
Abstract
1-Butyl-3-methylimidazolium chloride ([BMIM]Cl) was selected as co-solvent to dissolve cellulose and silk fibroin and the cellulose/silk fibroin blend fibers were fabricated with dry-jet wet spinning technology. The phase morphology of cellulose and silk fibroin in the blend fibers was studied by scanning electron microcopy and laser scanning confocal microscope. It is shown that the cellulose is in the continuous phase and silk fibroin exists as “fibril-like” in cellulose, in which the radial dimension of silk fibroin phase is 0.5–1.0 μm. The phase size of silk fibroin along the fiber axis increased with the increase of silk fibroin content and draw ratio. From the wide-angle X-ray scattering, it is found that the total crystallinity of the blend fibers decreased with increasing silk fibroin content. The hydrogen bond between cellulose and silk fibroin was observed from Fourier transform infrared spectra. Although the tensile strength and initial modulus of blend fibers decreased with increasing silk fibroin content, the tensile strength of blend fibers contain 35 wt% silk fibroin was up to 191 MPa.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Both cellulose and protein are abundant renewable resources, so the utilization of cellulose and silk fibroin has gained widely attention. The regenerated cellulose fiber such as viscose fiber and Lyocell fiber has been comprehensively developed (Fink et al. 2001). Protein material is biocompatible and non-irritation of human skin (Koller et al. 2007). However, the utilization of protein resource is still under various restrictions (Heslot 1998). The regenerated protein fibers have limitation as low strength (Um et al. 2001; Ki et al. 2007) or inefficiency (Zhou et al. 2009), which is caused by the severe degradation of protein during dissolution process (Vollrath et al. 2007). In order to improve the mechanical property of the regenerated protein fiber, the commercial regenerated protein fibers are prepared by grafting to synthetic polymer or dispersing in spinning dope. One of the commercial regenerated protein fiber is polyacrylonitrile-g-milk protein fiber which has excellent feeling and luster like silk. But the price of such fiber is very high (Morimoto 1970). Another protein grafting fiber is poly(vinyl alcohol)-g-soybean fiber which has disadvantages as pilling, not resistance to laundering and poor dimensional stability (Zhang et al. 2003). Liu et al. (2009) dispersed silk protein powders into polyurethane/dimethyl formamide dope and prepared blend fibers by wet spinning. However, the particle sizes of silk protein powders are too large for fiber forming and the dispersion of silk protein in the blend fiber is not good which makes it easy to fall off.
To make full use of the natural renewable resources, cellulose/protein blend fibers which have excellent miscibility and mechanical property can be expected to be prepared with solution blending method. Hirano et al. (2002) mixed the 10 % aqueous silk fibroin solution and 9 % viscose solution together to fabricate cellulose/silk fibroin blend fiber by wet spinning. The strength of the fiber is low (less than 1.5 cN/dtex) which is caused by the instability of silk fibroin aqueous LiBr/viscose solution mixture. As we know, the performance of polymer blends is strongly related to the miscibility of polymer components (Macosko 2000). Co-solvent, which can dissolve protein and other components under the same condition, is an effective method to improve the miscibility of protein blends. N,N-dimethylacetamide/LiCl was used as co-solvent for cellulose/silk fibroin blend fiber, the miscibility between cellulose and silk fibroin was enhanced (Marsano et al. 2007). Chen and Zhang (2004) dissolved cellulose and soybean protein with NaOH/thiourea aqueous solution as co-solvent and fabricated cellulose/soy bean blend membranes. The phase morphology of soybean protein and cellulose was studied by transmission electron microscope (TEM). The size distribution of soybean protein is about 50–100 nm. Marsano et al. (2008) made cellulose/silk fibroin blend membranes and fibers with N-methylmorpholine N-oxide hydrates (NMMO/H2O) as co-solvent. The phase size of silk fibroin is 2–10 μm in the blend membranes. Ionic liquid has also been found to be able to dissolve cellulose and silk fibroin under the same condition (Phillips et al. 2004; Swatloski et al. 2002). Shang et al. (2011) prepared cellulose/silk fibroin blend membranes using [BMIM]Cl as solvent. The surface morphology of the blend membranes was studied by atomic force microscopy. The surface morphology became smoother and the strength of the blend membrane was improved with the increase of cellulose content. The interaction between silk fibroin and cellulose in the blend has also been comprehensively discussed by Fourier transform infrared spectra (FTIR) (Shang et al. 2013). Otherwise, the mechanical strength of cellulose/silk fibroin blend membrane can be enhanced by improved coagulation technology as vapored methanol and cold pressing during vacuum dry (Zhou et al. 2013).
In our past work, the rheological behaviors of cellulose/silk fibroin solution using 1-butyl-3-methylimidazolium chloride ([BMIM]Cl) as co-solvent were studied. The change of microstructure in the solution was imaged according to the derivations of viscosity, dynamic modulus and shear rate. The phase morphology of cellulose and silk fibroin deduced from the solution property was agreed with the result from morphology of regenerated cellulose/silk fibroin film (Yao et al. 2014). As the continuous work, the regenerated cellulose/silk fibroin blend fibers were fabricated with [BMIM]Cl as co-solvent. The effect of cellulose/silk fibroin blend ratio, draw ratio on the phase morphology, structure and property of blend fibers were discussed.
Experiments
Materials
Cellulose pulp [degree of polymerization (DP) = 500] was supplied by Shandong Silver Eagle Chemical Fiber Co. Ltd, China. Silk fibroin was degummed from silk cocoon in boiling 0.5 % (w/w) Na2CO3 solution with a bath ratio of 1:50 and then washed with deionized water to remove silk sericin (Cao et al. 2009a).
1-Butyl-3-methylimidazolium chloride ([BMIM]Cl) was synthesized and purified in our laboratory as previously described (Csihony et al. 2002).
Solution preparation
The 8 wt% concentration of cellulose/silk fibroin/[BMIM]Cl solution were prepared by changing the weight ratio of cellulose to silk fibroin as 10/0 (Cell), 8/2 (CS82), 6/4 (CS64), 0/10 (SF). Cellulose and silk fibroin were dried under vacuum at 80 °C to remove water. Spinning solutions were obtained by kneading at 90 °C.
Spinning trials
The cellulose/silk fibroin blend fiber was spun through a laboratory-scale dry-jet wet spinning equipment. The cellulose/silk fibroin/[BMIM]Cl solution with 8 wt% was degassed and extruded through the spinneret orifices of 0.15 mm in diameter at 85 °C. The air gap distance is 50 mm and the air temperature is 25 °C. The coagulation bath was ethanol at the temperature of 10 °C. The blend fibers were stretched in the coagulation bath. The possible ranges of drawing ratios for this blend fibers is 1–5 according to the results of our experiments. The as-spun fiber was further washed in ethanol bath, and then dried and taken up. The spinning condition of the blend fibers are listed in Table 1. The viscosity of pure silk fibroin/[BMIM]Cl solution is too low to be used for dry-jet wet spinning.
Measurements
The zero shear rate viscosity of the solutions was measured by rotational rheometer (Physica MCR 301, Anton Paar). The 25-mm-diameter concentric parallel plate geometry was utilized. The chosen gap was 1 mm and the chosen temperature was 85 °C for all solutions.
The protein content in the blend fibers was obtained by testing nitrogen element (N) content with element analyzer (Elementar Vario EL III, German). The protein content was calculated according to the Eq. (1) (Yang et al. 2002):
where PSF is protein content, N is the nitrogen content in blend fibers.
The cross-section morphology of the fibers was measured on environment scanning electron microscope (SEM) (Quanta-250, FEI, USA). The fibers were cryogenically broken under liquid nitrogen and sputter-coated with a thin layer of gold.
The intrinsic fluorescence of silk fibroin in the blend fibers was captured by a laser scanning confocal microscope (LSCM) (TCS SP5, Leica, German), the excitation wavelength is 488 nm and the emission wavelength range is 500–550 nm.
Wide-angle X-ray scattering (WAXS) was measured at Beamline (BL16B) in Shanghai Synchrotron Radiation Facility with the wavelength of 0.124 nm. A bundle of blend fibers were put in a holder with the fiber direction perpendicular to the X-ray beam. The distance between the sample and Mar-CCD (165) was 107.8 mm. The 2D WAXS was processed with the software of Xpolar and Peakfit. The total crystallinity of the blend fibers was calculated according to Eq. (2):
where Wc,x is the total crystallinity, Ic and Ia are respectively the crystal peak area and amorphous phase area. The crystal orientation f c of cellulose along the longitudinal axis was calculated as previously described (Jiang et al. 2012b).
FTIR was measured by a Nicolet spectrometer (Nexus 670, Thermo Fisher, USA) at wavenumber of 700–4,000 cm−1 with a resolution of 0.09 cm−1. A bundle of blend fibers were pinched under total internal reflection accessory.
The mechanical property of the fibers was tested on a fiber mechanical strength tester (XQ-2, China). The stretching rate is 20 mm/min with a 20 mm gauge length. The diameter of the fiber was measured using microscope (XSZ–360AP, China). The tensile strength, initial modulus and elongation were calculated as the average of at least 20 measurements from stress–strain curves.
The moisture sorption curves of the blend fibers were determined by the following method. The blend fibers were dried at 105 °C to constant weight. The same fibers were then kept under standard condition (20 °C, 65 % RH). The mass change of each sample with time was measured to draw sorption curves. The moisture desorption curves were obtained as follows, the blend fibers were conditioned under 20 °C, 100 % RH. The same fibers were then kept under standard condition to get the weight change and draw desorption curves.
Results and discussion
Protein content of cellulose/silk fibroin blend fibers
The structure and property of regenerated silk fibroin fiber are greatly affected by coagulation solvents. Water, methanol, acetonitrile were chosen by Phillips et al. (2005) as coagulation solvents to prepare regenerated silk fiber. Only the fiber which is coagulated by methanol could be handled. So it is necessary to show that ethanol is the effective coagulation solvent for cellulose/silk fibroin blend fiber from the result of protein content measurement. The nitrogen content was measured by elemental analysis, the protein content was calculated according to Eq. (1). As shown in Table 2, there is only a slightly reduction of silk fibroin. Both of cellulose and silk fibroin were coagulated, which proved that ethanol is the effective coagulant for cellulose and silk fibroin. The viscosity of silk fibroin phase is low which might came to the coagulation bath with the solvent [BMIM]Cl together. With the increase of draw ratio, the diameters of the blend fibers decreased, the specific surface area between blend fibers and coagulation bath also decreased, more silk fibroin entered the coagulant with [BMIM]Cl, the weight loss of silk fibroin increased.
Morphology of cellulose/silk fibroin blend fibers
The cross-section morphology of the blend fibers was observed by SEM, as shown in Fig. 1. The typical fibrous and microporous structure exist in the blend fibers, which are similar as that of regenerated cellulose fiber (Fink et al. 2001; Cao et al. 2009b). These features appeared in all samples and were independent on the blend ratio and draw ratio.

SEM of the cross-section of cellulose/silk fibroin blend fibers
In order to study the phase morphology of cellulose and silk fibroin in the blend fibers, the blend fiber was observed by LSCM. Due to the existence of amino acids, the intrinsic fluorescence spectroscopy of silk fibroin has been found. Georgakoudi et al. (2007) characterized the intrinsic fluorescence spectra of silk fibroin from the excited range 250–335 nm and emission range 265–600 nm. Rice et al. (2008) used two photon excited fluorescence and second harmonic generation as non-invasive characterization to study the morphology of silk fibroin in solution, film, hydrogel and scaffold. The obvious silk fibroin morphology was obtained from emission range of 380–700 nm. The detection scale of LSCM varies from micro-scale to millimeter-scale which can be used as a new method to analyze the protein phase morphology in the blend material. As shown in Fig. 2, the figures of regenerated cellulose fibers are black, which means the absence of cellulose fluorescence. The green regions show the dispersion of silk fibroin, what exist as “fibril-like” in cellulose phase, in which the radial dimension of silk fibroin phase is 0.5–1.0 μm. Compare with sample CS64-3 and sample CS82-3, the radial dimension is a little larger with the increase of silk fibroin content. Along the fiber axis, the dispersion of silk fibroin in sample CS82-3 is not continuous. With the increase of draw ratio, the size of silk fibroin phase along the fiber axes become longer, as shown in sample CS82-4.

LSCM images of cellulose/silk fibroin blend fibers along the fiber axes
Crystalline structure of cellulose/silk fibroin blend fibers
As shown in Fig. 3a, the crystalline structure of regenerated cellulose fibers is cellulose II (Langan et al. 2001) with the characteristic reflections (\(\bar{1}\)10), (110), (012), (020), (103) (The corresponding peaks of 2θ at the wavelength of 0.124 nm are respectively 9.7°, 16.1°, 16.4°, 17.8°, 23.0°) (Jiang et al. 2012a). The crystalline structure of silk fibroin which is coagulated by ethanol is silk II, the characteristic reflections are (002) and (201/021) (the corresponding peaks are respectively 16.5°, 20.5° at the wavelength of 0.124 nm) (Sun et al. 2012). These peaks were chosen to calculate the total crystallinity of the blend fibers. As shown in Fig. 3b, c, the total crystallinity of blend fibers and the crystal orientation of cellulose decreased with increasing silk fibroin content. Because the ordering of cellulose and silk fibroin deduced owing to the interface of molecular chains (Zhou et al. 2013). During drawing process, the crystal orientation of cellulose can be enhanced with drawing. The separated silk fibroin domains are stretched. But the total crystallinity of the cellulose/silk fibroin blend fiber is hard to be enhanced with drawing owing to the low crystalline of silk fibroin (Um et al. 2001).

2D WAXS (a), total crystallinity and crystal orientation of cellulose (b, c) of cellulose/silk fibroin blend fibers (silk fibroin crystal planes are distinguished in italics)
Molecular structure of cellulose/silk fibroin blend fiber
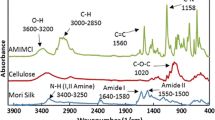
FTIR has been comprehensively used to study the interaction of polymer blends (Shang et al. 2009; Lü et al. 2010, 2011; Huang et al. 2013). The FTIR of cellulose/silk fibroin blend fiber is shown in Fig. 4. The conformation of silk fibroin includes random coil, α-Helix and β-Sheet structure (Drummy et al. 2005). The characteristic absorption peaks of β-Sheet silk fibroin are at 1,630 cm−1 (amide I), 1,530 cm−1 (amide II) and 1,260−1 (amide II) (Freddi et al. 1995; Hyoung-Joon et al. 2005). The adsorption peaks at 1,626 and 1,530 cm−1 appeared in the blend, which recommends that β-Sheet structure was formed through fiber forming process. The characteristic peak of OH stretching vibration is around 3,400 cm−1. There are three different hydroxyl groups of cellulose chain. The peak of OH stretching vibration is broader in sample Cell-3 and Cell-4, which is contributed to the intramolecular hydrogen bonding and intermolecular hydrogen bonding. The stretching vibration of NH in silk fibroin chain is around 3,288 cm−1 (Yao et al. 2014). As shown in Fig. 4, the peak around 3,288 cm−1 became narrow and the peak at 3,400 cm−1 became weaker with the increase of silk fibroin, which recommends that the new intermolecular hydrogen bonds exist between OH at the C2, C3 position of cellulose and NH in the amide groups of silk fibroin (Kondo et al. 1994; Yang et al. 2000).

FTIR of cellulose/silk fibroin blend fibers
Mechanical properties of cellulose/silk fibroin blend fibers
The mechanical properties of cellulose/silk fibroin blend fibers is shown in Table 3. The diameter of the fibers are between 20 and 30 μm, which decreased with drawing. The tensile strength (σb) and initial modulus (E) increased with increasing cellulose content. Cellulose is the continuous phase in the blend which contributes much to the mechanical strength. The dispersion of silk fibroin phase in the cellulose matrix component blend fibers is similar with increasing silk fibroin content. Hence, the tensile strength of the blend fiber decreased little from sample CS82 to CS64. The tensile strength increased with the drawing ratio, owing to the increase of crystal orientation of cellulose. The tensile strength of blend fiber which contains 35 wt% silk fibroin was still up to 191 MPa.
Moisture absorption and desorption of cellulose/silk fibroin blend fibers
The moisture absorption and desorption are related to the wearing comfort to human skins. As shown in Fig. 5, the moisture absorption and desorption process occur mainly within 120 min, the equilibrium moisture absorption regain of blend fiber is 11.8–13.1 % and the equilibrium desorption regain is 15.0–17.0 %. The equilibrium moisture of the blend fibers is not only related to the hydrophilic of cellulose and silk fibroin chains, but also the amorphous regions, crystallites and voids of the blend fibers (Okubayashi et al. 2004). The standard moisture absorption of degummed silk is 8.9 % (Charles 1938). The equilibrium moisture regain of Lyocell fiber at 60 % relative humidity and 20 °C is 9.2 % (Siroka et al. 2008). The regenerated cellulose fiber and silk have similar moisture ability. Hence, the moisture absorption and desorption regains of cellulose/silk fibroin blend fibers and regenerated cellulose fiber are almost same in this research.

Moisture absorption and desorption of cellulose/silk fibroin blend fibers
Conclusions
The cellulose/silk fibroin blend fibers were fabricated with [BMIM]Cl as solvent by co-dissolution method. Even though the tensile strength and initial modulus decreased with increasing silk fibroin, the tensile strength of blend fibers containing 35 wt% of silk fibroin was up to 191 MPa. This is related to “fibril-like” morphology of silk fibroin existed in the continuous phase of cellulose. The phase size of silk fibroin along the fiber axis increased with silk fibroin content and draw ratio, and the hydrogen bond interaction between cellulose and silk fibroin in the blend fibers was detected.
References
Cao H, Chen X, Huang L, Shao Z (2009a) Electrospinning of reconstituted silk fiber from aqueous silk fibroin solution. Mat Sci Eng C Mater 29(7):2270–2274
Cao Y, Wu J, Zhang J, Li H, Zhang Y, He J (2009b) Room temperature ionic liquids (RTILs): a new and versatile platform for cellulose processing and derivatization. Chem Eng J 147(1):13–21
Charles J (1938) Tests for physical properties of fibers. Text Res J 8:93–101
Chen Y, Zhang LN (2004) Blend membranes prepared from cellulose and soy protein isolate in NaOH/thiourea aqueous solution. J Appl Polym Sci 94(2):748–757
Csihony S, Fischmeister C, Bruneau C, Horvath IT, Dixneuf PH (2002) First ring-opening metathesis polymerization in an ionic liquid. Efficient recycling of a catalyst generated from a cationic ruthenium allenylidene complex. New J Chem 26(11):1667–1670
Drummy LF, Phillips DM, Stone MO, Farmer BL, Naik RR (2005) Thermally induced alpha-helix to beta-sheet transition in regenerated silk fibers and films. Biomacromolecules 6(6):3328–3333
Fink HP, Weigel P, Purz HJ, Ganster J (2001) Structure formation of regenerated cellulose materials from NMMO-solutions. Prog Polym Sci 26(9):1473–1524
Freddi G, Romano M, Massafra MR, Tsukada M (1995) Silk fibroin/cellulose blend films—preparation, structure, and physical-properties. J Appl Polym Sci 56(12):1537–1545
Georgakoudi I, Tsai I, Greiner C, Wong C, DeFelice J, Kaplan D (2007) Intrinsic fluorescence changes associated with the conformational state of silk fibroin in biomaterial matrices. Opt Express 15(3):1043–1053
Heslot H (1998) Artificial fibrous proteins: a review. Biochimie 80(1):19–31
Hirano S, Nakahira T, Zhang M, Nakagawa M, Yoshikawa M, Midorikawa T (2002) Wet-spun blend biofibers of cellulose–silk fibroin and cellulose–chitin–silk fibroin. Carbohydr Polym 47(2):121–124
Huang H, Wu J, Lin X, Li L, Shang S, Yuen M, Yan G (2013) Self-assembly of polypyrrole/chitosan composite hydrogels. Carbohydr Polym 95(1):72–76
Hyoung-Joon J, Park J, Karageorgiou V, Kim UJ, Valluzzi R, Cebe P, Kaplan DL (2005) Water-stable silk films with reduced beta-sheet content. Adv Funct Mater 15(8):1241–1247
Jiang G, Huang W, Li L, Wang X, Pang F, Zhang Y, Wang H (2012a) Structure and properties of regenerated cellulose fibers from different technology processes. Carbohydr Polym 87(3):2012–2018
Jiang G, Yuan Y, Wang B, Yin X, Mukuze KS, Huang W, Zhang Y, Wang H (2012b) Analysis of regenerated cellulose fibers with ionic liquids as a solvent as spinning speed is increased. Cellulose 19(4):1075–1083
Ki CS, Lee KH, Baek DH, Hattori M, Urn IC, Ihm DW, Park YH (2007) Dissolution and wet spinning of silk fibroin using phosphoric acid/formic acid mixture solvent system. J Appl Polym Sci 105(3):1605–1610
Koller D, Halmerbauer G, Böck A, Engstler G (2007) Action of a silk fabric treated with aegistm in children with atopic dermatitis: a 3-month trial. Pediatr Allergy Immunol 18(4):335–338
Kondo T, Sawatari C, Manley RS, Gray DG (1994) Characterization of hydrogen-bonding in cellulose synthetic-polymer blend systems with regioselectively substituted methylcellulose. Macromolecules 27(1):210–215
Langan P, Nishiyama Y, Chanzy H (2001) X-ray structure of mercerized cellulose II at 1 angstrom resolution. Biomacromolecules 2(2):410–416
Liu H, Xu W, Liu X, Xu J, Li W, Liu X (2009) Effects of superfine silk protein powders on mechanical properties of wet-spun polyurethane fibers. J Appl Polym Sci 114(6):3428–3433
Lü T, Shan G, Shang S (2010) Intermolecular interaction in aqueous solution of binary blends of poly (acrylamide) and poly (ethylene glycol). J Appl Polym Sci 118(5):2572–2581
Lü T, Shan G, Shang S (2011) Stability of two-phase polymerization of acrylamide in aqueous poly (ethylene glycol) solution. J Appl Polym Sci 122(2):1121–1133
Macosko CW (2000) Morphology development and control in immiscible polymer blends. Macromol Symp 149(1):171–184
Marsano E, Canetti M, Conio G, Corsini P, Freddi G (2007) Fibers based on cellulose–silk fibroin blend. J Appl Polym Sci 104(4):2187–2196
Marsano E, Corsini P, Canetti M, Freddi G (2008) Regenerated cellulose–silk fibroin blends fibers. Int J Biol Macromol 43(2):106–114
Morimoto S (1970) Silk-like fiber K-6 (Chinon). Ind Eng Chem 62(3):23–32
Okubayashi S, Griesser UJ, Bechtold T (2004) A kinetic study of moisture sorption and desorption on lyocell fibers. Carbohydr Polym 58(3):293–299
Phillips DM, Drummy LF, Conrady DG, Fox DM, Naik RR, Stone MO, Trulove PC, De Long HC, Mantz RA (2004) Dissolution and regeneration of Bombyx mori Silk fibroin using ionic liquids. J Am Chem Soc 126(44):14350–14351
Phillips DM, Drummy LF, Naik RR, De Long HC, Fox DM, Trulove PC, Mantz RA (2005) Regenerated silk fiber wet spinning from an ionic liquid solution. J Mater Chem 15(39):4206–4208
Rice WL, Firdous S, Gupta S, Hunter M, Foo CWP, Wang Y, Kim HJ, Kaplan DL, Georgakoudi I (2008) Non-invasive characterization of structure and morphology of silk fibroin biomaterials using non-linear microscopy. Biomaterials 29(13):2015–2024
Shang S, Zhu L, Chen W, Yi L, Qi D, Yang L (2009) Reducing silk fibrillation through MMA graft method. Fiber Polym 10(6):807–812
Shang S, Zhu L, Fan J (2011) Physical properties of silk fibroin/cellulose blend films regenerated from the hydrophilic ionic liquid. Carbohydr Polym 86(2):462–468
Shang S, Zhu L, Fan J (2013) Intermolecular interactions between natural polysaccharides and silk fibroin protein. Carbohydr Polym 93(2):561–573
Siroka B, Noisternig M, Griesser UJ, Bechtold T (2008) Characterization of cellulosic fibers and fabrics by sorption/desorption. Carbohydr Res 343(12):2194–2199
Sun M, Zhang Y, Zhao Y, Shao H, Hu X (2012) The structure–property relationships of artificial silk fabricated by dry-spinning process. J Mater Chem 22(35):18372–18379
Swatloski RP, Spear SK, Holbrey JD, Rogers RD (2002) Dissolution of cellose with ionic liquids. J Am Chem Soc 124(18):4974–4975
Um IC, Kweon HY, Park YH, Hudson S (2001) Structural characteristics and properties of the regenerated silk fibroin prepared from formic acid. Int J Biol Macromol 29(2):91–97
Vollrath F, Holland C, Terry AE, Porter D (2007) Natural and unnatural silks. Polymer 48(12):3388–3392
Yang G, Zhang LN, Liu YG (2000) Structure and microporous formation of cellulose/silk fibroin blend membranes I. Effect of coagulants. J Memb Sci 177(1–2):153–161
Yang G, Zhang LN, Cao XD, Liu YG (2002) Structure and microporous formation of cellulose/silk fibroin blend membranes Part II. Effect of post-treatment by alkali. J Memb Sci 210(2):379–387
Yao Y, Mukuze KS, Zhang Y, Wang H (2014) Rheological behavior of cellulose/silk fibroin blend solutions with ionic liquid as solvent. Cellulose 21(1):675–684
Zhang XF, Min BG, Kumar S (2003) Solution spinning and characterization of poly(vinyl alcohol)/soybean protein blend fibers. J Appl Polym Sci 90(3):716–721
Zhou G, Shao Z, Knight DP, Yan J, Chen X (2009) Silk fibers extruded artificially from aqueous solutions of regenerated Bombyx mori silk fibroin are tougher than their natural counterparts. Adv Mater 21(3):366–370
Zhou L, Wang Q, Wen J, Chen X, Shao Z (2013) Preparation and characterization of transparent silk fibroin/cellulose blend films. Polymer 54(18):5035–5042
Acknowledgments
This work is supported by Natural Science Foundation of China (51273041) and Chinese Universities Scientific Fund (CUSF-DH-D-2014026).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yao, Y., Zhang, E., Xia, X. et al. Morphology and properties of cellulose/silk fibroin blend fiber prepared with 1-butyl-3-methylimidazolium chloride as solvent. Cellulose 22, 625–635 (2015). https://doi.org/10.1007/s10570-014-0520-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-014-0520-z