
Abstract
The high crystallinity and the high microfibrils aspect ratio of tunicate cellulose (TC) indicate TC’s excellent chemical and material applications. However, its quantity and quality from different species have never been systematically reported and compared. In this study, the tunics of Ciona intestinalis (CI), Ascidia sp. (AS), Halocynthia roretzi (HR) and Styela plicata (SP) were processed to TC after an identical prehydrolysis-kraft cooking-bleaching sequence, while the tunicate fibrils were chemically and structurally characterized in situ and during the sequence. All tunics studied were composed of crystalline cellulose embedded with protein, lipids, sulfated glycans and mucopolysaccharides. The native composite structures are all very compact. However, the tunics from Phlebobranchia order (CI and AS) are soft, while those from Stolidobranchia, HR and SP, are hard. Fibrous cellulose could be prepared after removing the lipids, sulfated glycans and mucopolysaccharides through prehydrolysis, protein removal through kraft cooking and a final purification by bleaching. The final product is ~100 % pure cellulose which is in large molecular masses, composed of highly crystalline Iβ crystals, in elementary microfibrils form, with high specific surface area and thermal stability. There were lower TC yields from the soft tunics than from the hard ones. The cellulose fibrils had a section shape of lozenges with higher crystallinity. This study demonstrates that TC could be obtained in different yields and exhibited different chemical and morphological structures depending on the species. There is a great potential of tunicate resources for preparing excellent chemical and material cellulose.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose is a well-known and important structure for various applications in the society, including energy, chemicals and materials. Representative applications include ethanol production after cellulose hydrolysis and fermentation, pulp and paper production after kraft pulping of wood and regenerated cellulose from dissolving pulps. Recently cellulose has been highlighted for further development due to its abundance, renewability, sustainability and biocompatibility; these qualities are in high demand by consumers, industry and government. The production of new cellulose-based products has become a major research focus due to the high crystallinity and good processability, as well as the high specific strength and modulus, of cellulose. For example, after extracting nanoscale cellulose from natural resources, the produced nanocellulose will be a new “building block” for the next generation of cellulose-based composites. The applications of these materials would include barrier films, antimicrobial films, transparent films, flexible displays, reinforcing fillers for different polymers, biomedical implants, pharmaceuticals, drug delivery, fibers and textiles, templates for electronic components, separation membranes, batteries, supercapacitors, electroactive polymers and others (Moon et al. 2011).
Wood and annual plants are the commonly used natural resources for cellulose. However, the production would be limited by land resources. Although cellulose can also be produced from the cell walls of certain algae and bacteria (Habibi et al. 2010), tunicates, which live in the oceans, are the only known animal source for cellulose. The name ‘Tunicata’ is derived from its unique integumentary tissue called the ‘tunic’, which covers the entire epidermis of the animal. The tunic is created to perform various biological functions, such as phagocytosis, impulse conduction, tunic contractility, bioluminescence, photosynthetic symbiosis and allorecognition (Hirose 2009). The cellulose acts as a skeletal structure in the tunic tissues. Cellulose-synthesizing enzyme complexes have been found in the plasma membrane of their epidermal cells and are responsible for cellulose synthesis. The enzymes also have two other functions: proper formation of the tunic and a role in metamorphosis (Kimura and Itoh 2007).
Tunicates are a very plentiful resource; the subphylum Tunicata includes three classes, and all of the species in two of the classes, Ascidiacea and Thaliacea, possess tunics. The third class Appendicularia do not have tunic but secrete cellulosic materials (Kimura et al. 2001). There are over 2,300 species in Ascidiacea alone. The appearance and function of the tunic are very different within the class, depending on the species. Within Ascidiacea, 116 species have been investigated in detail for tunic morphological structures, specifically the cuticular protrusion structures, and their possible links with the ascidian phylogeny (Hirose et al. 1997). The earliest observation of highly oriented cellulose in the tests of the tunicates was conducted by Herzog and Gonell (1924), which was further confirmed by the observations of Wardrop (1970), Meyer et al. (1951) and Rånby (1952), indicating among many other things that the cellulose present in the tunic is chemically identical with plant cellulose. Tunicate cellulose (TC) is produced by the cellulose synthesizing enzyme complexes in the membrane of epidermis through different and poorly understood mechanisms; TC performs different functions in different tunicate families and species, resulting in differences in the TC structures. In the literature, 40 species of ascidian have been examined for their TC structural diversity. Based on the structural data (microfibril width, relative orientation factor and relative crystallinity for similarity and dissimilarity), a tree diagram of the studied ascidians has been drawn. Unfortunately, no detailed data have been released by the authors (Okamoto et al. 1996). However, TC is generally a highly crystalline and high aspect ratio cellulose (Larsson et al. 1995; Belton et al. 1989). Typically, it is composed of nearly pure cellulose Iβ allomorph (Nishiyama et al. 1999, 2003; Belton et al. 1989). About 10 % of cellulose Iα allomorph was found for the cellulose from Halocynthia sp. using CP/MAS 13C-NMR spectroscopy (Larsson et al. 1995). Hundreds of cellulose microfibrils are bundled in the tunic; the shape and dimensions of the microfibril bundle vary depending on the species. The microfibril bundles are deposited in a multilayered texture parallel to the surface of the epidermis. The tunicate cellulose microfibrils have a lozenge-shaped section (Habibi et al. 2006; Helbert et al. 1998) with two lattice parameters of a = 0.801 nm and b = 0.817 nm (Sugiyama et al. 1991b). An earlier study found that enzymatic degradation of the tunicin resulted in the isolation of microfibrils of 120–130 Å in width consisting of two sub-units and the microfibrils were found occurring in the tunicate as aggregates measuring 2,000–4,000 Å in diameter through staining and freeze-etching techniques (Wardrop 1970). From SEM study, the length of cellulose microfibrils ranges from 100 nm to several micrometers, while the width ranges from 15 to 30 nm; the aspect ratio falls between 3 and 67. TC has a high specific surface area ranging from 150 to 170 m2/g, a high crystallinity (95 %) and a reactive surface attributed to hydroxyl groups (Kimura and Itoh 2007). The degree of polymerization of tunicate cellulose was reported to be 700–3,500 (Krässig 1954). Therefore, tunicates should be excellent resources for chemical and material cellulose. Noticeably, the Ciona intestinalis tunicate species could be farmed at very high densities in the ocean, enabling TC production at a large scale and its practical use in chemical, materials or biofuel production (Troedsson et al. 2013).
To produce and use the cellulose from tunicates, the quantity or production yield is important. Historically, the tunic has been reported to contain approximately 60 % cellulose and 27 % nitrogen-containing components by dry weight (Berrill 1947). Many studies have been conducted to purify TC using different methods: (1) treatment with a KOH solution at an elevated temperature (Van Daele et al. 1992; De Leo et al. 1977); (2) the combined treatment with NaOH/KOH solution at an elevated temperature, followed by treatment with a NaClO2 solution (Nakashima et al. 2008; Yuan et al. 2006; Khandelwal and Windle 2013; Hirose et al. 2009; Sugiyama et al. 1991a; Kuga et al. 2002; Saito et al. 2013; Helbert et al. 1998; Kimura and Itoh 1997); and (3) treatment with acetic acid and nitric acid at an elevated temperature (so called Updegraff method; Nakashima et al. 2008; Kimura and Itoh 1997). Different species have been used; the most frequently studied species include Halocynthia roretzi (Yuan et al. 2006; Sugiyama et al. 1991a), Halocynthia papillosa (Helbert et al. 1998) and Metandroxarpa uedai (Moon et al. 2011). Ciona intestinalis was also reported in literature (De Leo et al. 1977). Additionally, 18 species from pyrosomas, doliolids and salps have been investigated (Hirose et al. 2009). Unfortunately, quantity values are rarely reported. In most cases, the cellulose preparation aimed only for the alkali-insoluble fibrous fraction from the tunic, which is called tunicin, as a specimen for SEM observation and XRD determination. The cellulose purity of the tunicin obtained was not of concern and was not determined. Noticeably, no systematic investigation has been conducted to reveal the quantity derived from different tunicate sources based on the purity value of the cellulose.
In this study, the tunics from four different tunicate species within two different orders and four different families in the class Ascidiacea (Fig. 1) were investigated for cellulose preparation using an identical prehydrolysis-kraft cooking-bleaching sequence. The sequence was originally reported by Korean researchers (Koo et al. 2002); the general idea involved was adopting the sequence for making dissolving pulp from wood after some reaction condition optimizations. This sequence is considered by us as a more suitable method than those mentioned above because the original dissolving pulp process has proven very effective and specific for cellulose preservation, resulting in limited damage to cellulose, particularly crystalline cellulose. In this study, the process principle was noticeably further clarified and corrected based on the chemical structures and structural changes to the tunicate. Our research focused on cellulose diversity among different tunicate species regarding production yield and quality characteristics for chemical (e.g., purity, molecular mass and specific surface area) and material applications (e.g., morphology, crystal structure, crystallinity index and thermal stability).

A hierarchical tree diagram for the taxonomic classification of the tunicates used in this study
Experimental
Tunicate samples and chemicals
Four tunicate species (Ciona intestinalis, Ascidia sp., Halocynthia roretzi and Styela plicata labeled CI, AS, HR, and SP, respectively) were used as raw materials, and their taxonomic classification is shown in Fig. 1. CI and AS were collected in the Norwegian Sea (Norway), while HR and SP came from the Korea Strait (North Korea) and the Yellow Sea (China), respectively. All chemicals were of analytical grade and were obtained from VWR International AB (Stockholm, Sweden).
Cellulose preparation process
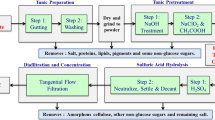
The process for TC preparation from HR is depicted in Fig. 2; the procedure for the other species was the same. Briefly, tunicate tunic was obtained by removing the interior organs of the animal with a knife; the wet tunicate tunic was freeze-dried and milled into powders using a Willey mill (<40 mesh). The purification process followed that of Koo et al. (2002), with some modifications. Briefly, the ground powders were mixed with an aqueous H2SO4 (1 %) solution in a glass vessel and heated at 180 °C for 2 h with occasionally shaking. This step is called prehydrolysis. The insoluble residue was collected by filtration, washed thoroughly with acetone/water (1:1, v/v) until the filtrate was neutral, and freeze dried. Afterwards, an alkaline treatment with aqueous solution of NaOH/Na2S (9/3, %) was conducted at 180 °C for 2 h with occasionally shaking, followed by filtration, washing and drying as described above. This step is called kraft cooking. After the alkaline treatment, a bleaching step was performed using aqueous NaClO solution (3 %) in the vessel with agitation at 75 °C for 1 h. This bleaching step was repeated 2–3 times until the product was completely white; the product was filtered, washed and dried using the same procedure described above.

Scheme of the tunicate cellulose preparation from Halocynthia roretzi (HR)
Characterization
Chemical composition analysis
The ash content of the tunics was determined by heating the sample at 600 °C for 4 h; this value was calculated based on the residue after gravimetric determination. The lipid content was expressed as the sum of the individual fatty acids that were identified using gas chromatography (GC). Briefly, a ~20 mg sample was weighed; afterwards, toluene (0.2 ml), methanol (1.5 ml) and 8 % HCl solution in methanol (0.3 ml) were added sequentially to the sample. This solution (~2 ml) was heated at 100 °C for 1 h. Next, 1 ml of saturated sodium chloride and 1 ml of hexane were added, and the solution was shaken for 1 min, followed by phase separation by letting the solution standing still for a while. The supernatant containing the fatty acid methyl esters was directly applied to GC analysis. The carbohydrate composition was determined using ionic chromatography (IC) of the monosugars after complete sample hydrolysis: grinding with 72 % H2SO4 at room temperature before autoclaving in 3 % H2SO4 at 120 °C for 1 h (cf. TAPPI Test Method T 249). Commercial monosaccharide’s including glucose, galactose, mannose, xylose and arabinose were used as standards and for calibration. The sample used for nitrogen content analysis was hydrolyzed using 6 M HCl for 24 h at 110 °C; the hydrolysate was analyzed using Shimadzu’s TNM-1 Total Nitrogen Module (Shimadzu, Japan). The system was calibrated using 1–100 ppm KNO3 standard solutions. The “crude protein” content was calculated by multiplying the total nitrogen content by 6.25.
Scanning electron microscope (SEM)
The sample was coated with gold using a Cressington 208HR high-resolution sputter coater. The thickness was controlled using a Cressington thickness monitor controller (3–5 nm). Afterwards, the sample was subjected to morphological analysis (Hitachi S-4800 FE-SEM). The diameter of the cellulose fibrils was calculated as the mean after examining 50 individual microfibrils using the Image J software.
Fourier transform infrared spectroscopy (FTIR)
The spectra were obtained using a Perkin-Elmer Spectrum 2000 FTIR spectrometer (Waltham, MA, USA) equipped with an ATR system, Spectac MKII Golden Gate (Creecstone Ridge, GA, USA). The sample was analyzed from 600 to 4,000 cm−1. The spectrum was obtained from a dry sample using 16 scans at a 4 cm−1 resolution and a 1 cm−1 interval at room temperature. Before data collection, a background scan was performed for background correction.
Size exclusive chromatography (SEC)
The obtained TC was first activated with deionized water at 4 °C for 1 h before the excess water was removed through filtration. Afterwards, a solvent exchange with methanol was applied three times before exchange with N,N-dimethylacetamide (DMAc). The activated sample was dissolved in 8 % LiCl/DMAc to a concentration of 0.8 % by sample weight by stirring at 4 °C for 5 days and then diluted with DMAc to a final concentration of 0.5 % relative to the sample weight. The SEC system was equipped with a Rheodyne injector, a DGU-20A3 degasser, a LC-20AD liquid chromatography and a RID-10A refractive index detector. The separations were achieved using four 20 µm Mixed-A columns with a guard column. The injection volume was 100 µL, and the separations were performed at 80 °C with a flow rate of 0.5 ml/min 0.5 % LiCl/DMAc. The calibration was conducted using pullulan standards with nominal masses from 320 to 800 kDa (Fluka/Riedel-de Haën, Seelze, Germany). The LC Solution software included with the SEC system (Shimadzu, Kyoto, Japan) was used for data acquisition and evaluation.
X-ray diffraction (XRD) analysis
A PANalytical X’Pert PRO Materials Research Diffractometer equipped with an X’Celerator detector was used to determine the crystallinity of the TCs. Briefly, powder TC samples (<40 mesh) were mounted on a low background silicon sample holder with a gentle pressing to make the surface as flat as possible and measured using a monochromatic Cu Kα radiation at 30 mA and 40 kV. The March–Dollase parameter of 2.0, with orientation to (001) preferred reflection, was set in the Mercury program to eliminate the contributions of the (012) and (102) reflection peaks to the crystallinity index of cellulose Iβ (French and Santiago Cintrón 2013; French 2014). After subtracting the background from the obtained spectrums, the crystallinity index was defined to evaluate the crystallinity and was calculated using the following equation:
where I200 is the intensity of the (200) lattice plane at 2θ = 22.8°, and Iam is the intensity from amorphous phase approximately 2θ = 18° (Reddy and Yang 2005; Segal et al. 1959). Furthermore, the crystal’s size of each TC was calculated out by Scherrer equation (Scherrer 1912; French and Santiago Cintrón 2013).
Specific surface area (SSA) analysis
The specific surface area was analyzed using the gas adsorption properties of the sample via BET analysis. First, approximately 0.1 g of sample was heated and degassed at 115 °C under vacuum for >300 min, removing all adsorbed molecules. Afterwards, controlled amounts of nitrogen were introduced and adsorbed. At the temperature of liquid nitrogen (−196 °C) under vacuum, the sample was exposed to varying pressures to generate adsorption isotherms. The adsorbed molecules were measured using the pressure variations caused by adsorption. A Micromeritics ASAP 2020 Surface Area and Porosity Analyzer (Micromeritics, USA) was used together with its software.
Thermo gravimetric analysis (TGA)
The thermo gravimetrical analyses were conducted on a Mettler Toledo TGA/SDTA 851e equipped with the STARe software for data analysis. The samples were subjected to a heating scan between 30 and 700 °C at 10 °C/min under an inert nitrogen atmosphere flowing at 50 ml/min.
Results and discussion
Outward similarity and dissimilarity of different tunicate species and their tunics
For the adult tunicate specimens from four different species, CI, AS, HR and SP from Cionidae, Ascidiidae, Pyuridae and Styeldae, respectively (Fig. 1), there were obvious outward diversities. The size of the animals, in decreasing order, was CI > HR > AS > SP, with a mean size of 13.3 × 4.3, 12.7 × 6.2, 7.5 × 4.4 and 3.6 × 2.9 cm (length × width), respectively (Table 1). However, the mean individual weight, in decreasing order, was HR at 70.4 g, CI at 24.5 g, AS at 6.95 g and SP at 1.96 g. The cuticular layers of the animals, or their tunics, are an external supportive and protective skeleton that account for more than 50 % of the mean weight percentages over the entire animal weight (57–73 %), except for HR (only 30.3 %). The tunics were composed of mostly water (81–95 %; Table 1). All tunics were constructed from homogenous tissues, while in contrast, the two tunicates from the order Phlebobranchia, CI and AS, possessed soft, flexible, thin and gelatinous tunics, in light yellow transparent and slightly white almost transparent forms, respectively. The two from Stolidobranchia (HR and SP) had hard, thick and opaque tunics that varied from red to brown and white in color, respectively (Fig. 3).

Outward appearance of the whole animal and the tunic and produced cellulose morphologies of different tunicates observed by SEM
The SEM view of the surface layer of the tunics showed microfibrils deposited in a multi-layered texture with a bundled structure parallel to the epidermis (Fig. 3). All microfibrils had similar mean diameters (19–22 nm; Table 2), which is in agreement with the data reported in the literature that there are two types of membrane subunits present in tunicates: one major type with large particles (14.5 nm in diameter) on the periphery and one with small subunits of 7.2 nm that fill the center; the latter was hypothesized to be an intermediate structure involved in cellulose synthesis (Kimura and Itoh 1996). The microfibrils were somewhat bundled, which was more obvious for the hard tunics from HR and SP than for the soft ones from CI and AS; the microfibrils were embedded in a continuous amorphous matrix (more obvious in CI and SP), and their lateral mutual orientations were random except for HR. HR had an ordered orientation (Fig. 3). Our results are consistent with the previous study results on the morphology of HR tunic which showed that the tunic surface was entirely covered with a cuticular layer that had a very dense structure of bundled fibres and the tunic matrix was distributed between the fibres (Hirose et al. 2009). To study the structural compaction of the tunics, their specific surface areas (SSAs) were measured using freeze-dried samples. During freeze-drying, the water evaporated directly from the ice without or with few collapse of the structure of the network. Therefore, the SSA reflects the “original” compaction of the tunics. All tunics from the 4 different tunicate species had very similar and low SSAs, ranging from 1.7 to 3.2 m2/g (Table 2), indicating very compact networks; therefore, these structures provided good physical protection against environmental influences, such as sea waves and various biological attacks.
Principle of cellulose production process
Initial chemical structure of the tunic
Chemically, the principal constituents of the tunics are proteins with amino groups and tryptophan, scleroproteins, collagen and elastic fibers as well as high quantities of acid mucopolysaccharides and neutral polysaccharides (Lunetta 1983). In our study, around half of the dry weight of the tunics from CI and AS (43–57 %) from the Norwegian Sea were ash, while HR from the Korea Strait and SP from the Yellow Sea had relatively lower ash contents of 11.55 and 16.32 %, respectively (Table 3). Because sea salt and tunicate body inorganics were the main components of the ash and all tunics had similar and high moisture contents, the differences might be attributable to the salinity difference in different sea water (Reid 1979; Wang et al. 2003) in addition to the different metal accumulation capacity of the tunicates (Carlisle and Bernal 1968; Swinehart et al. 1974). Additionally, 0.28–4.25 % fatty acids (representing lipids) were detected in the tunics. Assuming that the nitrogen content originated from proteins (see experimental part and below), protein accounted for 25.82–38.08 % of the dry weights of SP, CI and HR, while the lowest protein content (17.74 %) was found in AS. The microfibrils present in the tunics are cellulose-protein fibrils (Van Daele et al. 1992). The carbohydrate compositions shown in Table 3 indicate that the thick, hard HR and SP tunics contained a high concentration of carbohydrates (46.52 and 48.27 % dry weight) while the thin and soft CI and AS tunics showed much lower carbohydrate contents (21.28 and 16.12 % respectively). It can be seen that for CI, AS and HR, the ratio between protein and carbohydrate was approximately 1:1 which agreed quite well with the data of 50 % carbohydrate and 50 % protein in the tunic of Halocynthia aurantium reported by Smith and Dehnel (1970) though the ratio for SP was higher than 1:1. Furthermore, most of the monosugars after acid hydrolysis were glucose with relative percentages of 94.22 and 86.95 in the HR and SP tunics, respectively. The relative glucose percentages for CI and AS were much lower but still exceeded 75 %. Other monosugars were observed, including arabinose, galactose, xylose and mannose. Correspondingly to the lower glucose contents, the soft tunics had higher contents of these other sugars than the hard tunics, especially the relative galactose contents (~20 vs. <11.4 %). These other sugars did not originate from any hemicelluloses, as they do in wood samples; they arise from sulfated mucopolysaccharides (composing of hexosamine, uronic acid and sulfate; Cassaro and Dietrich 1977; Lunetta 1983), sulfated glycans (Albano and Mourão 1986; Mourao and Perlin 1987) or sulfated O-linked glycans (Baginski et al. 1999). In the literature describing SP tunics, sulfated glycans were mainly found in the forms of sulfated galactans, while glucose and mannose were also involved in the sulfated glycans structures. Together with proteins, they are essential for the maintenance of tunic structural integrity (Albano and Mourão 1986). Additionally, sulfated xylan was reported in green seaweed (Percival and Wold 1963). Reasonably, the higher contents of other sugars in the soft tunics implied higher contents of sulfated glycans, making the structures more flexible than the hard tunics. Notably, some of the glucoses in the tunics were not from cellulose structures but were from these sulfated structures (Albano and Mourão 1986).
Specifically, the tunic structures could be described as cellulose-protein fibrils cemented by these sulfated mucopolysaccharides or sulfated glycans and lipids. These composite structures were verified by FTIR (Fig. 4). The peak at 1,161 cm−1 arose from the C–O anti-symmetric bridge stretching. The shoulder band at 1,112 cm−1 was attributed to the C–OH skeletal vibration. The C–O–C pyranose ring skeletal vibration gave prominent bands at 1,057 and 1,032 cm−1. These four peaks are characteristic of cellulose (Nakashima et al. 2008). The small peak at 895 cm−1 corresponded to the glycosidic –CH deformation with a ring vibration and –OH bending, which is characteristic of β-glycosidic linkages between glucoses in cellulose (Nakashima et al. 2008; Sun et al. 2004). The broad band at 1,220–1,260 cm−1 revealed a sulfate ester (Mohamed and Agili 2013) with a S=O band at 1,240 cm−1 (Albano and Mourão 1986). The intense bands at 1,610 and 1,540 cm−1 were mistakenly assigned as aromatic rings or lignin in the literature (Koo et al. 2002); these bands should be attributed to the mucin materials, specifically carboxylate ions, water and amides (Pancake and Karnovsky 1971). This assignation could be further supported by the FTIR spectra for N-acetyl glucosamine in the literature (Kovács et al. 2008).

FT-IR spectra changes of Ciona intestinalis (CI) during chemical processing. Similar trends of FT-IR spectra changes were also observed for all of the other samples (data not shown)
Prehydrolysis step
This prehydrolysis step, as depicted in Fig. 2, caused the largest weight loss for all tunics. For AS, only 20.85 % of the solid material was recovered; the recovery yields were 39.80–53.05 % for CI, HR and SP (Table 3). This step removed all lipids and ash without largely reducing the protein content. Additionally, the highest total carbohydrate losses occurred with reduction percentages of 7.35, 12.18, 23.33 and 19.31 for CI, AS, HR and SP, respectively. Almost all of the sulfated mucopolysaccharides or sulfated glycans were removed during this step as indicated firstly by the removal of most of the other sugars than glucose. This conclusion is supported by the results from the literature using HR (Koo et al. 2002); the sulfur content dropped from 3.58 % to zero. In this study, this change was verified using its FTIR spectrum; the broad band at 1,220–1,260 cm−1 and the intense bands at 1,610 and 1,540 cm−1 were eliminated (Fig. 4). The small reductions in nitrogen content were in fact attributable to the removal of those hexosamines from the mucopolysaccharides and perhaps not from the proteins. This is supported by the presence of 5 % hexosamine by dry weight found in the tunic of Halocynthia aurantium (Smith and Dehnel 1970) and high quantity of acid mucopolysaccharides in the tunic of Molgula impura and Styela partita (Lunetta 1983), which were involved directly in protein-polysaccharide linkages in the tunic. Therefore, almost all of the components other than the cellulose-protein fibrils were removed. The majority of the remaining carbohydrates were from cellulose because no additional loss of carbohydrates was observed during the following kraft cooking treatment or bleaching step.
Kraft cooking step
After this alkaline treatment, approximately 12–22 % more of the dry weight was removed for CI, HR and SP, while only 12.03 % was removed for AS. This weight loss was mainly attributed to the protein eliminations because corresponding protein losses were observed (Table 3). Although the treatment with high concentrations of alkali and sulfide was very harsh, 3.60–9.75 % proteins were resistant. Certain proteins were difficult to hydrolyze using the alkaline treatment. The protein removal was verified by FTIR analysis, where the band at 1,540 cm−1 (due to N–H stretching from amide groups) was reduced (Fig. 4). The sugars other than glucose specifically arabinose (from mucopolysaccharides) were nearly completely removed, leaving only mannose.
Bleaching step
Complete protein removal was achieved using this last bleaching step, and no protein was present in the bleached samples (Table 3). The protein band at 1,540 cm−1 in the FTIR vanished after bleaching, verifying the elimination of the proteins (Fig. 4). Apparently, the alkaline resistant proteins were sensitive toward NaClO oxidation. The further weight losses (4–12 %) in this step were assigned to the elimination of some chromophoric structures initially present in the tunics or generated during the previous steps. A small reduction in glucose was also observed for all samples. All of the final products were highly pure celluloses because the glucose accounted for ~100 % of the weight for the TCs (Table 3).
Cellulose quantity from different tunicate species
The production yields of chemically pure cellulose following the sequence were 12.35, 2.54, 20.99 and 23.92 % for CI, AS, HR and SP, respectively. Apparently, the yields from the harder and thicker Stolidobranchia (the investigated HR and SP) tunics were much higher than the soft and thin Phlebobranchia ones. However, after recalculation by removing the ash content from the starting sample weights, which was much higher in CI, CI was a comparable source of cellulose, resulting in a comparable yield of 21.64 vs. 23.72 % (HR) and 28.59 % (SP), much higher than 5.87 % (AS). Here the yield comparison is based on pure cellulose and after an identical three-step production sequence.
In the literature, much lower cellulose purification yields were reported for CI (6.48 ± 0.55 % (w/w) of the dry weight of the whole tunic; Nakashima et al. 2008), but different methods were used in that study. A higher cellulose yield (32 %) for HR was reported using the same sequence used in this study, while no purity data were given in the literature (Koo et al. 2002). There were also data reported for other tunicate species: 17.29 % for Cnemidocarpa finmarkiensis, 18.99 % for Pyura haustor, 31.22 % for Corella willmeriana and 54.47 % for Ascidia paratropa (Nakashima et al. 2008). The cellulose yield is different between the different tunicate species, and the absolute yield depends on the methods used, similar to the cellulose purity.
Cellulose characteristics from different tunicate species
Molecular masses
Although the TCs were ~100 % pure, there were variations in their molecular masses depending on the original species. The highest weight average degree of polymerization (DPw) (4,200) was found for CI, followed by those for SP and HR (DPw of 3,910 and 3,520, respectively); the cellulose from AS had the lowest DPw of 1,650 (Table 4). Additionally, the molecular mass distributions display different patterns. AS cellulose is a single fraction with a relatively uniform molecular mass distribution (Fig. 5), which is reflected by its low polydispersity index (PDI) value (5.2) (Table 4). HR cellulose consists of one main fraction with a long tail and a higher PDI (10.3), implying that a wide range of molecular masses were present in the sample. There were two major fractions in the CI cellulose: a broader one in the higher Mw region (DPw > 911) and a narrower one in the lower Mw region (DPw < 911), with the highest PDI (10.6). Two fractions were also observed for SP cellulose, although the major fraction is in the lower Mw region (DPw < 2,452), and the PDI was 6.4. The DP of 1,650–4,200 for tunicate cellulose in this work agrees quite well with the DP of 700–3,500 reported by Krässig (1954). The literature indicates that the Mw of the cellulose from HR was 4.13 × 106 Da when using a viscometric determination method (Tamai et al. 2004), which was higher than the corresponding HR sample in our study. However, the production methods were not described (Tamai et al. 2004). Different production methods and different molecular mass determination methods can result in different values. The differences might also arise from the differences in sampling location, season, animal age, etc. Compared to the DPw values of 1,615 and 1,408 for dissolving pulp from softwood and hardwood, respectively (Li et al. 2012), the DPw value of AS cellulose was comparable, while the other TCs in this study exhibited much higher molecular masses than the woody celluloses, implying better chemical and mechanical properties.

Molecular mass distributions of cellulose from different tunicates
Microfibril structure
The ultrastructures of the TC microfibrils also vary depending on the original species. According to the SEM images shown in Fig. 3, the microfibril structures from the original tunics were preserved after the production sequence. After removing the other tunic components, the composite structures of cellulose-protein fibrils cemented by sulfated mucopolysaccharides/glycans no longer existed. The pure cellulose obtained presented as smaller microfibrils than those in the tunics, with mean widths of 16.04 ± 0.64, 12.70 ± 1.36, 12.59 ± 1.15 and 15.05 ± 2.30 nm for CI, AS, HR and SP, respectively (Table 2). The celluloses from the soft and thin tunics of CI and AS were less aggregated than those from the hard and thick HR and SP tunics. Though all tunicate cellulose microfibrils had a section shape of lozenges, the former were in a random orientation within an interwoven loose mesh, while the latter were arranged in randomly orientated poly-lamellate deposits (Fig. 3). Wide bundles were also observed in the literature (Kimura and Itoh 1996, 2007; Kim et al. 2013). The obtained TC was still aggregated in the form of microfibrils, and hundreds of cellulose microfibrils were still bundled, similar to the original tunics; the shape and dimension of the TC microfibril bundles varied depending on the original tunicate species (Okamoto et al. 1996). The lengths of the obtained TCs could not be accurately measured using SEM because no obvious breakpoints in the microfibrils could be found in the field of vision. These microfibrils should be several micrometers or longer, revealing their very high ratios of length to diameter (aspect ratios). The literature indicated that the TC fibrils have a cross-sectional diameter around 10–20 nm with a high aspect ratio, and our results agree (Koo et al. 2002). Compared to woody cellulose, e.g., from Cryptomeria japonica [3.6 nm wide and 272 ± 127 nm long (Saito et al. 2013)], the obtained TCs have larger diameters and aspect ratios.
Specific surface area (SSA)
The SSAs of the obtained celluloses after freeze-drying (Table 2) are different between species: 133, 95.8, 79.9 and 81.4 m2/g for CI, AS, HR and SP, respectively. The cellulose from CI was also subjected to supercritical CO2 drying (Sehaqui et al. 2011) and the SSA was found as 154 m2/g, indicating that the freeze-drying did not cause very significant collapse of the network. Compared to the SSAs (1.7–3.2 m2/g) of the starting tunics, the chemical processing procedure significantly increased the SSAs of the samples due to the purification of the cellulose fibrils from the other cementing components in the native composite structure, the removal of the small filling subunits between the large particles and the retention of the pure cellulose network. Apparently, the celluloses from the hard-tunic Stolidobranchia tunicates (HR and SP) had much lower SSAs than the soft structures from the Phlebobranchia species (CI and AS). Figure 3 shows that the larger cellulose microfibril bundles were preserved from the Stolidobranchia tunics, while the loose network was a remaining feature for the Phlebobranchia species (see above), giving the TCs from HR and SP lower SSAs than those from CI and AS. Generally, SSAs ranging from 81.4 to 133 m2/g agree with the reported data (100–150 m2/g) for tunicate cellulose (Kuga et al. 2002). These high SSAs reveal their excellent adsorption capacity and chemical reactivity at the surfaces, suggesting good application potentials as functional or “smart” materials and as chemical cellulose for derivatization.
Crystallinity
From the X-ray diffraction (XRD) patterns shown in Fig. 6, all the TCs obtained had a cellulose I structure: a doublet peak at 2θ 14.7° and 2θ 16.8° associated with the (1ī0) and (110) reflections, respectively, and a singlet peak at 2θ 22.8° attributed to (200) diffraction. No representative cellulose type II peaks were observed, including those at 2θ 12° (1ī0), 20° (110) and 22° (200) (Nakashima et al. 2008; Oh et al. 2005; Kim et al. 2013). Therefore, the TCs were nearly pure cellulose I allomorphs, which is consistent with other studies (Koo et al. 2002; Nakashima et al. 2008). Noticeably, as observed in the FTIR analyses conducted during the production sequence (highlighted square regions in Fig. 4), they are cellulose Iβ allomorph structures which were originally present in the tunics and were not generated during the production sequence; no shifts were observed from 3,240 to 750 cm−1 (for Iα allomorphs) to 3,270 and 710 cm−1 (for Iβ allomorphs), respectively (Sugiyama et al. 1991a). An alkaline treatment at an elevated temperature could change Iα allomorphs into Iβ allomorphs (Sugiyama et al. 1991b).

X-ray diffractograms of tunicate celluloses from different tunicates
The crystallinity index calculated from the XRD spectra of TCs was 89, 70, 95 and 95 % for CI, AS, HR and SP celluloses, respectively (Table 4). The two celluloses from the hard and thick tunic species from the Stolidobranchia order showed higher indexes than those from the Phlebobranchia order, indicating the diversity of the TCs between the different species. Kim et al. measured the crystallinity index of the cellulose from HR, and the value (74 %; Kim et al. 2010) was lower than that from the same species in this work. Again, different TC could be obtained from an identical species when different production methods were used. The crystallinity index of the TCs obtained is comparable to that of algal cellulose (>80 %), larger than that of bacterial cellulose (65–79 %) and much larger than the celluloses from wood or other plants (44–65 %; Klemm et al. 2005), indicating that TCs have very compact cellulose molecules packages, which implies excellent mechanical properties and great potential for applications in the next generation of cellulose-based materials, such as those used for reinforcing composite films. In addition, the calculated crystal’s size of (200) diffraction was 60.6, 60.2, 73.2 and 81.0 Å for CI, AS, HR and SP celluloses, respectively (Table 4), and this size range is consistent with the crystal’s size of 72.3 Å reported for the cellulose from tunicate Styela clava (Pullawan 2012).
Thermal stability
From the thermogravimetric analyses, the degradation onset temperature (To) increased in the following order: AS < CI < SP ~ HR (207, 226, 261 and 269 °C; Table 4; Fig. 7). This order of thermal stability for TCs is consistent with that of their crystallinity (Table 4). The higher the crystallinity index of the cellulose, the better the packing of the cellulose chains will be; thus, the initiation of decomposition will require more energy. However, all celluloses had a similar peak decomposition temperature (Tmax) (354–369 °C). Therefore, once decomposition began, similar mechanisms will be followed because their purities and fundamental structures were almost identical. Apparently, all TCs showed very good thermal stability, especially those from HR and SP, making them good candidates for producing thermally stable materials.

Thermogravimetric analysis diagrams of the celluloses obtained from different tunicates
Conclusions
Cellulose synthesis is a characteristic common to all tunicates. In the investigated tunics, a supramolecular organization was observed that involves cellulose-based microfibrils containing or associating with other components, such as proteins, lipids and sulfated mucopolysaccharides or sulfated glycans. The microfibrils are initially bundled, and the dimensions of the microfibril bundles and their orientation are different between species. The prehydrolysis-kraft cooking-bleaching sequence is very effective at removing lipids, sulfated mucopolysaccharides, sulfated glycans, proteins and chromophores, resulting in chemically pure cellulose. This sequence is generally applicable for all tunicate species. Both the chemical and morphological structures of the cellulose from the initial tunics are largely preserved following the sequence. For example, the celluloses are almost entirely Iβ allomorphs. The elementary β-1,4 glucan chains are still packed in crystalline states, while the width, arrangement and crystalline states of fibrils still differ between species. Tunicate celluloses are excellent for various chemical and material applications because they have high purities, large molecular masses, low polydispersity indexes, high surface areas, high crystallinities, large widths and high aspect ratios. Almost all of the above structural and morphological characteristics of tunicate celluloses are better than those of the celluloses from wood or annual plants. Tunicates are therefore better cellulose sources; additionally, they will not compete with agriculture or forestry for land resources. With the knowledge acquired here regarding the removal of lipids, proteins and sulfated glycans or mucopolysaccharides during the cellulose production sequence, the recovery of other useful components from tunicate becomes possible during the production of tunicate cellulose on a large scale, thus expanding the applications of tunicate resources.
References
Albano RM, Mourão P (1986) Isolation, fractionation, and preliminary characterization of a novel class of sulfated glycans from the tunic of Styela plicata (Chordata Tunicata). J Biol Chem 261:758–765
Baginski T, Hirohashi N, Hoshi M (1999) Sulfated O-linked glycans of the vitelline coat as ligands in gamete interaction in the ascidian, Halocynthia roretzi. Dev Growth Differ 41:357–364
Belton PS, Tanner SF, Cartier N, Chanzy H (1989) High-resolution solid-state carbon-13 nuclear magnetic resonance spectroscopy of tunicin, an animal cellulose. Macromolecules 22:1615–1617
Berrill NJ (1947) The structure, development and budding of the ascidian, Eudistoma. J Morphol 81:269–281
Carlisle DB, Bernal JD (1968) Vanadium and other metals in ascidians [and discussion]. Proc R Soc Lond B Biol Sci 171:31–42
Cassaro CM, Dietrich CP (1977) Distribution of sulfated mucopolysaccharides in invertebrates. J Biol Chem 252:2254–2261
De Leo G, Patricolo E, Lunetta GDA (1977) Studies on the fibrous components of the test of Ciona intestinalis Linnæus. I. Cellulose-like polysaccharide. Acta Zool 58:135–141
French A (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896
French A, Santiago Cintrón M (2013) Cellulose polymorphy, crystallite size, and the Segal crystallinity index. Cellulose 20:583–588
Habibi Y, Chanzy H, Vignon M (2006) TEMPO-mediated surface oxidation of cellulose whiskers. Cellulose 13:679–687
Habibi Y, Lucia LA, Rojas OJ (2010) Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem Rev 110:3479–3500
Helbert W, Nishiyama Y, Okano T, Sugiyama J (1998) Molecular imaging of Halocynthia papillosa cellulose. J Struct Biol 124:42–50
Herzog RO, Gonell HW (1924) Ueber den feinbau der kunstseide. Kolloid-Zeitschrift 35:201–202
Hirose E (2009) Ascidian tunic cells: morphology and functional diversity of free cells outside the epidermis. Invertebr Biol 128:83–96
Hirose E, Lambert G, Kusakabe T, Nishikawa T (1997) Tunic cuticular protrusions in ascidians (Chordata, Tunicata): a perspective of their character-state distribution. Zool Sci 14:683–689
Hirose E, Ohtake SI, Azumi K (2009) Morphological characterization of the tunic in the edible ascidian, Halocynthia roretzi (Drasche), with remarks on ‘soft tunic syndrome’ in aquaculture. J Fish Dis 32:433–445
Khandelwal M, Windle AH (2013) Self-assembly of bacterial and tunicate cellulose nanowhiskers. Polymer 54:5199–5206
Kim U-J, Eom SH, Wada M (2010) Thermal decomposition of native cellulose: influence on crystallite size. Polym Degrad Stab 95:778–781
Kim SM, Park JM, Kang TY, Kim YS, Lee SK (2013) Purification of squirt cellulose membrane from the cystic tunic of Styela clava and identification of its osteoconductive effect. Cellulose 20:655–673
Kimura S, Itoh T (1996) New cellulose synthesizing complexes (terminal complexes) involved in animal cellulose bio-synthesis in the tunicate Metandrocarpa uedai. Protoplasma 194:151–163
Kimura S, Itoh T (1997) Cellulose network of hemocoel in selected compound styelid ascidians. J Electron Microsc 46:327–335
Kimura S, Itoh T (2007) Biogenesis and function of cellulose in the tunicates. In: Brown RM Jr, Saxena I (eds) Cellulose: molecular and structural biology. Springer, Netherlands, pp 217–236
Kimura S, Ohshima C, Hirose E, Nishikawa J, Itoh T (2001) Cellulose in the house of the appendicularian Oikopleura rufescens. Protoplasma 216:71–74
Klemm D, Heublein B, Fink H-P, Bohn A (2005) Cellulose: fascinating biopolymer and sustainable raw material. Angew Chem Int Ed Engl 44:3358–3393
Koo Y-S, Wang Y-S, You S-H, Kim H-D (2002) Preparation and properties of chemical cellulose from ascidian tunic and their regenerated cellulose fibers. J Appl Polym Sci 85:1634–1643
Kovács A, Nyerges B, Izvekov V (2008) Vibrational analysis of N-acetyl-alpha-d-glucosamine and beta-d-glucuronic acid. J Phys Chem B 112:5728–5735
Krässig VH (1954) Untersuchungen zur konstitution des Tunicins. 408. Mitteilung über makromolekulare Verbindungen. Die Makromolekulare Chemie 13:21–29
Kuga S, Kim D-Y, Nishiyama Y, Brown RM (2002) Nanofibrillar carbon from native cellulose. Mol Cryst Liqid Cryst 387:13–19
Larsson PT, Westermark U, Iversen T (1995) Determination of the cellulose Iα allomorph content in a tunicate cellulose by CP/MAS 13C-NMR spectroscopy. Carbohydr Res 278:339–343
Li D, Sevastyanova O, Ek M (2012) Pretreatment of softwood dissolving pulp with ionic liquids. Holzforschung 66:935–943
Lunetta GD (1983) Comparative study of the tunics of two ascidians: Molgula impura and Styela partita. Acta Embryol Morphol Exp 43:137–149
Meyer K, Huber L, Kellenberger E (1951) La texture de la cellulose animale. Experientia 7:216–217
Mohamed SF, Agili FA (2013) Antiviral sulphated polysaccharide from brown algae Padina pavonia characterization and structure elucidation. Int J ChemTech Res 5:1469–1476
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941–3994
Mourao PA, Perlin AS (1987) Structural features of sulfated glycans from the tunic of Styela plicata (Chordata-Tunicata). A unique occurrence of l-galactose in sulfated polysaccharides. Eur J Biochem 166:431–436
Nakashima K, Sugiyama J, Satoh N (2008) A spectroscopic assessment of cellulose and the molecular mechanisms of cellulose biosynthesis in the ascidian Ciona intestinalis. Mar Genomics 1:9–14
Nishiyama Y, Okano T, Langan P, Chanzy H (1999) High resolution neutron fibre diffraction data on hydrogenated and deuterated cellulose. Int J Biol Macromol 26:279–283
Nishiyama Y, Sugiyama J, Chanzy H, Langan P (2003) Crystal structure and hydrogen bonding system in cellulose I(alpha) from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 125:14300–14306
Oh SY, Yoo DI, Shin Y, Kim HC, Kim HY, Chung YS, Park WH, Youk JH (2005) Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr Res 340:2376–2391
Okamoto T, Sugiyama J, Itoh T (1996) Structural diversity of ascidian crystalline celluloses. Wood Res 83:27–29
Pancake SJ, Karnovsky ML (1971) The isolation and characterization of a mucopolysaccharide secreted by the snail, Otella lactea. J Biol Chem 246:253–262
Percival E, Wold JK (1963) 1040. The acid polysaccharide from the green seaweed Ulva lactuca. Part II. The site of the ester sulphate. J Chem Soc 5459–5468
Pullawan T (2012) Interfacial micromechanics of all-cellulose nanocomposites using Raman spectroscopy. PhD thesis, University of Manchester
Rånby BG (1952) Physico-chemical investigations on animal cellulose (tunicin). Arkiv Kemi 4:241–248
Reddy N, Yang Y (2005) Structure and properties of high quality natural cellulose fibers from cornstalks. Polymer 46:5494–5500
Reid JL (1979) On the contribution of the Mediterranean Sea outflow to the Norwegian-Greenland Sea. Deep Sea Res Part A Oceanogr Res Pap 26:1199–1223
Saito T, Kuramae R, Wohlert J, Berglund LA, Isogai A (2013) An ultrastrong nanofibrillar biomaterial: the strength of single cellulose nanofibrils revealed via sonication-induced fragmentation. Biomacromolecules 14:248–253
Scherrer P (1912) Bestimmung der inneren struktur und der größe von kolloidteilchen mittels röntgenstrahlen. In: Kolloidchemie Ein Lehrbuch. Chemische Technologie in Einzeldarstellungen. Springer, Berlin, pp 387–409
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29:786–794
Sehaqui H, Zhou Q, Ikkala O, Berglund LA (2011) Strong and tough cellulose nanopaper with high specific surface area and porosity. Biomacromolecules 12:3638–3644
Smith MJ, Dehnel PA (1970) The chemical and enzymatic analyses of the tunic of the ascidian Halocynthia aurantium (pallas). Comp Biochem Physiol 35:17–30
Sugiyama J, Persson J, Chanzy H (1991a) Combined infrared and electron diffraction study of the polymorphism of native celluloses. Macromolecules 24:2461–2466
Sugiyama J, Vuong R, Chanzy H (1991b) Electron diffraction study on the two crystalline phases occurring in native cellulose from an algal cell wall. Macromolecules 24:4168–4175
Sun JX, Sun XF, Zhao H, Sun RC (2004) Isolation and characterization of cellulose from sugarcane bagasse. Polym Degrad Stab 84:331–339
Swinehart JH, Wilton RB, Halko DJ, Schroeder NC (1974) The vanadium and selected metal contents of some ascidians. Biol Bull 146:302–312
Tamai N, Tatsumi D, Matsumoto T (2004) Rheological properties and molecular structure of tunicate cellulose in LiCl/1,3-dimethyl-2-imidazolidinone. Biomacromolecules 5:422–432
Troedsson C, Thompson E, Bouquet J-M, Magnesen T, Schander C, Li J (2013) Tunicate extract for use in animal feeds. WO2013088177A1
Van Daele Y, Revol J-F, Gaill F, Goffinet G (1992) Characterization and supramolecular architecture of the cellulose-protein fibrils in the tunic of the sea peach (Halocynthia papillosa, Ascidiacea, Urochordata). Biol Cell 76:87–96
Wang B-d, Wang X-l, Zhan R (2003) Nutrient conditions in the Yellow Sea and the East China Sea. Estuar Coast Shelf Sci 58:127–136
Wardrop AB (1970) The structure and formation of the test of Pyura stolonifera Tunicata. Protoplasma 70:73–86
Yuan H, Nishiyama Y, Wada M, Kuga S (2006) Surface acylation of cellulose whiskers by drying aqueous emulsion. Biomacromolecules 7:696–700
Acknowledgments
The China Scholarship Council (CSC) is acknowledged for supporting Yadong Zhao’s PhD study at KTH. Christofer Troedsson, Eric Thompson, and Jean-Marie Bouquet from University of Bergen are acknowledged for the tunicate sample collections in Norway.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhao, Y., Li, J. Excellent chemical and material cellulose from tunicates: diversity in cellulose production yield and chemical and morphological structures from different tunicate species. Cellulose 21, 3427–3441 (2014). https://doi.org/10.1007/s10570-014-0348-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-014-0348-6