials (Fig. 23). Intuitively it is thus clear that the lowest thermal conductivity can be reached for low-density mesoporous materials. For silica and resorcinol–formaldehyde aerogels it was demonstrated that the dependence of thermal conductivity on density has a U-shape, as shown in Fig. 23 (Lu et al. 1992): higher density leads to conductivity increase because of λsolid input, and lower density leads to λgas increase because of the presence of large pores which do not contribute to Knudsen effect.

Total thermal conductivity (1), solid phase conductivity (2), gaseous conductivity (3) and calculated radiative conductivity (4) of resorcinol–formaldehyde aerogels as a function of density at ambient conditions. Reprinted with permission from [Lu X, Arduini-Schuster MC, Kuhn J, Njilsson O, Fricke J, Pekala RW (1992) Thermal conductivity of monolithic organic aerogels. Science 255:971–972]. Reprinted with permission from AAAS
Not much is known about the thermal conductivity of bio-aerogels. Low thermal conductivity, around 0.015–0.022 W/m K, was reported for aerogels based on pectin (Rudaz et al. 2014; Groult and Budtova 2018a), nanofibrillated cellulose (Jiménez-Saelices et al. 2017; Kobayashi et al. 2014; Seantier et al. 2016), alginate (Raman et al. 2015) and starch (Druel et al. 2017). Recently, U-shape conductivity-density dependence was obtained for low-methylated pectin aerogels, see Fig. 24 (Groult and Budtova 2018a), showing that bio-aerogels obey classical trends.

Reprinted from Groult S, Budtova T (2018a) Thermal conductivity/structure correlations in thermal super-insulating pectin aerogels. Carbohydr Polym 196:73–81, Copyright 2018, with permission from Elsevier
Thermal conductivity versus density for low methylated pectin aerogels, made at various pH and cross-linked with calcium and not. For more details see Groult and Budtova (2018a).
Thermal conductivity of cellulose II aerogels was also studied in details and many efforts were made to find the conditions in which these aerogels should become a thermal superinsulating material. None resulted in conductivity lower than that of air, 0.025 W/m K (Fig. 25). When cellulose was dissolved in Ca(SCN)2·4H2O, aerogel conductivity was either 0.044–0.055 W/m K (Laskowski et al. 2015) or even higher, 0.04–0.075 W/m K (Karadagli et al. 2015). The conductivity of aerogels from cellulose/LiOH/urea/water was 0.025 W/m K (Cai et al. 2012) and from cellulose/NaOH/water and cellulose/[Emim][OAc] around 0.03–0.032 W/m K (Rudaz 2013) (Table S1). Freeze-dried cellulose aerogels possessed similar conductivity of around 0.029–0.032 (Nguyen et al. 2014). Cellulose cross-linking with epichlorohydrin (ECH) in NaOH/water was performed in the view of decreasing pore size and thus decreasing gaseous phase conduction, see Eq. 3 (Rudaz 2013). This allowed decreasing the conductivity to 0.026 W/m K (Fig. 25), however, the “barrier” of air conductivity was not overcome.

Thermal conductivity versus bulk density of cellulose II aerogels (solvents: NaOH/water and [Emim][OAc]), tritylcellulose and tritylcellulose/hydrophobised silica aerogels. Solid line corresponds to the conductivity of air, dashed line is given to guide the eye.
Cellulose is hydrophilic and adsorbs humidity from air which results in densification and increase of conductivity. Tritylcellulose was synthesized and aerogels prepared; their conductivity was lower than that of non-modified cellulose (0.027–0.029 W/mK, see Fig. 25) but again it was higher than the one of air (Demilecamps et al. 2016). Thermal conductivity of freeze-dried cellulose aerogels hydrophobised with plasma treatment was 0.03–0.033 W/m K (Shi et al. 2013a).
In the view of decreasing the conductivity of cellulose aerogels, composite aerogels based on cellulose/silica were prepared. When cellulose/Ca(SCN)2·4H2O solutions were mixed with granular superinsulating silica aerogel, the conductivity of composite aerogels slightly decreased as compared to neat cellulose aerogel counterpart but was still high, 0.04–0.05 W/m K (Laskowski et al. 2015). When “wet” cellulose was impregnated with silica sol (TEOS) in order to fill the pores of cellulose matrix and thus decrease the conduction of the gas (air) phase, surprisingly the conductivity of composite aerogels increased from 0.025 to 0.04–0.05 W/m K (Cai et al. 2012). The reason could be a strong increase in the density of composite aerogel (0.14 g/cm3 for neat cellulose aerogel, 0.19 for neat silica aerogel and 0.3–0.6 g/cm3 for composite aerogels) which means that the formation of silica aerogel inside the pores of cellulose matrix was perturbed and did not lead to a formation of a superinsulating material. Similar impregnation approach was used by Demilecamps et al. (2015): cellulose was dissolved in [Emim][OAc]/DMSO, coagulated in ethanol and impregnated with polyethoxydisiloxane. Thermal conductivity decreased from 0.033 W/m K for neat cellulose aerogel to 0.026 W/m K for composite cellulose-silica (Demilecamps et al. 2015). A similar decrease in conductivity was reported for cellulose matrix filled with TEOS, freeze-dried and hydrophobised via plasma treatment: from 0.03 W/m K for the neat cellulose to 0.026 W/m K for cellulose-silica composite (Shi et al. 2013b).
The only way which resulted in cellulose-based thermal superinsulating aerogels was making fully hydrophobic cellulose/silica interpenetrated network. “Wet” (coagulated) tritylcellulose was impregnated with polyethoxydisiloxane which was gelled inside cellulose matrix, silica was hydrophobised and all was dried with supercritical CO2. As a result, tritylcellulose matrix was filled with superinsulating hydrophobic silica aerogel which led to conductivities of 0.021–0.022 W/m K (Demilecamps et al. 2016). The evolution of morphology from macroporous tritylcellulose to mesoporous tritylcellulose/silica composite aerogel is shown in Fig. 26 which demonstrates that pores of cellulosic matrix were homogeneously filled with silica aerogel. A strong increase in the fraction of mesopores was confirmed by specific surface area: from 250–330 m2/g for tritylcellulose to 610–750 m2/g for composite aerogels (Demilecamps et al. 2016).

Reprinted from Demilecamps A, Alves M, Rigacci A, Reichenauer G, Budtova T (2016) Nanostructured interpenetrated organic-inorganic aerogels with thermal superinsulating properties. J Non Cryst Solids 452:259–265, Copyright 2016, with permission from Elsevier
SEM images of aerogels based on a tritylcellulose, b polyethoxydisiloxane and c tritylcellulose/polyethoxydisiloxane interpenetrated network.
Why it is not possible to make thermal superinsulating cellulose II aerogels while this is feasible with nanocellulose and several other polysaccharides such as pectin, starch and alginate? The reason is, probably, in “unfavourable” cellulose II aerogel morphology (see “Morphology and specific surface area” section), with too thick pore walls and too many large macropores. Molecular modelling could be very helpful to answer how cellulose chains are packing during non-solvent exchange and why this is different from other polysaccharides that form finely nanostructured thermal superinsulating aerogels.
Potential applications of cellulose II aerogels
Cellulose aerogels are said to be versatile materials suitable for numerous applications, and indeed they are, especially if considering additional properties coming from different options in cellulose functionalization and making composite and hybrid materials. Some applications of cellulose II aerogels are already tested and they will be overviewed below. Each section will start with a very brief description of the application area focusing on cellulose materials (other than aerogels) and on aerogels (other than cellulose-based). Cellulose I aerogels and freeze-dried cellulose II will also be considered as there are not many publications devoted to cellulose II aerogels’ applications. The reader will see that still there is a lot of room for improvements and applications to explore.
Bio-medical applications
Until now, the largest potential pharma- and/or bio-medical applications of bio-aerogels is suggested to be a carrier for the release of active substances, mainly drugs. There are different ways of drug incorporation in a bio-aerogel: either mixing with polysaccharide solution, or impregnating into “wet” aerogel precursor, or during drying (recall Fig. 1). The choice of the route mainly depends on drug solubility in the corresponding fluid. In order to have high drug loading, it should be obviously not washed out during subsequent processing steps. Aerogels based on alginate, pectin, starch and chitosan have been reported as drug carriers (García-González et al. 2011), and the majority of literature describes drug loading in bio-aerogels using adsorption in supercritical carbon dioxide. The kinetics of drug release depends on several parameters such as the state of the drug (crystalline or amorphous) and matrix behaviour (swelling or contraction, dissolution) during mass transport in/from the matrix. Potentially, the release can be tuned by functionalization of the matrix and by making a composite or hybrid matrix, for example, organic–inorganic or organic-organic (based on different polymers). It was also demonstrated that bio-aerogels (based on alginate, alginate/lignin, pectin/xanthan, starch and starch/caprolactone) are non-cytotoxic, feature good cell adhesion and some can be used for bone regeneration (Martins et al. 2015; Quraishi et al. 2015; Horvat et al. 2017; Goimil et al. 2017).
Cellulose I aerogels and foams (based on bacterial and nanofibrillated cellulose) have been widely studied in the view of their use in bio-medical applications as they are biocompatible, non-toxic and support growth and proliferation of cells (for more details, see Liebner et al. 2016). They can be used as matrices for drug delivery as release kinetics can be controlled by pore size and functionalization. The addition of silver or zinc oxide particles makes cellulose I aerogels antibacterial materials.
Surprisingly, only few publications demonstrate the potential use of cellulose II aerogels in bio-medical applications, despite that many suggest it. Biocompatibility was demonstrated for cellulose II aerogels with dual porosity, with large pores made by porogens that are leached out and smaller pores coming from supercritical drying of coagulated cellulose matrix (Pircher et al. 2015). Aerogels made from phosphorylated cellulose, with low degree of substitution, showed good hemocompatibility (Liebner et al. 2012).
Absorption and adsorption
Absorption of oils and organic solvents is an important environmental problem to solve with one example being spilled hydrocarbons in seawater. Various “sorption” (in the large sense of the term) approaches exist involving physical, chemical, biological treatments and their combinations. Porous materials with high efficiency, selectivity and allowing multiple reuse (many cycles) and easy degradation (better biodegradation) are in the focus of research with low cost also being an important criterion. Non-modified natural materials such as sugar cane bagasse, rice and coconut husk, natural fiber mats and fabrics have traditionally been used for absorption purposes, however their absorption capacity is not high (below 10 g/g) and selectivity (capability to absorb only oil) is rather poor as they are hydrophilic.
Aerogels are discussed in literature for their potential applicability in oil and organic fluids absorption as they are highly porous and with high specific surface area which can be chemically tuned to increase the selectivity. An overview of the advantages (performance) and disadvantages (price) of using aerogels for environmental applications is given by Maleki (2016). Carbon and graphene aerogels seem to have the highest absorption capacity of oils and organic solvents, up to 200 times their own weight (Maleki 2016).
Cellulose aerogels, both from cellulose I and cellulose II, are good candidates to be used for absorption provided they are chemically modified to increase the selectivity. Here only non-pyrolysed freeze-dried and supercritically dried cellulose will be discussed as absorption and adsorption applications of the corresponding carbons are presented in “Carbon cellulose aerogels” section. Only few studies report the use of cellulose II aerogels for absorption of organic fluids: for example, cellulose was coated with TiO2 and showed five times higher absorption capacity of oil, up to 28 g/g, as compared to non-coated counterpart (Chin et al. 2014, see details on aerogel preparation in Table S1 of the Supporting Information). Most of the studies on absorption of oils and organic solvents are performed on freeze-dried cellulose II and all use silylation (with trimethylchlorosilane, octadecyltrimethoxysilane, etc.) or plasma treatment (Lin et al. 2015) or chemical vapour deposition (Zhang et al. 2016; Liao et al. 2016). The absorption capacity of oils is usually within 20–25 g/g (Lin et al. 2015; Zhang et al. 2016) with the best result, up to 59 g/g, obtained for cellulose dissolved in NaOH/urea/water, cross-linked with N,N′-methylenebisacrylamide, freeze-dried and functionalized with trimethylchlorosilane (Liao et al. 2016). The absorption of organic solvents is around 40–50 g/g (Zhang et al. 2016). As expected, the absorption capacity decreases with the increase of fluid viscosity. Most of the publications cited above report good cellulose hydrophobicity and recyclability with up to 10–15 cycles tested. Similar absorption capacities were reported for nanofibrillated freeze-dried cellulose either TiO2 functionalised (Korhonen et al. 2011) or silylated (Cervin et al. 2012) or surface-grafted and cross-linked (Mulyadi et al. 2016). Higher values were obtained for nanofibrillated cellulose treated with methyltrimethoxysilane and freeze-dried: the absorption capacity of oil was up to 50–60 g/g and of organic solvents up to 100 g/g (Zhang et al. 2014). Finally, very high absorption values were reported for surface-modified bacterial cellulose with trimethylchlorosilane in liquid phase and freeze-dried: absorption of oils was up to 100–120 g/g and of organics up to 185 g/g (Sai et al. 2015). Interestingly, high absorption of oils (up to 95 g/g), even better than by many of cellulose I and cellulose II cryo- and aerogels, was obtained for “simple” recycled cellulose fibers, cross-linked with Kymene, freeze-dried and coated with methyltrimethoxysilane (Feng et al. 2015).
The analysis of the absorption capacity obtained for various cellulose I and cellulose II porous materials and of the reasons of different results reported can be a topic of another review article. What are the main driving forces of high absorption by cellulose-based materials: porosity, way and type of functionalization, surface area, type of cellulose? Should cellulose be dissolved or “delaminated” to obtain aero- or cryogels with fine structure for having high absorption capacity? These questions need answers if willing to use cellulose I and/or cellulose II aero- and cryogels in absorption applications.
Another important environmental problem to solve is water pollution with heavy metal ions. Adsorption is considered to be one of the efficient methods among others such as membrane filtration and separation, precipitation, ion exchange, etc. The price, re-use, metal recovery and adsorbent regeneration (if possible) and degradation are playing an important role as in the case of absorption. Cellulose, when chemically modified (etherification, esterification, oxydation, grafting of various ligands) is considered as an alternative to synthetic adsorbents, with adsorption capacity being as high as 45 mg/g for Cr(VI), 105 mg/g for Pb(II), 169 mg/g for Cd (II), 188 mg/g for Ni (II) and 246 mg/g for Cu (II) (O’Connell et al. 2008). Similar and even better values are reported for chitosan and its composites (Wan Ngah et al. 2011). It should be noted that adsorption capacity significantly depends on solution pH and initial concentration of metal ions.
For the same reasons as for the absorption of organic pollutants, aerogels are considered to be promising materials due to their high adsorption performance, with carbon aerogels being the leaders: for example, 68 mg/g for Cr(VI) and 240 mg/g for Pb(II) (Maleki 2016). As far as cellulose I and cellulose II cryo- and aerogel are concerned, cellulose modification is needed to make them efficient adsorbents. Cellulose II aerogels were prepared via the dissolution of microcrystalline cellulose in NaOH/urea/water, immersed in FeCl3 and MnCl2 solutions to obtain MnFe2O4 cellulose aerogel which adsorbed Cu (II) up to 90 mg/g (Cui et al. 2018). Another way to get high adsorption is to make composites: cellulose was dissolved in NaOH/urea/water, mixed with graphene oxide, cross-linked with N,N′-methylene bisacrylamide and freeze-dried; the adsorption of methylene blue was 138 mg/g and of Cu (II) 85 mg/g (Geng 2018). Nanocellulose is often chemically modified and tested for the adsorption of heavy metals. For example, freeze-dried nanofibrillated cellulose was grafted with poly(methacylic acid-co-maleic acid) and Pb was adsorbed at around 95 mg/g and Cd at around 90 mg/g (Maatar and Boufi 2015). However, high adsorption of metal ions can also be obtained on never-dried nanocellulose: for example, cellulose nanocrystals and nanofibers were enzymatically phosphorylated and the adsorption of Cu (II) was 117 mg/g and 114 mg/g, respectively (Liu et al. 2015).
High specific surface area of cellulose I and cellulose II aerogels should, theoretically, promote high adsorption capacity of heavy metal ions. Is the drying of coagulated cellulose or of nanocellulose needed to get high adsorption? Is there any advantage of nanocellulose versus cellulose II or it is cellulose modification which counts the most? Depending on the answers to these questions the price of cellulose adsorbent will be very different. A detailed analysis of results reported in literature taking into account the “state” of cellulose (polymorph type, wet or dry, way of drying, surface area and chemical modification) would be helpful to provide the best recipe in terms of performance.
Composite aerogels with metal (nano)particles and quantum dots
Polymer/metal nanoparticles composites is a quickly developing area due to their various applications in optics, electronics, medical, catalysis and sensors. One of the challenges is to prevent nanoparticle self-aggregation and this is the reason why they are often immobilized or loaded in/on polymers, graphene or carbons. Another challenge is not to modify particles’ activity and selectivity due to their immobilization. Cellulose (in various forms: fibers, fabric and nanocellulose) is often used as metal nanoparticle support as cellulose is biodegradable and biocompatible and also binds metal nanoparticles minimizing the risk of contamination. The latter is possible if the surface of cellulose material is modified in the adequate way to immobilize the nanoparticle, usually by electrostatic interactions. Many publications report on making antimicrobial cellulose fibers and fabrics with Ag, Cu and Zn nanoparticles. Different forms of nanocellulose were shown to be very promising in catalysis applications: nanocellulose can be metal nanoparticle support, stabilizer and also a reducing agent in the in situ synthesis of metal nanoparticles (Kaushik and Moores 2016).
Whatever is the matter of nanoparticles’ support, porosity is one of the pre-requisites for having an efficient incorporation of metal nanoparticles. Aerogels are thus excellent candidates for metal nanoparticles’ support. Aerogels based on β-lactoglobulin amyloid fibrils loaded with gold nanoparticles were demonstrated to be a promising catalyst (Nyström et al. 2016). Often carbon-based aerogels are employed as metal nanoparticles support. For example, carbons from pyrolysed resorcinol–formaldehyde aerogels and loaded with Pt or Ru nanoparticles can be used as electrode materials and catalyst support in proton-exchange membrane fuel cells (Biener et al. 2011). Fe3O4 nanoparticles supported by freeze-dried nitrogen-doped graphene was demonstrated to be efficient cathode catalysts for oxygen reduction reaction (Wu et al. 2012).
Cellulose II aerogels were used as support of metal (nano)particles (Cai et al. 2009; Chin et al. 2014; Schestakow et al. 2016b; Cui et al. 2018). Schestakow et al. (2016b) and Cai et al. (2009) report the deposition of noble metal (silver, gold and platinum) nanoparticles into cellulose aerogel network. Metals were impregnated into “wet” aerogel precursor, and nanoparticles were attached to cellulose via reduction reaction. In these cases composite aerogel density slightly increased and specific surface area remained the same as in the corresponding neat cellulose II aerogels (Table S1). The potential in using Ag-doped cellulose II aerogel as catalyst was demonstrated (Schestakow et al. 2016b).
Magnetic cellulose II aerogels were synthesized by adding either Fe2O3 nanoparticles (Chin et al. 2014) or MnFe2O4 (Cui et al. 2018). In the first case aerogel was coated with TiO2 and used for the absorption of oil which was up to 25 g/g. The magnetic property of aerogel was used to extract the sample with absorbed oil from the container. When magnetic cellulose/MnFe2O4 aerogels were synthesised, composite aerogel density increased and specific surface area increased slightly (Cui et al. 2018). These composites showed the adsorption of copper ions up to 63 mg/g, and magnetic property was used, as in the previous case, for the separation of the sample from water.
Freeze-dried bacterial cellulose (density 0.015 g/cm3, specific surface area 103 m2/g) was used as a support of ferrite crystal nanoparticles (size from 40 to 60 nm and up to 120 nm at high FeSO4/CoCl2 concentrations) (Olsson et al. 2010). Nanoparticles were deposited on freeze-dried bacterial cellulose from solution followed by heating at 90 °C, immersing in NaOH/KNO3 at 90 °C and then freeze-dried again. Flexible magnetic samples (density 0.3 g/cm3) were obtained; they were suggested to be used as electronic actuators (Olsson et al. 2010).
Quantum dots are semiconductor nanocrystals of a size of few nanometers, with a new generation of quantum dots based on carbon and graphene. Due to their unique electro-optical properties, so-called “quantum confinement”, they have superior brightness and photostability compared to conventional fluorescent dyes, well suited for multicolour applications, for biological imaging (bioassays, bioprobes and biosensors) and various energy related devices (LED, photodetectors, solar cells, etc.).
Organic and inorganic aerogels were used as a support of quantum dots. Pyrolysed resorcinol–formaldehyde aerogels were used as carriers of carbon quantum dots and this composite supercapacitor showed excellent stability over 1000 charge–discharge cycles and 20 times higher specific capacitance as compared to its neat counterpart (Lv et al. 2014). Silica aerogels were functionalized with polyethylenimine-capped quantum dots and new NO2 gas sensor was obtained (Wang et al. 2013b). CdSe–ZnS quantum dots were covalently immobilized on tetramethylorthosilicate and luminescent aerogels were obtained (Sorensen et al. 2006).
It seems there is only one publication reporting on cellulose/quantum dots aerogels. Cellulose was dissolved in [Hmim][Cl] and mixed with 3-(trimethoxysilyl)-propyl-functionalized (ZnS)x(CuInS2)1−x core/ZnS shell suspended in toluene (Wang et al. 2013a). 1-mercapto-3-(trimethoxysilyl)-propyl ligands allowed the migration of quantum dots from toluene to cellulose/ionic liquid solutions which resulted in quantum dots covalently bonded to cellulose. Fluorescent aerogels were obtained. Depending on the thickness of quantum dot shell, specific surface area of aerogels increased almost twice and mechanical properties of composite aerogels were also improved as compared to their neat cellulose counterpart (Wang et al. 2013a).
Carbon cellulose aerogels
Carbon aerogels are mesoporous and microporous materials with high electrical conductivity. They are made by pyrolysis of organic aerogels in an inert atmosphere. Since resorcinol–formaldehyde aerogels were synthesized in the 1990s of the last century (Pekala et al. 1995), their carbon counterparts (Pekala et al. 1998), including those made from xerogels, remain the most popular and studied. Other systems, such as phenolic–furfural, melamine–formaldehyde, polyacrylonitrile, and polyurethane were also used for making carbon aerogels. Carbon aerogels usually have high specific surface area, around 500–1000 m2/g; density can be higher than that of non-pyrolysed counterparts, around 0.1–0.5 g/cm3. Pyrolysis is usually performed during 8–10 h at temperature rising up to 1000 °C which leads to volume shrinkage and mass loss (for resorcinol–formaldehyde aerogels, linear shrinkage is around 30% and mass loss around 50%) (Shen and Guan 2011). Carbon aerogels are proposed to be used in electrochemical and energy applications: in double layer capacitors (supercapacitors), lithium-ion batteries, for hydrogen storage, adsorption, as catalyst supports when metal-doped and for thermal insulation at high temperatures.
The information on carbon aerogels from bio-aerogels is scarce with very few systematic studies correlating the morphology and properties of neat aerogels with processing parameters and resulting structure and properties of carbons. A significant input was made by the team from the university of York, UK. Starch-based mesoporous carbons, so-called Starbons, were obtained from pyrolysed dissolved-retrograded high amylose corn starch, doped with acid and dried at ambient pressure (Budarin et al. 2006). The increase of temperature from 150 to 700 °C led to the increase of specific surface area, from 200 to 500 m2/g, respectively. These carbons were suggested to be used as an alternative to acid catalysts (White et al. 2009). Similar approach was applied to other types of starches, and carbons with specific surface area around 170–370 m2/g were synthesised (Bakierska et al. 2014). Carbons were obtained by pyrolysis of alginate aerogels: specific surface area of the starting aerogel was 320 m2/g and it first increased with the increase of temperature (up to 388 m2/g at 500 °C) and then decreased to 300 m2/g at 1000 °C (White et al. 2010a). These carbon aerogels were suggested to be used for the separation of some low molecular weight carbohydrates (White et al. 2010a). Carbons from pectin aerogels were also prepared by the same team (White et al. 2010b). As for alginate carbon aerogels, specific surface area of pectin-based carbons increased with the increase of temperature, from 200 m2/g for pectin aerogel to 377 m2/g for carbon aerogel made at 450 °C, and then decreased to 298 m2/g at 700 °C (White et al. 2010b). The mass loss at 700 °C was around 70%.
As far as carbons from cellulose aerogels are concerned, the majority are made from pyrolysed freeze-dried either “nanocellulose” or dissolved-coagulated cellulose aerogels. Most of the work is focused not on cellulose matrix and its transformations, but on testing the properties of new porous biomass-based carbons for various applications. For example, bacterial cellulose was freeze-dried, pyrolysed and nitrogen-doped (specific surface area 916 m2/g) for making metal-free oxygen reduction electrocatalyst, for fuel cells and metal-air batteries (Liang et al. 2015). The same group used pyrolysed freeze-dried bacterial cellulose for the absorption of organic fluids (100–300 times its weight) (Wu et al. 2013). Interestingly, this carbon showed a high shape recovery under compression. The absorption of oils was also tested on carbonised freeze-dried cross-linked microfibrillated cellulose; from 40 to 60 g/g of oil was absorbed within few minutes (Meng et al. 2015). As mentioned in “Absorption and adsorption” section, efficient oil absorption can be obtained for functionalised, but not pyrolysed, freeze-dried nanofibrillated cellulose [for example, 20–40 g/g when coated with TiO2 (Korhonen et al. 2011) or up to 45 g/g when hydrophobised with octyltrichlorosilanes (Cervin et al. 2012)]. Pyrolysed freeze-dried bacterial cellulose was also explored as anode material in lithium ion batteries (Wang et al. 2014b).
Similar approaches were applied to pyrolysed freeze-dried dissolved-coagulated cellulose. For example, cellulose was dissolved in LiOH/urea/water, freeze-dried from TBA and pyrolysed; specific surface area of neat cellulose was 149 m2/g, of pyrolysed counterpart 500 m2/g and absorption capacity of hydrocarbons and oil was up to 25 g/g (Wang et al. 2014a). Similar absorption was reported for pyrolysed freeze-dried cellulose from cellulose/NaOH/urea/water (Lei et al. 2018). Carbons from freeze-dried cellulose dissolved in NaOH/water based solvent were reported as supercapacitor electrodes when nitrogen-doped (Hu et al. 2016), KOH activated (Yang et al. 2018) and CO2 activated (Zhuo et al. 2016, Zu et al. 2016). These carbons were also shown to possess high CO2 adsorption capacity (Zhuo et al. 2016; Hu et al. 2016) and be suitable as monolithic catalysts when MnOx/N doped (Zhou et al. 2018).
Very few is reported on carbons from cellulose II supercritically dried aerogels. They were made by pyrolysis of cellulose aerogels prepared by cellulose dissolution in [Emim][OAc] (Sescousse 2010) and in NaOH/water (Gavillon 2007; Guilminot et al. 2008; Sescousse 2010; Rooke et al. 2011, 2012). Mass and volume loss after pyrolysis was around 80% and 90%, respectively (Gavillon 2007). The interesting point is that despite a severe shrinkage, the samples kept their initial shape (Fig. 27). This means that the volume and shape of carbon aerogel can be predicted and controlled from the very first steps of preparation (here, gelation of cellulose/NaOH/water solutions).

Representative photos of cellulose aerogels and its carbon counterpart and the corresponding SEM images. Aerogel was made from 7 wt% cellulose/NaOH/water solution, gelled and coagulated in ethanol. Photos are courtesy of R. Sescousse (Sescousse 2010), SEM images are adapted with permission of Electrochemical Society, from [Rooke J, de Matos Passos C, Chatenet M, Sescousse R, Budtova T, Berthon-Fabry S, Mosdale R, Maillard F (2011) Synthesis and properties of platinum nanocatalyst supported on cellulose-based carbon aerogel for applications in PEMFCs. J Electrochem Soc 158:B779–B789]; permission conveyed through Copyright Clearance Center, Inc
SEM images in Fig. 27 show densification of carbon aerogel as compared to its non-pyrolysed counterpart; bulk density of carbons is usually higher by 50 to 100% reaching the values of 0.25–0.35 g/cm3 (Gavillon 2007; Sescousse 2010). As well as for carbons from other organic aerogels, the number of macropores in carbons from porous cellulose seems to be reduced as compared to their counterparts before pyrolysis, possibly due to shrinkage. This can be deduced from the increasing specific surface area: 299 m2/g in aerogel versus 892 m2/g in CO2 activated carbon (Zu et al. 2016); 149 m2/g in freeze-dried from TBA versus 500 m2/g in carbon (Wang et al. 2014a, b), 130 m2/g in TBA vacuum dried cellulose tunicate nanocrystals versus 667 m2/g (pyrolysis under nitrogen) and 549 m2/g (pyrolysis under HCl) in carbons (Ishida et al. 2004) and 145 m2/g in aerogel versus 244 m2/g in carbon (Sescousse 2010). It should be noted that activation step may strongly increase specific surface area.
The microstructure of carbon cellulose aerogels determines the application to be selected. For example, carbons from aerogels based on cellulose dissolved in NaOH/water turned out to be very promising as cathodes in Li/SOCl2 primary batteries. Typical electrodes for this type of batteries are made from carbon black powders with polytetrafluoroethylene binder. While classical batteries have solid cathode and anode and the discharge is limited by the amount of oxidant or reductant, in liquid cathode cells it is limited by the porosity of carbon current collector. The optimal collector should have the highest possible pores volume with pore size in the range of mesopores up to small macropores (< 100 nm). The discharge properties of carbon aerogels from cellulose/8 wt%NaOH/water gelled solutions are presented in Fig. 28 and compared with the reference material used by French company SAFT. “Green” carbon aerogels are excellent current collectors and in some cases their capacity is higher than that of the reference material (Rooke et al. 2012). The difference in the performance [Fig. 28, case (a) vs. case (b)] is related to the size of the pores in the carbon aerogel: while the volume of mesopores is practically the same, 2.9 and 3.2 cm3/g, it is the mean size of the pores which is different, 92 nm (a) versus 61 nm (b) (Rooke et al. 2012). Pore size distributions are shown in the insets of Fig. 28a, b.

Discharge at 0.4 mA of Li/SOCl2 button-type battery with a thin carbon aerogels collector disk. Carbons are made from pyrolysed aerogels prepared from a 5 wt% cellulose/NaOH/water gelled solution and b 7 wt% cellulose/NaOH/water gelled solution. The discharge time was in the range of 300–400 h. Data are taken from (Sescousse 2010) and (Rooke et al. 2012). Insets correspond to pore size distributions
The same carbon aerogels, doped with platinum, were shown to have suitable properties to be used in proton exchange membrane fuel cell (PEMFC) (Guilminot et al. 2008; Rooke et al. 2012). Platinum nanoparticles can be homogeneously deposited on carbon aerogel matrix and this “green” electrocatalyst compares well with standard Pt/carbon black materials.
The evolution of cellulose mass and composition during pyrolysis is known to be a complex process: briefly, after the loss of water cellulose depolymerisation occurs, levoglucosan is formed which is then decomposed into various anhydrosugars, which in turn can react and form unstable intermediates (furanes, volatile substances) and char (Li et al. 2001; Lin et al. 2009b). While pyrolysis of cellulose has been extensively studied, practically nothing is known on the evolution of porous cellulose (aerogel or freeze-dried) mass, volume, porosity, density and pore size during pyrolysis as a function of temperature profile. How does the structure of carbon aerogel correlate with that of the corresponding porous cellulose? How all parameters, which control structure formation in cellulose II aerogels, influence the structure and properties of their carbons? These questions need to be answered if willing to make carbons with added value from cellulose II aerogels. It is a huge area worth exploring as the properties these carbons are very promising for various applications.
Conclusions and prospects
This review presented the main results obtained, till now, on cellulose II aerogels made via dissolution-solvent exchange-drying with supercritical CO2. The main trends, not always well established, are analysed and discussed. The properties and morphology of cellulose II aerogels are compared, when possible, when those of classical (inorganic, synthetic polymer) aerogels and bio-aerogels.
As mentioned in the Introduction, there are numerous open questions that remain to be answered, and this is the leitmotif of practically each topic discussed, on the trends in cellulose II aerogels structure and properties, and on aerogels’ applications. Cellulose aerogels, and bio-aerogels in general, are very “young” materials and are at the interface of different disciplines that were previously non crossing: polymer/cellulose physics and chemistry and aerogels/porous materials. If adding all other various disciplines related to applications (controlled release, electro-chemistry, sorption, pharma, bio-medical), it is clear that cellulose aerogels need a multidisciplinary approach and common efforts of the experts from different scientific fields.
The understanding of the formation of cellulose II aerogel structure during coagulation is one of fundamental questions to be looked at. How cellulose chains are packing? What is the influence of non-solvent? Does the state of the matter before coagulation, solution or gel, influence aerogel structure? How cellulose derivatization may influence structure formation and properties of aerogels? The advantage of supercritical drying is that it preserves, to a large extent, the morphology of coagulated cellulose and thus allows answering these questions in future. As a consequence, this may help the understanding why, for example, cellulose II aerogels are not thermal superinsulating materials while cellulose I and some other bio-aerogels are.
Cellulose II aerogels are materials with high added value. At least two application domains seem to be very promising now, but the list is certainly far incomplete: in pharma/bio-medical and as carbons. In the first case biocompatible materials with controlled and hierarchical porosity can be made. Controlled release and scaffolds are thus the potential areas. The first results on pyrolysed cellulose aerogels turned out to show excellent discharge properties. The possibility of making cellulose II “wet” (coagulated) and “dry” (aerogel, cryogel) objects of complex shapes using “direct ink writing” (or 3D printing) technique is not well explored yet. This approach can be very attractive in making, for example, cellulose II aerogels of individualized shape and controlled porosity for both application areas mentioned above. Finally, numerous options of cellulose derivatization and/or functionalization may definitely open new cellulose aerogel applications which remain unexplored.
References
Aaltonen O, Jauhiainen O (2009) The preparation of lignocellulosic aerogels from ionic liquid solutions. Carbohydr Polym 75:125–129
Aegerter MA, Leventis N, Koebel MM (2011) Aerogels handbook. Springer, New York
Alaoui AH, Woignier T, Scherer GW, Phalippou J (2008) Comparison between flexural and uniaxial compression tests to measure the elastic modulus of silica aerogel. J Non Cryst Solids 354:4556–4561
Bakierska M, Molenda M, Majda D, Dziembaj R (2014) Functional starch based carbon aerogels for energy applications. Proc Eng 98:14–19
Biener J, Stadermann M, Suss M, Worsley MA, Biener MM, Rose KA, Baumann TF (2011) Advanced carbon aerogels for energy applications. Energy Environ Sci 4:656–667
Biesmans G, Randall D, Francais E, Perrut M (1998) Polyurethane-based organic aerogels’ thermal performance. J Non Cryst Solids 225:36–40
Biganska O, Navard P (2005) Kinetics of precipitation of cellulose from cellulose–NMMO–water solutions. Biomacromolecules 6:1948–1953
Borisova A, De Bruyn M, Budarin VL, Shuttleworth PS, Dodson Mateus JR, Segatto L, Clark JH (2015) A sustainable freeze-drying route to porous polysaccharides with tailored hierarchical meso- and macroporosity. Macromol Rapid Commun 36:774–779
Buchtova N, Budtova T (2016) Cellulose aero-, cryo- and xerogels: towards understanding of morphology control. Cellulose 23:2585–2595
Budarin V, Clark JH, Hardy JJA, Luque R, Milkowski K, Tavener SJ, Wilson AJ (2006) Starbons: new starch-derived mesoporous carbonaceous materials with tunable properties. Angew Chem Int Ed 45:3782–3786
Budtova T, Navard P (2016) Cellulose in NaOH–water based solvents: a review. Cellulose 23:5–55
Cai J, Kimura S, Wada M, Kuga S, Zhang L (2008) Cellulose aerogels from aqueous alkali hydroxide–urea solution. ChemSusChem 1:149–154
Cai J, Kimura S, Wada M, Kuga S (2009) Nanoporous cellulose as metal nanoparticles support. Biomacromolecules 10:87–94
Cai J, Liu S, Feng J, Kimura S, Wada M, Kuga S, Zhang L (2012) Cellulose-silica nanocomposite aerogels by in situ formation of silica in cellulose gel. Angew Chem Int Ed 51:2076–2079
Cai H, Sharma S, Liu W, Mu W, Liu W, Zhang X, Deng Y (2014) Aerogel microspheres from natural cellulose nanofibrils and their application as cell culture scaffold. Biomacromolecules 15:2540–2547
Cervin NT, Aulin C, Larsson PT, Wagberg L (2012) Ultra porous nanocellulose aerogels as separation medium for mixtures of oil/water liquids. Cellulose 19:401–410
Cervin NT, Andersson L, Ng JBS, Olin P, Bergström L, Wågberg L (2013) Lightweight and strong cellulose materials made from aqueous foams stabilized by nanofibrillated cellulose. Biomacromolecules 14:503–511
Chin SF, Romainor ANB, Pang SC (2014) Fabrication of hydrophobic and magnetic cellulose aerogel with high oil absorption capacity. Mater Lett 115:241–243
Chtchigrovsky M, Primo A, Gonzalez P, Molvinger K, Robitzer M, Quignard F et al (2009) Functionalized chitosan as a green, recyclable, biopolymer-supported catalyst for the [3 + 2] Huisgen cycloaddition. Angew Chem Int E. 48(32):5916–5920
Cross J, Goswin R, Gerlach R, Fricke J (1989) Mechanical properties of SiO2–aerogels. Revue de physique appliquée, Colloque c4, Supplement au n° 4, tome 24, c4-184-c4-195
Cui S, Wang X, Zhang X, Xia W, Tang X, Lin B, Qi W, Zhang X, Shen X (2018) Preparation of magnetic MnFe2O4-cellulose aerogel composite and its kinetics and thermodynamics of Cu(II) adsorption. Cellulose 25:735–751
De Cicco F, Russo P, Reverchon E, García-González CA, Aquino RP, Del Gaudio P (2016) Prilling and supercritical drying: a successful duo to producecore-shell polysaccharide aerogel beads for wound healing. Carbohydr Polym 147:482–489
De France KJ, Hoare T, Cranston ED (2017) Review of hydrogels and aerogels containing nanocellulose. Chem Mater 29:4609–4631
De Oliveira W, Glasser WG (1996) Hydrogels from polysaccharides. 1. Cellulose beads for chromatographic support. J Appl Polym Sci 60:63–73
Demilecamps A (2015) Synthesis and characterization of polysaccharide-silica composite aerogels for thermal superinsulation. PhD thesis, Mines ParisTech, France
Demilecamps A, Reichenauer G, Rigacci A, Budtova T (2014) Cellulose–silica composite aerogels from “one-pot” synthesis. Cellulose 21:2625–2636
Demilecamps A, Beauger C, Hildenbrand C, Rigacci A, Budtova T (2015) Cellulose–silica aerogel. Carbohydr Polym 122:293–300
Demilecamps A, Alves M, Rigacci A, Reichenauer G, Budtova T (2016) Nanostructured interpenetrated organic-inorganic aerogels with thermal superinsulating properties. J Non Cryst Solids 452:259–265
Diascorn N, Calas S, Sallée H, Achard P, Rigacci A (2015) Polyurethane aerogels synthesis for thermal insulation–textural, thermal and mechanical properties. J Supercrit Fluids 106:76–84
Druel L, Bardl R, Vorwerg W, Budtova T (2017) Starch aerogels: a member of the family of thermal superinsulating materials. Biomacromolecules 18:4232–4239
Druel L, Niemeyer P, Milow B, Budtova T (2018) Rheology of cellulose-[DBNH][CO2Et] solutions and shaping into aerogel beads. Green Chem 20:3993–4002
Egal M, Budtova T, Navard P (2007) Structure of aqueous solutions of microcrystalline cellulose-sodium hydroxide below 0 °C and the limit of cellulose dissolution. Biomacromolecules 8:2282–2287
Escudero RR, Robitzer M, Di Renzo F, Quignard F (2009) Alginate aerogels as adsorbents of polar molecules from liquid hydrocarbons: hexanol as probe molecule. Carbohydr Polym 75:52–57
Feng J, Nguyen ST, Fan Z, Duong HM (2015) Advanced fabrication and oil absorption properties of super-hydrophobic recycled cellulose aerogels. Chem Eng J 270:168–175
Fink HP, Weigel P, Purz HJ, Ganster J (2001) Structure formation of regenerated cellulose materials from NMMO solutions. Prog Polym Sci 26:1473–1524
Firgo H, Rüf H, Hainbucher KM, Weber H (2004) Method for the production of a porous cellulose body. WO/2004/065424
Fischer F, Rigacci A, Pirard R, Berthon-Fabry S, Achard P (2006) Cellulose-based aerogels. Polymer 47:7636–7645
Fumagalli M, Ouhab D, Molina Boisseau S, Heux L (2013) Versatile gas-phase reactions for surface to bulk esterification of cellulose microfibrils aerogels. Biomacromolecules 14:3246–3255
Fumagalli M, Sanchez F, Molina-Boisseau S, Heux L (2015) Surface-restricted modification of nanocellulose aerogels in gas-phase esterification by di-functional fatty acid reagents. Cellulose 22:1451–1457
Ganesan K, Dennstedt A, Barowski A, Ratke L (2016) Design of aerogels, cryogels and xerogels of cellulose with hierarchical porous structures. Mater Des 92:345–355
Ganesan K, Budtova T, Ratke L, Gurikov P, Baudron V, Preibisch I, Niemeyer P, Smirnova I, Milow B (2018) Review on the production of polysaccharide aerogel particles. Materials 11:2144–2181
García-González CA, Alnaief M, Smirnova I (2011) Polysaccharide-based aerogels-promising biodegradable carriers for drug delivery systems. Carbohydr Polym 86:1425–1438
García-González CA, Uy JJ, Alnaief M, Smirnova I (2012) Preparation of tailor-made starch-based aerogel microspheres by the emulsion-gelation method. Carbohydr Polym 88:1378–1386
Gavillon R (2007) Preparation et caracterisation de materiaux cellulosiques ultra poreux. PhD Thesis. Mines ParisTech, France
Gavillon R, Budtova T (2007) Kinetics of cellulose regeneration from cellulose–NaOH–water gels and comparison with cellulose-N-methylmorpholine-N-oxide-water solutions. Biomacromolecules 8:424–432
Gavillon R, Budtova T (2008) Aerocellulose: new highly porous cellulose prepared from cellulose–NaOH aqueous solutions. Biomacromolecules 9:269–277
Geng H (2018) Preparation and characterization of cellulose/N,N′-methylene bisacrylamide/graphene oxide hybrid hydrogels and aerogels. Carbohydr Polym 196:289–298
Gericke M, Trygg J, Fardim P (2013) Functional cellulose beads: preparation, characterization, and applications. Chem Rev 113:4812–4836
Gibson LJ, Ashby MF (1997) Cellular solids. Structure and properties, 2nd edn. Cambridge University Press, Cambridge
Glenn GM, Irving DW (1995) Starch-based microcellular foams. Cereal Chem 72:155–161
Goimil L, Braga MEM, Dias AMA, Gómez-Amoza JL, Concheiro A, Alvarez-Lorenzo C, de Sousa HC, García-González CA (2017) Supercritical processing of starch aerogels and aerogel-loaded poly(e-caprolactone) scaffolds for sustained release of ketoprofen for bone regeneration. J CO2 Util 18:237–249
Groult S, Budtova T (2018a) Thermal conductivity/structure correlations in thermal super-insulating pectin aerogels. Carbohydr Polym 196:73–81
Groult S, Budtova T (2018b) Tuning structure and properties of pectin aerogels. Eur Polym J 108:250–261
Guilminot E, Gavillon R, Chatenet M, Berthon-Fabry S, Rigacci A, Budtova T (2008) New nanostructured carbons based on porous cellulose: elaboration, pyrolysis and use as platinum nanoparticles substrate for oxygen reduction electrocatalysis. J Power Sources 185:717–726
Guizard C, Leloup J, Deville S (2014) Crystal templating with mutually miscible solvents: a simple path to hierarchical porosity. J Am Ceram Soc 97:2020–2023
Hall CA, Le KA, Rudaz C, Radhi A, Lovell CS, DamionRA Budtova T, Ries ME (2012) Macroscopic and microscopic study of 1-ethyl-3-methyl-imidazolium acetate–water mixtures. J Phys Chem B 116:12810–12818
Hansen CM (2007) Hansen solubility parameters: a user’s handbook, 2nd edn. CRC Press, Boca Raton
Hedlund A, Kohnke T, Theliander H (2017) Diffusion in ionic liquid–cellulose solutions during coagulation in water: mass transport and coagulation rate measurements. Macromolecules 50:8707–8719
Hoepfner S, Ratke L, Milow B (2008) Synthesis and characterisation of nanofibrillar cellulose aerogels. Cellulose 15:121–129
Horvat G, Xhanari K, Finsgar M, Gradisnik L, Maver U, Knez Z, Novak Z (2017) Novel ethanol-induced pectin–xanthan aerogel coatings for orthopedic applications. Carbohydr Polym 166:365–376
Hu Y, Tong X, Zhuo H, Zhong L, Peng W, Wang S, Sun R (2016) 3D hierarchical porous N-doped carbon aerogel from renewable cellulose: an attractive carbon for high-performance supercapacitor electrodes and CO2 adsorption. RSC Adv 6:15788–15795
Hwang K, Kwon G-J, Yang J, Kim M, Hwang WJ, Youe W, Kim D-Y (2018) Chlamydomonas angulosa (Green Alga) and Nostoc commune (Blue-Green Alga) microalgae-cellulose composite aerogel beads: manufacture, physicochemical characterization, and Cd (II) adsorption. Materials 11:562–581
Innerlohinger J, Weber HK, Kraft G (2006a) Aerocellulose: aerogels and aerogel-like materials made from cellulose. Macromol Symp 244:126–135
Innerlohinger J, Weber HK, Kraft G (2006b) Aerocell Aerogels from cellulosic materials. Lenzing Ber 86:137–143
Ishida O, Kim D-Y, Kuga S, Nishiyama Y, Brown RM (2004) Microfibrillar carbon from native cellulose. Cellulose 11:475–480
IUPAC (2014) Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by McNaught AD, Wilkinson A. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006–) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. https://doi.org/10.1351/goldbook. Last update 2014-02-24; version: 2.3.3
Jiménez-Saelices C, Seantier B, Cathala B, Grohens Y (2017) Spray freeze-dried nanofibrillated cellulose aerogels with thermal superinsulating properties. Carbohydr Polym 157:105–113
Jin H, Nishiyama T, Wada M, Kuga S (2004) Nanofibrillar cellulose aerogels. Colloids Surf A Physicochem Eng Asp 240:63–67
Karadagli I, Schulz B, Schestakow M, Milow B, Gries T, Ratke L (2015) Production of porous cellulose aerogel fibers by an extrusion process. J Supercrit Fluids 106:105–114
Katti A, Shimpi N, Roy S, Lu H, Fabrizio EF, Dass A, Capadona LA, Leventis N (2006) Chemical, Physical, and Mechanical Characterization of Isocyanate Cross-linked Amine-Modified Silica Aerogels. Chem Mater 18:85–296
Kaushik M, Moores A (2016) Review: nanocelluloses as versatile supports for metal nanoparticles and their applications in catalysis. Green Chem 18:622–637
Kistler SS (1931) Coherent expanded aerogels and gellies. Nature 127(3211):741
Knez Z, Markocic E, Leitgeb M, Primozic M, Hrncic MK, Skerget M (2014) Industrial applications of supercritical fluids: a review. Energy 77:235–243
Kobayashi Y, Saito T, Isogai A (2014) Aerogels with 3D Ordered Nanofiber Skeletons of Liquid-Crystalline Nanocellulose Derivatives as Tough and Transparent Insulators. Angew Chem Int Ed 53:10394–10397
Köhnke T, Lund K, Brelid H, Westman G (2010) Kraft pulp hornification: a closer look at the preventive effect gained by glucuronoxylan adsorption. Carbohydr Polym 81:226–233
Korhonen JT, Kettunen M, Ras RHA, Ikkala O (2011) Hydrophobic Nanocellulose Aerogels as Floating, Sustainable, Reusable, and Recyclable Oil Absorbents. ACS Appl Mater Interfaces 3:1813–1816
Laity PR, Glover PM, Hay JN (2002) Composition and phase changes observed by magnetic resonance imaging during non-solvent induced coagulation of cellulose. Polymer 43:5827–5837
Laskowski J, Milow B, Ratke L (2015) The effect of embedding highly insulating granular aerogel incellulosic aerogel. J Supercrit Fluids 106:93–99
Lavoine N, Bergstrom L (2017) Nanocellulose-based foams and aerogels: processing, properties, and applications. J. Mater. Chem. A 5:16105–16117
Lei E, Li W, Ma C, Liu S (2018) An ultra-lightweight recyclable carbon aerogel from bleached softwood kraft pulp for efficient oil and organic absorption. Mater Chem Phys 214:291–296
Leventis N, Sotiriou-Leventis C, Zhang G, Rawashdeh A-MM (2002) Nanoengineering strong silica aerogels. Nano Lett 2:957–960
Li S, Lyons-Hart J, Banyasz J, Shafer K (2001) Real-time evolved gas analysis by FTIR method: an experimental study of cellulose pyrolysis. Fuel 80:1809–1817
Liang H-W, Wu Z-Y, Chen L-F, Li C, Yu S-H (2015) Bacterial cellulose derived nitrogen-doped carbon nanofiber aerogel: an efficient metal-free oxygen reduction electrocatalyst for zinc-air battery. Nano Energy 11:366–376
Liao Q, Su X, Zhu W, Hu W, Qian Z, Li L, Yao J (2016) Flexible and durable cellulose aerogels for highly effective oil/water separation. RSC Adv 6:63773–63781
Liebert T (2010) Cellulose solvents – remarkable history, bright future. In: Liebert et al (eds) Cellulose solvents: for analysis, shaping and chemical modification. ACS symposium series. American Chemical Society, Washington
Liebner F, Potthast A, Rosenau T, Haimer E, Wendland M (2008) Cellulose aerogels: highly porous, ultra-lightweight materials. Holzforschung 62:129–135
Liebner F, Haimer E, Potthast A, Loidl D, Tschegg S, Neouze MA (2009) Cellulosic aerogels as ultra-lightweight materials. Part 2: synthesis and properties. Holzforschung 63:3–11
Liebner F, Dunareanu R, Opietnik M, Haimer E, Wendland M, Werner C, Maitz M, Seib P, Neouze M-A, Potthast A, Rosenau T (2012) Shaped hemocompatible aerogels from cellulose phosphates: preparation and properties. Holzforschung 66:317–321
Liebner F, Pircher N, Schimper C, Haimer E, Rosenau T (2016) Aerogels: cellulose-based. In: Encyclopedia of biomedical polymers and polymeric biomaterials. Taylor and Francis, New York, pp 37–75
Lin C, Zhan H, Liu M, Fu S, Lucia LA (2009a) Novel preparation and characterization of cellulose microparticles functionalized in ionic liquids. Langmuir 25:10116–10120
Lin Y-C, Cho J, Tompsett GA, Westmoreland PR, Huber GW (2009b) Kinetics and mechanism of cellulose pyrolysis. J Phys Chem C 113:20097–20107
Lin R, Li A, Zheng T, Lu L, Cao Y (2015) Hydrophobic and flexible cellulose aerogel as an efficient, green and reusable oil sorbent. RSC Adv 5:82027–82033
Litschauer M, Neouze M-A, Haimer E, Henniges U, Potthast A, Rosenau T, Liebner F (2011) Silica modified cellulosic aerogels. Cellulose 18:143–149
Liu W, Budtova T, Navard P (2011) Influence of ZnO on the properties of dilute and semi-dilute cellulose–NaOH–water solutions. Cellulose 18:911–920
Liu S, Yu T, Hu N, Liu R, Liu X (2013) High strength cellulose aerogels prepared by spatially confined synthesis of silica in bioscaffolds. Colloids Surf A Physicochem Eng Asp 439:159–166
Liu P, Borrell PF, Bozic M, Kokol V, Oksman K, Mathew AP (2015) Nanocelluloses and their phosphorylated derivatives for selective adsorption of Ag+, Cu2+ and Fe3+ from industrial effluents. J Hazard Mater 294:177–185
Lozinsky VI, Galaev IYu, PlievaFM Savina IN, Jungvid H, Mattiasson B (2003) Polymeric cryogels as promising materials of biotechnological interest. Trends Biotechnol 21:445–451
Lozinsky VI, Damshkaln LG, Bloch KO, Vardi P, Grinberg NV, Burova TV, Grinberg VY (2008) Cryostructuring of polymer systems. XXIX. Preparation and characterization of supermacroporous (spongy) agarose-based cryogels used as three-dimensional scaffolds for culturing insulin-producing cell aggregates. J Appl Polym Sci 108:3046–3062
Lu X, Arduini-Schuster MC, Kuhn J, Njilsson O, Fricke J, Pekala RW (1992) Thermal conductivity of monolithic organic aerogels. Science 255:971–972
Lu A, Liu Y, Zhang L, Potthast A (2011) Investigation on metastable solution of cellulose dissolved in NaOH/urea aqueous system at low temperature. J Phys Chem B 115:12801–12808
Luo X, Zhang L (2010) Creation of regenerated cellulose microspheres with diameter ranging from micron to millimeter for chromatography applications. J Chromatogr A 1217:5922–5929
Lv L, Fan Y, Chen Q, Zhao Y, Hu Y, Zhang Z, Chen N, Qu L (2014) Three-dimensional multichannel aerogel of carbon quantum dots for high-performance supercapacitors. Nanotechnology 25:235401
Maatar W, Boufi S (2015) Poly(methacylic acid-co-maleic acid) grafted nanofibrillated cellulose as a reusable novel heavy metal ions adsorbent. Carbohydr Polym 126:199–207
Mäki-Arvelaa P, Anugwoma I, Virtanena P, Sjöholma R, Mikkola JP (2010) Dissolution of lignocellulosic materials and its constituents using ionic liquids—a review. Ind Crops Prod 32:175–201
Maleki H (2016) Recent advances in aerogels for environmental remediation applications: a review. Chem Eng J 300:98–118
Maleki H, Duraes L, Portugal A (2014) An overview on silica aerogels synthesis and different mechanical reinforcing strategies. J Non Cryst Solids 385:55–74
Markevicius G, Ladj R, Niemeyer P, Budtova T, Rigacci A (2017) Ambient-dried thermal superinsulating monolithic silica-based aerogels with short cellulosic fibers. J Mater Sci 52:2210–2221
Martins M, Barros AA, Quraishi S, Gurikov P, Raman SP, Smirnova I, Duarte ARC, Reis RL (2015) Preparation of macroporous alginate-based aerogels for biomedical applications. J Supercrit Fluids 106:152–159
Martoïa F, Cochereau T, Dumont PJJ, Orgéas L, Terrien M, Belgacem MN (2016) Cellulose nanofibril foams: links between ice-templating conditions, microstructures and mechanical properties. Mater Des 104:376–391
Meador MAB, Alemn CR, Hanson K, Ramirez N, Vivod SL, Wilmoth N, McCorkle L (2015) Polyimide aerogels with amide cross-links: a low cost alternative for mechanically strong polymer aerogels. ACS Appl Mater Interfaces 7:1240–1249
Meador MAB, Agnello M, McCorkle L, Vivod SL, Wilmoth N (2016) Moisture-resistant polyimide aerogels containing propylene oxide links in the backbone. ACS Appl Mater Interfaces 8:29073–29079
Meng Y, Young TM, LiuP ContescuCI, Huang B, Wang S (2015) Ultralight carbon aerogel from nanocellulose as a highly selective oil absorption material. Cellulose 22:435–447
Mi Q-Y, Ma S-R, Yu J, He J-S, Zhang J (2016) Flexible and transparent cellulose aerogels with uniform nanoporous structure by a controlled regeneration process. ACS Sustain Chem Eng 4:656–660
Mohamed SMK, Ganesan K, Milow B, Ratke L (2015) The effect of zinc oxide (ZnO) addition on the physical and morphological properties of cellulose aerogel beads. RSC Adv 5:90193–90201
Mulik S, Sotiriou-Leventis C, Leventis N (2007) Time-efficient acid-catalyzed synthesis of resorcinol-formaldehyde aerogels. Chem Mater 19:6138–6144
Mulyadi A, Zhang Z, Deng Y (2016) Fluorine-free oil absorbents made from cellulose nanofibril aerogels. ACS Appl Mater Interfaces 8:2732–2740
Nguyen ST, Feng J, Ng SK, Wong JPW, Tan VBC, Duong HM (2014) Advanced thermal insulation and absorption properties of recycled cellulose aerogels. Colloids Surf A Physicochem Eng Asp 445:128–134
Nyström G, Fernández-Ronco MP, Bolisetty S, Mazzotti M, Mezzenga R (2016) Amyloid templated gold aerogels. Adv Mater 28:472–478
O’Connell DW, Birkinshaw C, O’Dwyer TF (2008) Heavy metal adsorbents prepared from the modification of cellulose: a review. Biores Technol 99:6709–6724
Olsson RT, Samir MASA, Salazar-Alvarez G, Belova L, LA StromV Berglund, Ikkala O, Nogues J, Gedde UW (2010) Making flexible magnetic aerogels and stiff magnetic nanopaper using cellulose nanofibrils as templates. Nat Nanotechnol 5:584–588
Ookuna S, Igarashi K, Hara M, Aso K, Yoshidone H, Nakayama H, Suzuki K, Nakajima K (1993) Porous ion-exchanged fine cellulose particles, method for production thereof, and affinity carrier. USOO5196527A
Pekala RW (1989) Organic aerogels from the polycondensation of resorcinol with formaldehyde. J Mater Sci 24:3221–3227
Pekala RW, Alviso CT, LeMay JD (1990) Organic aerogels: microstructural dependence of mechanical properties in compression. J Non Cryst Solids 125:67–75
Pekala RW, Alviso CT, Lu X, Gross J, Fricke J (1995) New organic aerogels based upon a phenolic-furfural reaction. J Non Cryst Solids 188:34–40
Pekala RW, Farmer JC, Alviso CT, Tran TD, Mayer CT, Miller JM, Dunn B (1998) Carbon aerogels for electrochemical applications. J Non Cryst Solids 225:74–80
Pierre AC (2011) History of aerogels. In: Aegerter MA et al (eds) Aerogels handbook, advances in sol–gel derived materials and technologies. Springer, New York, pp 813–831
Pinkert A, Marsh KN, Pang S, Staiger MP (2009) Ionic liquids and their interaction with cellulose. Chem Rev 109:6712–6728
Pinnow M, Fink HP, Fanter C, Kunze J (2008) Characterization of highly porous materials from cellulose carbamate. Macromol Symp 262:129–139
Pircher N, Fischhuber D, Carbajal L, Strau C, Nedelec J-M, Kasper C, Rosenau T, Liebner F (2015) Preparation and reinforcement of dual-porous biocompatible cellulose scaffolds for tissue engineering. Macromol Mater Eng 300:911–924
Pircher N, Carbajal L, Schimper C, Bacher M, Rennhofer H, Nedelec J-M, Lichtenegger HC, Rosenau T, Liebner F (2016) Impact of selected solvent systems on the pore and solid structure of cellulose aerogels. Cellulose 23:1949–1966
Plappert SF, Nedelec J-M, Rennhofer H, Lichtenegger HC, Liebner FW (2017) Strain hardening and pore size harmonization by uniaxial densification: a facile approach toward superinsulating aerogels from nematic nanofibrillated 2,3-dicarboxyl cellulose. Chem Mater 29:6630–6641
Pour G, Beauger C, Rigacci A, Budtova T (2015) Xerocellulose: lightweight, porous and hydrophobic cellulose prepared via ambient drying. J Mater Sci 50:4526–4535
Quignard F, Valentin R, Di Renzo F (2008) Aerogel materials from marine polysaccharides. New J Chem 32:1300–1310
Quraishi S, Martins M, Barros AA, Gurikov P, Raman SP, Smirnova I, Duarte ARC, Reis RL (2015) Novel non-cytotoxic alginate–lignin hybrid aerogels as scaffolds fortissue engineering. J Supercrit Fluids 105:1–8
Raman SP, Gurikov P, Smirnova I (2015) Hybrid alginate based aerogels by carbon dioxide induced gelation: novel technique for multiple applications. J Supercrit Fluids 106:23–33
Rege A, Schestakow M, Karadagli I, Ratke L, Itskov M (2016) Micro-mechanical modelling of cellulose aerogels from molten salt hydrates. Soft Matter 12:7079–7088
Rein DM, Cohen Y (2011) Aeropolysaccharides, composites and preparation thereof. EP 2 354 165 A1
Robitzer M, Di Renzo F, Quignard F (2011) Natural materials with high surface area. Physisorption methods for the characterization of the texture and surface of polysaccharide aerogels. Microporous Mesoporous Mater 140:9–16
Rooke J, de Matos Passos C, Chatenet M, Sescousse R, Budtova T, Berthon-Fabry S, Mosdale R, Maillard F (2011) Synthesis and properties of platinum nanocatalyst supported on cellulose-based carbon aerogel for applications in PEMFCs. J Electrochem Soc 158:B779–B789
Rooke J, Sescousse R, Budtova T, Berthon-fabry S, Simon B, Chatenet M (2012) Cellulose-based nanostructured carbons for energy conversion and storage devices. In: Rufford T, Hulicova-Jurcakova D, Zhu J (eds) Green carbon materials: advances and applications. Pan Stanford Publishing Pte Ltd, Singapore, pp 89–111
Rosenberg P, Suominen I, Rom M, Janicki J, Fardim P (2007) Tailored cellulose beads for novel applications. Cellul Chem Technol 41:243–254
Roy C, Budtova T, Navard P (2003) Rheological properties and gelation of aqueous cellulose–NaOH solutions. Biomacromolecules 4:259–264
Rudaz C (2013) Cellulose and pectin aerogels: towards their nano-structuration. PhD thesis, MINES ParisTech
Rudaz C, Courson R, Bonnet L, Calas-Etienne S, Sallée H, Budtova T (2014) Aeropectin: fully biomass-based mechanically strong and thermal superinsulating aerogel. Biomacromolecules 15:2188–2195
Sai H, Fu R, Xing L, Xiang J, Li Z, Li F, Zhang T (2015) Surface modification of bacterial cellulose aerogels’ web-like skeleton for oil/water separation. ACS Appl Mater Interfaces 7:7373–7381
Schestakow M, Karadagli I, Ratke L (2016a) Cellulose aerogels prepared from an aqueous zinc chloride salt hydrate melt. Carbohydr Polym 137:642–649
Schestakow M, Muench F, Reimuth C, Ratke L, Ensinger W (2016b) Electroless synthesis of cellulose–metal aerogel composites. Appl Phys Lett 108:213108
Seantier B, Bendahou D, Bendahou A, Grohens Y, Kaddami H (2016) Multi-scale cellulose based new bio-aerogel composites with thermalsuper-insulating and tunable mechanical properties. Carbohydr Polym 138:335–348
Sehaqui H, Zhou Q, Ikkala O, Berglund LA (2011) Strong and tough cellulose nanopaper with high specific surface area and porosity. Biomacromolecules 12:3638–3644
Sehaqui H, Zimmermann T, Tingaut P (2014) Hydrophobic cellulose nanopaper through a mild esterification procedure. Cellulose 21:367–382
Sescousse R (2010) Nouveaux matériaux cellulosiques ultra-poreux et leurs carbones à partir de solvants verts. PhD thesis, Mines ParisTech, France
Sescousse R, Budtova T (2009) Influence of processing parameters on regeneration kinetics and morphology of porous cellulose from cellulose–NaOH–water solutions. Cellulose 16:417–426
Sescousse R, Smacchia A, Budtova T (2010) Influence of lignin on cellulose-NaOH-water mixtures properties and on Aerocellulose morphology. Cellulose 17:1137–1146
Sescousse R, Gavillon R, Budtova T (2011a) Aerocellulose from cellulose–ionic liquid solutions: preparation, properties and comparison with cellulose–NaOH and cellulose–NMMO routes. Carbohydr Polym 83:1766–1774
Sescousse R, Gavillon R, Budtova T (2011b) Wet and dry highly porous cellulose beads from cellulose–NaOH–water solutions: influence of the preparation conditions on beads shape and encapsulation of inorganic particles. J Mater Sci 46:759–765
Shen J, Guan DY (2011) Preparation and application of carbon aerogels. In: Aegerter MA et al (eds) Aerogels handbook, advances in sol–gel derived materials and technologies. Springer, New York, pp 813–831
Shi J, Lu L, Guo W, Sun Y, Cao Y (2013a) An environment-friendly thermal insulation material from cellulose and plasma modification. J Appl Polym Sci 130:3652–3658
Shi J, Lu L, Guo W, Zhang J, Cao Y (2013b) Heat insulation performance, mechanics and hydrophobic modification of cellulose–SiO2 composite aerogels. Carbohydr Polym 98:282–289
Shi Z, Huang J, Liu C, Ding B, Kuga S, Cai J, Zhang L (2015) Three-dimensional nanoporous cellulose gels as a flexible reinforcement matrix for polymer nanocomposites. ACS Appl Mater Interfaces 7:22990–22998
Sorensen L, Strouse GF, Stiegman AE (2006) Fabrication of stable low-density silica aerogels containing luminescent ZnS capped CdSe quantum dots. Adv Mater 18:1965–1967
Svensson A, Larsson PT, Salazar-Alvarez G, Wågberg L (2013) Preparation of dry ultra-porous cellulosic fibres: characterization and possible initial uses. Carbohydr Polym 92:775–783
Tan C, Fung B, Newman JK, Vu C (2001) Organic aerogels with very high impact strength. Adv Mater 13:644–646
Teichner SJ (1986) Aerogels of inorganic oxides. In: Frick J (ed) Aerogels, Springer proceedings in physics 6, proceedings of the first international symposium, Worzburg, Fed. Republic of Germany, September 23–25. 1985 Springer, Heidelberg, pp 22–30
Tejado A, Chen WC, Alam MN, van de Ven TGM (2014) Superhydrophobic foam-like cellulose made of hydrophobized cellulose fibres. Cellulose 21:1735–1743
Trygg J, Fardim P, Gericke M, Mäkilä E, Salonen J (2013) Physicochemical design of the morphology and ultrastructure of cellulose beads. Carbohydr Polym 93:291–299
Trygg J, Yildir E, Kolakovic R, Sandler N, Fardim P (2014) Anionic cellulose beads for drug encapsulation and release. Cellulose 21:1945–1955
Tsioptsias C, Stefopoulos A, Kokkinomalis I, Papadopoulou L, Panayiotou C (2008) Development of micro- and nano-porous composite materials by processing cellulose with ionic liquids and supercritical CO2. Green Chem 10:965–971
Veronovski A, Tkalec G, Knez Z, Novak Z (2014) Characterisation of biodegradable pectin aerogels and their potential use as drug carriers. Carbohydr Polym 113:272–278
Voon LK, Pang SC, Chin SF (2016) Highly porous cellulose beads of controllable sizes derived from regenerated cellulose of printed paper wastes. Mater Lett 164:264–266
Voon LK, Pang SC, Chin SF (2017) Porous cellulose beads fabricated from regenerated cellulose as potential drug delivery carriers. J Chem 2017:1–11
Wan Ngah WS, Teong LC, Hanafiah MAKM (2011) Adsorption of dyes and heavy metal ions by chitosan composites: a review. Carbohydr Polym 83:1446–1456
Wang Z, Liu S, Matsumoto Y, Kuga S (2012) Cellulose gel and aerogel from LiCl/DMSO solution. Cellulose 19:393–399
Wang H, Shao Z, Bacher M, Liebner F, Rosenau T (2013a) Fluorescent cellulose aerogels containing covalently immobilized (ZnS)x(CuInS2)12x/ZnS (core/shell) quantum dots. Cellulose 20:3007–3024
Wang R, Li G, Dong Y, Chi Y, Chen G (2013b) Carbon quantum dot-functionalized aerogels for NO2 gas sensing. Anal Chem 85:8065–8069
Wang H, Gong Y, Wang Y (2014a) Cellulose-based hydrophobic carbon aerogels as versatile and superior adsorbents for sewage treatment. RSC Adv 4:45753–45759
Wang L, Schutz C, Salazar-Alvarez G, Titirici M-M (2014b) Carbon aerogels from bacterial nanocellulose as anodes for lithium ion batteries. RSC Adv 4:17549–17554
Weigold L, Reichenauer G (2014) Correlation between mechanical stiffness and thermal transport along the solid framework of a uniaxially compressed polyurea aerogel. J Non Cryst Solids 406:73–78
White RJ, Budarin V, Luque R, Clark JH, Macquarrie DJ (2009) Tuneable porous carbonaceous materials from renewable resources. Chem Soc Rev 38:3401–3418
White RJ, Antonio C, Budarin VL, Bergstrom E, Thomas-Oates J, Clark JH (2010a) Polysaccharide-derived carbons for polar analyte separations. Adv Funct Mater 20:1834–1841
White RJ, Budarin VL, Clark JH (2010b) Pectin-derived porous materials. Chem Eur J 16:1326–1335
Wong JCH, Kaymak H, Brunner S, Koebel MM (2014) Mechanical properties of monolithic silica aerogels made from polyethoxydisiloxanes. Microporous Mesoporous Mater 183:23–29
Wu Z-S, Yang S, Sun Y, Parvez K, Feng X, Müllen K (2012) 3D nitrogen-doped graphene aerogel-supported Fe3O4 nanoparticles as efficient electrocatalysts for the oxygen reduction reaction. J Am Chem Soc 134:9082–9085
Wu Z-Y, Li C, Liang H-W, Chen J-F, Yu S-H (2013) Ultralight, flexible, and fire-resistant carbon nanofiber aerogels from bacterial cellulose. Angew Chem 125:2997–3001
Yang X, Fei B, Ma J, Liu X, Yang S, Tian G, Jiang Z (2018) Porous nanoplatelets wrapped carbon aerogels by pyrolysis of regenerated bamboo cellulose aerogels as supercapacitor electrodes. Carbohydr Polym 180:385–392
Zhang Z, Sèbe G, Rentsch D, Zimmermann T, Tingaut P (2014) Ultralightweight and Flexible silylated nanocellulose sponges for the selective removal of oil from water. Chem Mater 26:2659–2668
Zhang H, Li Y, Xu Y, Lu Z, Chen L, Huang L, Fan M (2016) Versatile fabrication of a superhydrophobic and ultralight cellulose-based aerogel for oil spillage clean-up. Phys Chem Chem Phys 18:28297–28306
Zhang M, Dou M, Wang M, Yu Y (2017) Study on the solubility parameter of supercritical carbon dioxide system by molecular dynamics simulation. J Mol Liq 248:322–329
Zhang DY, Zhang N, Song P, Hao JY, Wan Y, Yao XH, Chen T, Li L (2018) Functionalized cellulose beads with three dimensional porous structure for rapid adsorption of active constituents from Pyrola incarnate. Carbohydr Polym 181:560–569
Zhou S, Chen G, Feng X, Wang M, Song T, Liu D, Lu F, Qi H (2018) In situ MnOx/N-doped carbon aerogels from cellulose as monolithic and highly efficient catalysts for the upgrading of bioderived aldehydes. Green Chem 20:3593–3603
Zhuo H, Hu Y, Tong X, Zhong L, Peng W, Sun R (2016) Sustainable hierarchical porous carbon aerogel from cellulose forhigh-performance supercapacitor and CO2 capture. Ind Crops Prod 87:229–235
Zu G, Shen J, Zou L, Wang F, Wang X, Zhang Y, Yao X (2016) Nanocellulose-derived highly porous carbon aerogels for supercapacitors. Carbon 99:203–211
Acknowledgments
I would like to devote this review and warmly thank my PhD and Master students and post-doctoral researchers without whom the progress in bio-aerogels in general and this article in particular would not have been possible.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Budtova, T. Cellulose II aerogels: a review. Cellulose 26, 81–121 (2019). https://doi.org/10.1007/s10570-018-2189-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-2189-1