
Abstract
Autophagy is a lysosomal degradation pathway of eukaryotic cells that is highly conserved from yeast to mammals. During this process, cooperating protein complexes are recruited in a hierarchic order to the phagophore assembly site (PAS) to mediate the elongation and closure of double-membrane vesicles called autophagosomes, which sequester cytosolic components and deliver their content to the endolysosomal system for degradation. As a major cytoprotective mechanism, autophagy plays a key role in the stress response against nutrient starvation, hypoxia, and infections. Although numerous studies reported that impaired function of core autophagy proteins also contributes to the development and progression of various human diseases such as neurodegenerative disorders, cardiovascular and muscle diseases, infections, and different types of cancer, the function of this process in human diseases remains unclear. Evidence often suggests a controversial role for autophagy in the pathomechanisms of these severe disorders. Here, we provide an overview of the molecular mechanisms of autophagy and summarize the recent advances on its function in human health and disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The term autophagy (“self-eating” from Greek) describes lysosome-dependent degradative routes of eukaryotic cells, during which cytosolic material is delivered to the endo-lysosomal system for subsequent degradation. Its three major forms involve different mechanisms, which are responsible for the sequestration of different cargo. Chaperone-mediated autophagy is a selective delivery pathway accomplished by lysosomal-associated membrane protein 2A (LAMP2A) and chaperon complexes while cytoplasmic constituents are directly engulfed by the invaginations of lysosomes during microautophagy. The process of macroautophagy involves the formation of double-membrane autophagosomes to sequester cytosolic cargo either in a selective or in a non-selective way. Matured autophagosomes undergo fusion with late endosomes or lysosomes (vacuole in yeast), and their degraded content is recycled. Macroautophagy (hereafter referred to as autophagy) is the most prevalent and therefore the best-characterized self-degradative pathway. It not only provides nutrients for the cell but also has an essential housekeeping function: by the elimination of excessive and damaged organelles, protein aggregates, and invading microorganisms it contributes to the organism’s health and longevity.
The molecular mechanism of autophagy
Autophagy is a complex intracellular process involving sequential steps of nucleation, elongation, and closure of a so-called phagophore or isolation membrane to form the autophagosome and subsequent membrane fusing events. Although various organelles have already been reported as primary membrane sources, origin of autophagosomes is still unclear. Three-dimensional reconstruction studies carried out on different mammalian cell types showed that specific subdomains of the endoplasmic reticulum (ER) are intimately associated with cup-shaped autophagic membranes called omegasomes and presumably serve as an assembly site for them (Hayashi-Nishino et al. 2009). However, Hailey and colleagues found in a normal rat kidney cell line (NRK58B) that mitochondria can also provide membrane for forming autophagic structures (Hailey et al. 2010). In addition, a more recent study using COS-7 fibroblast-like cells suggested that ER-mitochondria contact sites may also play a role in the formation of autophagosomes (Hamasaki et al. 2013). In parallel with these studies, Rubinsztein and his colleagues found that during periods of increased autophagosome formation, the plasma membrane can also serve as an important membrane source in HeLa cells (Ravikumar et al. 2010). Moreover, experiments in both yeast and human embryonic kidney (HEK293) cells showed that ATG9-positive, Golgi-derived small vesicles play a key role in the elongation of the phagophore membrane (Yamamoto et al. 2012). According to recent studies, recycling endosomes also provide membrane for autophagosomes during starvation-induced autophagy in HEK293A, in HeLa cells, and in the larval fat body of Drosophila melanogaster (Longatti et al. 2012; Puri et al. 2013). Together, these data suggest that more different organelles can serve as membrane source for autophagic structures at the same time, and the level of their contribution might vary among cell types and organisms.
Although autophagy itself was originally described in mammalian cells, the underlying molecular mechanisms were identified in the yeast Saccharomyces cerevisiae (Tsukada and Ohsumi 1993). The core autophagic machinery consists of a subset of autophagy-related (ATG) proteins, which were shown to be essential for autophagosome formation. ATG proteins form several complexes, which are recruited to the PAS in a hierarchical order (Itakura and Mizushima 2010).
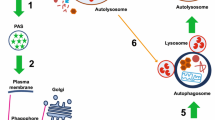
Autophagosome formation
Following autophagy induction, an initiation complex (ULK1 complex) is recruited to the PAS (Fig. 1). The complex is named after the serine/threonine kinase ULK1/2 (Atg1 in yeast), which is the only known ATG protein with kinase activity (Yan et al. 1998). This complex also contains ATG13 (Chang and Neufeld 2009; Funakoshi et al. 1997), the scaffold protein RB1CC1/FIP200—Atg17 in yeast—(Hara et al. 2008; Nagy et al. 2014) and ATG101 as well (Hosokawa et al. 2009). Activated ULK1/2 not only phosphorylates ATG13 and RB1CC1/FIP200 but also enhances the activity of the autophagy nucleation complex (see below) by phosphorylation (Kim et al. 2013a). In addition, a recent study suggested that—through the phosphorylation of a myosin light-chain kinase-like protein—ULK1/Atg1 indirectly regulates the activity of myosin II motor protein, thereby affecting the delivery of membrane source to the elongating PAS (Tang et al. 2011). Similarly, yeast ATG17 was found to directly bind ATG9, thereby recruiting it to the PAS (Sekito et al. 2009). ATG9 is a transmembrane protein, which has a key role in the transport of Golgi-derived vesicles to the PAS, providing membrane source for its expansion (Yamamoto et al. 2012). Shuttling of ATG9 between PAS and different membrane sources is mediated by ATG2 and (WIPIs) (Atg18 in yeast) (Nagy et al. 2013).

The major molecular mechanisms involved in autophagy. Upon autophagy induction, ULK1 complex and BECN1 complex appear at the phagophore assembly site (PAS) to orchestrate the recruitment of further autophagy-related (ATG) proteins and the elongation of the phagophore membrane. After cleavage by the protease ATG4 and subsequent conjugation to phosphatidylethanolamine (PE), ATG8/LC3 is attached to the phagophore membrane. Following autophagosome closure, ATG8/LC3 is recycled from the outer membrane again by the help of ATG4. However, ATG8/LC3 bound to inner membrane is sequestered into the lumen of the autophagosome and degraded—together with other cytosolic material—by lysosomal hydrolases. Closed autophagosomes undergo fusion with late endosomes and lysosomes, thereby acquiring lysosomal enzymes and membrane proteins required for degradation. The fusion events are mediated by the multiunit tethering factor HOPS complex and the autophagosome-specific SNARE syntaxin 17. Following autophagosome-lysosome fusion, lysosomal enzymes are activated due to acidic pH provided by v-ATPases. Lysosomal associated membrane proteins (LAMPs) control fusion and protect the autolysosomal membrane against degradation. During selective autophagy, cargo—such as bacteria containing vacuole (BCV)—is specifically bound by adaptor proteins. These adaptors are called autophagy receptors since they also interact with ATG8/LC3, thus attaching cargo to the phagophore membrane
The nucleation complex (together with ULK1 complex) plays an essential role in the early stages of autophagy; it is involved in the elongation of the phagophore. The complex comprises of the class III phosphatidylinositol 3-kinase (PI3K) vacuolar protein sorting 34 (VPS34) and its regulatory subunit Vps15. These proteins are responsible for the elevated phosphatidylinositol 3-phosphate (PI3P) level of autophagic membranes (Lindmo et al. 2008). The inositol head group phosphorylated at position 3 is recognized by distinctive protein domains, such as FYVE and PX, so the presence of PI3P is a key factor in the subsequent protein recruitment to the phagophore. As it also contains Beclin 1 (BECN1), the mammalian homolog of the yeast Atg6 protein (Furuya et al. 2005), the nucleation complex is called BECN1 complex as well. VPS34, VPS15, and BECN1 proteins constitute the core which can form, depending on the fourth subunit, two different complexes in yeast, fruit fly, and mammals. The BECN1 complex containing UV radiation resistance associated (UVRAG) protein plays an essential role in endosomal maturation, whereas that containing ATG14L is required for autophagy (Lőrincz et al. 2014; Zhong et al. 2009). Presumably, ATG14L has a primary role in recruiting the BECN1 core complex to the PAS (Matsunaga et al. 2010).
An essential step of phagophore elongation is the lipidation of the ubiquitin-like protein Atg8/LC3. For this process, two ubiquitin-like conjugation systems are required (reviewed in Nakatogawa 2013). First, owing to the E1 enzyme-like protein activating activity of ATG7 and the E2 enzyme-like protein conjugating function of ATG10, the ubiquitin-like ATG12 protein forms covalent bond with ATG5. The ATG5–12 conjugate is subsequently organized into a multimeric protein complex containing ATG16L1 (Atg16 in yeast) as well. This large complex acts as a ubiquitin ligase-like enzyme and mediates the covalent binding of an ubiquitin-like Atg8 family protein (Atg8 in yeast, MAP1LC3 or GABARAP in mammals) to phosphatidyl-ethanolamine (PE) (Romanov et al. 2012). However, before this step, ATG8 must be activated by the abovementioned E1-like ATG7 and the E2 enzyme-like protein ATG3 and also by a specific cleavage carried out by ATG4 protease. The result of this elaborate process is a lipidated Atg8, which is subsequently attached to both the inner and outer membrane of the expanding phagophore. Before autophagosome completion, all ATG proteins except for Atg8 dissociate from the isolation membrane and are recycled to the PAS. Following phagophore closure, Atg8 is also recycled from the outer membrane, again mediated by ATG4 (Satoo et al. 2009), whereas inner membrane proteins are trapped inside the autophagosome and will be degraded by lysosomal enzymes. Although the exact function of Atg8 in the process of autophagy remains unclear, it is a widely used autophagy marker, since it localizes to autophagic structures from the early steps till degradation (Klionsky et al. 2016).
Maturation process
Following completion and the removal of LC3/Atg8 from the outer membrane, autophagosomes undergo homo- and heterotypical fusion events (Fig. 1). Growing evidence shows that autophagosomes can fuse not only with lysosomes to form autolysosomes but also with various populations of endosomes leading to the formation of hybrid organelles called amphisomes. In many cell types, this latter is an essential step for subsequent lysosomal fusion and degradation. The convergence of endosomal and autophagic routes might underlie the phenomenon that vesicles with cytosolic content (and thus interpreted as autophagosomes) were found to be positive for UVRAG or RAB7, proteins essential for endosomal maturation and fusion (Hyttinen et al. 2013). Presumably, these structures are amphisomes containing both autophagic and heterophagic membrane portions.
Notably, several studies found that the abovementioned small GTPase RAB7 (Ypt7 in yeast), a widely used marker of late endosomes (for a review see Wang et al. 2011), not only is present on autophagic/amphisomal structures but also required for the late stages of autophagy. In mammalian cells, dynein-dependent movement of autophagosomes was observed toward the perinuclear region where lysosomes are localized (Kimura et al. 2008); and RAB7, together with its effectors RILP (RAB interacting lysosomal protein) and FYCO1 (FYVE and coiled-coil domain containing 1), plays a key role in this microtubular transport (Bains et al. 2011; Pankiv et al. 2010).
Furthermore, RAB7 recruits additional proteins required for the late endosomal or lysosomal tethering and fusion. Recent studies found that depletion of RAB7 or overexpression of its inactive GDP-locked form resulted in the accumulation of enlarged autophagosomes, probably due to their impaired fusion with lysosomes (Gutierrez et al. 2004). Homotypic fusion and vacuole protein sorting (HOPS) complex, which was identified as an evolutionarily conserved hexameric tethering factor required for lysosomal/vacuolar fusion from yeast to mammals (for a review see Balderhaar and Ungermann 2013), is a well-known interacting partner of RAB7. Several studies based on various model organisms found that recruitment of HOPS complex to autophagosomes, presumably via binding to RAB7, is required for autophagic clearance (Jiang et al. 2014; Takáts et al. 2014; Wartosch et al. 2015). HOPS not only functions as a bridge connecting the opposing membranes before fusion but also directly mediates the fusion events through interaction with soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs). In yeast, several SNARE proteins, for instance, Vam3p and Vam7p, were reported to participate in autophagosome-vacuole fusion (Darsow et al. 1997; Sato et al. 1998). In fruit fly and mammals, recent studies revealed the existence of an autophagosome-specific SNARE called syntaxin 17, which—together with its partners SNAP-29 and VAMP7 or VAMP8—is responsible for the autophagosome-lysosome fusion (Itakura et al. 2012; Takáts et al. 2013). Furuta and colleagues also showed that VAMP8 together with VTI1B has an important role in autophagic fusion events (Furuta et al. 2010).
Ultimately, the outer membrane of autophagosomes or amphisomes fuse with lysosomes to form autolysosomes, a process often referred to as autophagosome maturation. Following the fusion, autophagic content is degraded by lysosomal hydrolases specific to a wide range of targets, e.g., cathepsins. Numerous studies showed that lack of these enzymes or their reduced activity led to impaired autophagic degradation and autophagosome accumulation (Maruzs et al. 2015; Tatti et al. 2012). These hydrolases are usually produced in pro-form and activated in lysosomes by the highly acidic pH (between 4.5 and 5.0) generated and maintained by lysosomal proton pumps called v-ATPases. Although it was previously presumed that lysosomal acidification is essential for autophagosome-lysosome fusion, a recent study revealed that fusion events are independent of lysosomal pH: while v-ATPase dysfunction resulted in an impaired degradation, autolysosome formation was not disrupted (Mauvezin et al. 2015). Autolysosomal membrane also contains a significant amount of integral lysosome-associated membrane glycoproteins (LAMPs), which probably play a key role in membrane protection against proteolytic enzymes. Several studies suggested that LAMP-2 is required for autophagosome-lysosome fusion, as well, since lack of this protein led to the accumulation of autophagosomes (Huynh et al. 2007).
Eventually, the content of autolysosomes is degraded, and the resulting molecules are released into the cytoplasm by permeases where they are recycled in anabolic processes (Yang et al. 2006). Increased amino acid level is already sensed in the lysosomal lumen by v-ATPases and, subsequently, results in the activation of mechanistic/mammalian target of rapamycin complex 1 (mTORC1) at the cytosolic side. As mTORC1 is the master negative regulator of autophagy (see below), this interaction is a part of a negative feedback loop downregulating the autophagic process (for a review see Jewell et al. 2013). Reactivated mTORC1 signaling also regulates the recycling of lysosomal membrane proteins and hydrolases through a process called lysosome reformation (Yu et al. 2010).
Selective autophagy
In most cell types, autophagy occurs at low basal levels as a housekeeping process to maintain cellular homeostasis by eliminating old and damaged organelles and misfolded proteins. However, upon stress conditions, such as nutrient starvation, hypoxia, growth factor deprivation, or oxidative stress, the autophagic degradation is rapidly upregulated and provides the cell with recycled nutrients and energy. Although stress-induced autophagy is mostly regarded as a non-specific (or bulk) degradative pathway, growing evidence shows that during basal or housekeeping autophagy, cargo can be sequestered selectively (for more detailed information, see Zaffagnini and Martens 2016). Various forms of selective autophagy are named after their specific targets, which can be invading pathogens (xenophagy or antimicrobial autophagy), mitochondria (mitochondrial autophagy or mitophagy), peroxisomes (pexophagy), cilia (ciliophagy), ER and ribosomes (reticulo- and ribophagy), or protein aggregates (aggrephagy).
Numerous studies suggest that ubiquitination might be a general tag designating the cargo to selective autophagic degradation (Kirkin et al. 2009). During selective autophagy, targeted proteins or organelles are recognized by adaptor proteins, such as SQSTM1/P62 (Pankiv et al. 2007), NBR1 (Deosaran et al. 2012), optineurin (Korac et al. 2013), nix (Novak et al. 2010), CALCOCO2/NDP52 (Heo et al. 2015), or Alfy (Isakson et al. 2013). A common structural feature of these autophagy receptors is the presence of canonical or non-canonical Atg8/LC3 interacting regions (LIRs)—also known as Atg8-interacting motifs (AIMs). LIRs specifically recognize and bind Atg8 family proteins; thereby, these autophagy receptors proteins are able to attach the cargo to the phagophore membrane (von Muhlinen et al. 2012; Pankiv et al. 2007). Interestingly, Ref(2)P, the Drosophila homolog of SQSTM1 was recently reported to interact with other proteins of the core autophagy machinery—for instance ATG101, ATG18, ATG14, and VPS34—raising the possibility that it has a role not only in cargo selection but also in autophagosome formation (Nagy et al. 2014).
Regulation of autophagy
Autophagy is a complex process fine tuned by several environmental signals through various signaling pathways (Fig. 2). One of the major regulators is the abovementioned mTOR, an evolutionarily conserved serine/threonine kinase, which integrates metabolic signals including amino acids and growth factors as well as oxygen and energy level to coordinate cell growth, proliferation, and metabolic processes in order to maintain cellular homeostasis. mTOR forms two complexes with distinct functions, but only mTOR complex 1 (mTORC1) has been described in the regulation of autophagy: active mTORC1 performs an inhibitory effect on autophagy induction by phosphorylating the initiation complex members (reviewed in Meijer et al. 2015).

Regulation of autophagy. Upon receptor tyrosine kinase (RTK) activation in the presence of insulin-like or other growth factors, AKT kinase is activated and inhibits tuberous sclerosis complex (TSC1/2) via phosphorylation. TSC1/2 functions as a GTPase activating protein (GAP) for the small GTPase Rheb; thus, active TSC1/2 has an inhibitory effect on Rheb. Inactivated TSC1/results in a GTP-bound Rheb which enables it to localize to lysosomal membranes and there to activate mTORC1, master regulator of cell growth, survival, and catabolic processes. mTORC1 is recruited to this place by the RAG small GTPases, which are translocated to the lysosome membrane following their activation by the guanosine exchange factor (GEF) Ragulator complex. Once localized to the lysosome, mTORC1 can be activated by Rheb. mTORC1 activity is also enhanced upon nutrient sufficiency. Elevated amino acids are already sensed in the lysosomal lumen by the transmembrane protein v-ATPase, which subsequently activates mTORC1 at the cytosolic surface. Active mTORC1 phosphorylate members of the autophagy initiation complex, thereby, prevents induction of autophagy. 5′-AMP-activated protein kinase (AMPK) is activated by high intracellular AMP/ATP ratio and by the LKB1 kinase and induces autophagy via direct phosphorylation of ULK1 or inhibition of mTORC1. During oxidative stress, death-associated protein Ser/Thr kinase (DAPK) is activated and induces autophagy via BECN1 complex activation by protein kinase D (PKD) phosphorylation. Upon hypoxia, HIF1α translocates to the nucleus and activates the transcription of genes necessary for metabolic adaptation to low oxygen levels, such as BNIP3. BNIP3 is a pro-apoptotic Bcl-2 family protein, which binds Bcl-2, thereby releasing BECN1 protein resulting in subsequent induction of autophagy. Bcl-2-BECN1 interaction is sustained by ER-associated IP(3) receptor (IP3R) and disrupted by c-Jun N-terminal protein kinase 1 (JNK1)-mediated phosphorylation of Bcl-2
mTORC1 activity is regulated by multiple signaling pathways and molecules. Upon receptor tyrosine kinase activation in the presence of insulin-like and other growth factors, the serine/threonine kinase AKT phosphorylates and thus inhibits tuberous sclerosis complex (TSC1/TSC2), a GTPase-activating protein (GAP) for the small GTPase Rheb (Zhang et al. 2003). Consequently, Rheb is stabilized in an active, GTP-bound form and activates mTORC1, thereby repressing autophagy (Long et al. 2005). Phosphatase and tensin homolog (PTEN) is a phosphoinositide-3 phosphatase, which activates autophagy by antagonizing the above signaling pathway (Arico et al. 2001). Subcellular localization of mTORC1 has an essential role in its regulation, as well. Amino acid sufficiency promotes the activation of RAG GTPases in a Ragulator complex-dependent manner (Sancak et al. 2010). Next, active RAGs recruit mTORC1 to the lysosome membrane, where it can be activated owing to both elevated amino acid levels and direct binding to Rheb.
5′-AMP-activated protein kinase (AMPK), activated upon low cellular energy levels, regulates autophagy positively both by direct phosphorylation of ULK1 (Bach et al. 2011) and indirectly by repressing mTORC1 activity (Gwinn et al. 2008). A recent study revealed that upon nutrient deprivation, AMPK promotes autophagy via inhibitory phosphorylation of non-autophagic BECN1 complexes (Kim et al. 2013a).
Autophagy regulation is not confined to the modulation of the induction. Upon hypoxia, HIF1α transcription factor activates expression of several genes necessary for metabolic adaptation to low oxygen levels. Among these hypoxia responsive genes, there are BNIP3 and BNIP3L, which were found to increase mitochondrial autophagy (Zhang et al. 2008). While BNIP3 is able to suppress the activity of mTORC1 by binding and inhibiting Rheb (Li et al. 2007), BNIP3 and BNIP3L also contribute to the formation of the BECN1 complex by displacing BECN1 from its inhibitory binding partner Bcl-2 (Bellot et al. 2009). Similarly, other proteins containing Bcl-2 homology 3 (BH3) domain play a positive regulatory role by disrupting the Bcl-2/Bcl-XL inhibition of BECN1 (Maiuri et al. 2007). Among others, the ER-associated IP(3) receptor functions as an autophagy inhibitor by forming a complex with BECN1 and probably Bcl-2 (Vicencio et al. 2009). In addition, upon nutrient starvation, c-Jun N-terminal protein kinase 1 (JNK1) contributes to the induction of autophagy by phosphorylating Bcl-2 and thereby disrupting its interaction with BECN1 (Wei et al. 2008).
Oxidative stress promotes autophagy in another VPS34-dependent manner as well. Under these conditions, death-associated protein serine/threonine kinase (DAPK), a well-known regulator of cell death activates protein kinase D (PKD), which subsequently phosphorylates VPS34 leading to increased autophagosome formation (Eisenberg-Lerner and Kimchi 2012).
Autophagy in diseases
As autophagy maintains cellular homeostasis and acts as a major cytoprotective mechanism upon different stress conditions, it is essential for the organismal survival under physiological conditions. Regarding this importance, it is not surprising that defects or deregulation of this process were observed in various human diseases, such as infections, neurodegenerative, cardiovascular and immune disorders, diabetes, or cancer (Fig. 3). Here, we summarize in more detail how autophagy is relevant to disease pathogenesis.

Autophagy is deregulated in several human diseases. Pathogens are able to inhibit or induce autophagy or to exploit the autophagic machinery in order to support their own survival in host cells during infections (orange). Neurodegenerative disorders (blue) impair distinct steps of autophagy contributing to the pathogenesis of disease. Autophagy is often upregulated in cardiovascular diseases (green) as a cytoprotective response; however, excessive autophagy may exacerbate some pathological conditions. Deregulation of the autophagic process may contribute to metabolic diseases (brown) as well. In cancer (purple), autophagy also seems to play a dual role: as a surveillance mechanism, it protects normal cells from oncogenic transformation, whereas the process is able to promote tumor cell survival upon stress conditions. Abbreviations in the figure are the following: AD Alzheimer’s disease, ALS amyotrophic lateral sclerosis, CM cardiomyopathy, HD Huntington’s disease, HF heart failure, HIV human immunodeficiency virus, KSHV Kaposi sarcoma-associated herpesvirus, LMNA-related DCM lamin A/C mutation-related dilated cardiomyopathy, PD Parkinson’s disease, SENDA static encephalopathy of childhood with neurodegeneration in adulthood (color figure online)
Autophagy in infections and immunity
Autophagy contributes to the defense against infectious diseases via selective removal of intracellular pathogens (bacteria, parasites, and viruses) and by activation of innate immune response. Already inside the cell, invading pathogens are targeted by SQSM1-like receptors (SLRs) of the selective autophagic machinery and are directly eliminated via xenophagy; extracellularly, they are attacked by ATG8/LC3-associated phagocytosis (LAP), a phagocytotic process which also engages parts of the autophagic machinery (Henault et al. 2012). The most studied examples of antibacterial autophagy are host response against group A Streptococcus and Mycobacterium tuberculosis, but autophagy also has an important role in the defense against other bacteria, such as Salmonella, Shigella, and Listeria (for further details see Yuk et al. 2012). In addition to its role in targeting and degradation of bacteria, autophagy also promotes the elimination of bacterial toxins, for instance, the α-toxin of Staphylococcus aureus (Maurer et al. 2015). Moreover, autophagy functions as an antiviral process—by targeting HIV (Campbell et al. 2015), hepatitis C (Dash et al. 2016), or Sindbis virus (Orvedahl et al. 2010)—and it contributes to the elimination of protozoans, as well (Cervantes et al. 2014; Choi et al. 2014).
Given the significance of xenophagy as a major antimicrobial response of host cells, several microorganisms have developed defense mechanisms to escape autophagic degradation. As a result of co-evolution, several pathogens are able to evade autophagy recognition. For example, Shigella flexneri masks its surface by secreting a virulence protein (Ogawa et al. 2005). In contrast, Salmonella enterica subsp. enterica serovar Typhimurium triggers autophagy in infected cells by depletion of amino acids. However, during the prolonged starvation caused by this, mTORC1 is recruited to the Salmonella-containing vacuole and reactivated in a RAG-Ragulator-dependent pathway. By this strategy, autophagy is locally inhibited and bacteria avoid the elimination by xenophagy (Tattoli et al. 2012). M. tuberculosis also modulates autophagy in a reactive oxygen species (ROS)-dependent manner (Shin et al. 2010).
Other microorganisms subvert or exploit the core autophagic machinery. For instance, Coxiella burnetii and Yersinia pseudotuberculosis are able to hijack the autophagic machinery. Presumably, increased autophagic activity provides membrane source for the expansion of intracellular vacuoles, which function as replication site for these bacteria (Moreau et al. 2010). With a slightly different strategy, Legionella pneumophila fine tunes the host autophagy response by secreting proteins which promote the transformation of bacteria-containing vacuoles into autophagic structures; at the same time, however, it also inhibit autophagosome formation and maturation into autolysosomes. It seems that autophagy is beneficial to the replication of Legionella, and this complex mechanism is used probably to avoid elimination and to gain more time for its replication (Choy et al. 2012; Khweek et al. 2013; Rolando et al. 2016). Similarly, Niu and colleagues reported that Anaplasma phagocytophilum induces autophagy independently of mTORC1 via the secretion of Ats-1, a BECN1-binding protein and uses the cytoplasmic components nutrients sequestered in the autophagosomes for its own growth (Niu et al. 2012).
Recent studies identified various viral factors which also target the autophagic machinery to modulate autophagic response. Proteins produced by HIV interact with BECN1 and ATG8 leading to impaired maturation of autophagosomes, thereby protecting viruses from degradation (Borel et al. 2015). Influenza A virus deploys a similar strategy to block autophagosome-lysosome fusion (Beale et al. 2014). Gamma herpesvirus also inhibits autophagy through the production of Bcl-2 like proteins (vBcl-2), which bind BECN1 via its BH3 domain (Su et al. 2014). Although BECN1 has a central role in autophagy regulation, it is not the only target of viral proteins. Kaposi’s sarcoma-associated herpesvirus encodes viral factors, which not only block VPS34 activity but also prevent the E3 enzyme-like ATG3 from interacting with ATG8, thus inhibiting autophagy (Lee et al. 2009; Liang et al. 2013).
Autophagy is able to activate adaptive immunity as an effector process of Toll-like receptor (TLR) signaling, playing an important role in the trafficking of pathogens to the lysosomes (for a detailed review see Deretic et al. 2013). TLR4 was recently shown to activate the ubiquitin-ligase TNF receptor-associated factor 6 (TRAF6), which subsequently induces autophagy via ubiquitination (and subsequent activation) of ULK1 and BECN1 (Shi and Kehrl 2010). In response of pathogen-associated molecular patterns (PAMPs), TLR7 was also shown to stimulate autophagy (Delgado et al. 2008).
Autophagy can also be activated—probably in BECN1-dependent manner—by proinflammatory cytokines (IL-1β and IFN-γ) in some effector cells, such as macrophages (Pilli et al. 2012). Thereafter, induced autophagy contributes not only to the elimination of microorganisms but also to the generation of antigenic peptides. These peptides appear on the cell surface in association with MHC class II molecules and are presented to CD4+ T cells, thereby promoting the development of a self-tolerant T cell repertoire (Aichinger et al. 2013). In line with these observations, Lee and colleagues found that ATG5-deficient dendritic cells (DCs) failed to activate herpes simplex virus-specific CD4+ T cells (Lee et al. 2010a). Another study showed that in ATG5−/− chimeric mice, both CD4+ and CD8+ T cell proliferation was impaired (Pua et al. 2007). Furthermore, autophagy seems to affect T cell polarization, as IL-1α and IL-1β released from autophagy-deficient macrophages enhanced polarization and duration of Th17 responses (Castillo et al. 2012). In a mouse model, both ATG5 and ATG7 were required for the maintenance of memory B cells against influenza (Chen et al. 2014). These findings together strongly suggest that autophagy is essential for the proper function of adaptive immunity.
Autophagy in neurodegenerative diseases
Numerous early reports described that autophagosomes accumulate in the brains of patients and model organisms with various neurodegenerative disorders suggesting a crucial role for autophagy in the pathogenesis of these diseases (Boland et al. 2008; Ravikumar et al. 2004). Other studies highlighted the importance of autophagy in neurodegenerative diseases by demonstrating that fruit flies or mice lacking ATG7 or ATG5 show symptoms of neurodegeneration in the central nervous system (Hara et al. 2006; Juhász et al. 2007).
Proteinopathies are late-onset neurodegenerative diseases featured by the accumulation of protein aggregates. Autophagy was found to be essential for the clearance of cytosolic aggregate-prone proteins underlying the symptoms of such diseases. These include mutant α-synuclein in Parkinson’s disease, polyglutamine-expanded Huntingtin in Huntington’s disease, wild-type and mutant forms of Tau in various dementias, such as Alzheimer’s disease or mutant TDP-43 in amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD).
Huntington’s disease (HD) is characterized by the perinuclear accumulation of polyQ-rich extension containing mutant Huntingtin (htt) protein (for detailed information see Labbadia and Morimoto 2013). A recent study of Heng and colleagues observed elevated levels of autophagy proteins in HD patients suggesting that autophagy—as a cytoprotective process—might struggle against the disease (Heng et al. 2010). A plausible explanation for autophagy induction in HD can be that mTORC1, the major inhibitor of this process, is also sequestered into htt aggregates (Ravikumar et al. 2004). Supporting the role of autophagy in htt clearance, Metzger and colleagues reported that polymorphism in ATG7 is associated with an early onset in HD (Metzger et al. 2013). However, more recent studies found that accumulated autophagosomes fail to selectively recognize and isolate abnormally accumulated htt, so they do not contribute significantly to protein degradation (Martinez-Vicente et al. 2010). In line with this, Pryor and colleagues found that htt enhances mTORC1 activity thereby contributing to the pathogenesis of HD (Pryor et al. 2014). Moreover, proteins required for autophagy such as BECN1 were found to be also trapped into htt aggregates (Shibata et al. 2006). Interestingly, direct participation of normal htt in autophagy regulation was described in cell culture and in mice and Drosophila models of HD as well. Both full-length htt (lacking polyQ region), and its interacting proteins (RAB5 and Rhes) were found to induce autophagy, thus defending cells against toxicity caused by mutant htt (Mealer et al. 2014; Ravikumar et al. 2008; Zheng et al. 2010).
Alzheimer’s disease (AD) involves the extracellular presence of amyloid-β (Aβ) plaques derived from the cleavage of amyloid precursor protein (APP) and also the intracellular accumulation of the microtubule-associated protein Tau, leading to the formation of neurofibrillary tangles (reviewed in Kumar et al. 2015). Autophagy seems to have a major role in the clearance and secretion of Aβ (Nilsson et al. 2013; Tian et al. 2011; Vingtdeux et al. 2011). Supporting these findings, both APP and presenilin 1 (a protein required for Aβ production and whose mutations were described in familial autosomal-dominant AD) were observed inside of accumulated autophagic structures in AD neurons (Yu et al. 2005). Furthermore, a recent study showed that autophagy induction via trehalose decreases Tau levels (Krüger et al. 2012). However, the involvement of autophagy in the pathogenesis of AD is more complex. Although previously it was thought that increased autophagy during AD is a cytoprotective response, more recent studies revealed evidence showing that the observed autophagosome accumulation is rather caused by impaired autophagic degradation. It is supported by the fact that presenilin-1 not only is essential for Aβ production but it also promotes the lysosomal trafficking of V0a1 subunit of v-ATPase, thus enabling the acidification of lysosomes (Lee et al. 2010b). Consequently, presenilin 1 mutations can be responsible for Aβ and autophagosome accumulation independently. Interestingly, restoration of lysosomal function in a mouse AD model by deletion of the lysosomal cysteine protease inhibitor cystatin B facilitated the clearance of Aβ and improved cognitive performance (Yang et al. 2011a). Other studies suggested that—in parallel with autophagic maturation—autophagosome formation may also be affected in AD neurons and non-neuronal cells, as a significant reduction was observed in the levels of BECN1 and protein in them (Lucin et al. 2013; Pickford et al. 2008; Rohn et al. 2011).
Parkinson’s disease (PD) is accompanied by the intracellular accumulation of mutant α-synuclein protein (for a review see Beitz 2014). The close relationship between PD and autophagy is underlined by the findings that loss of PTEN-induced putative kinase 1 (PINK1/PARK6) and Parkin (PARK2)—proteins playing crucial role in mitophagy (Geisler et al. 2010)—cause autosomal-recessive and sporadic juvenile-onset PD. Although the mitophagic role of PARK2 was questioned in an in vivo mouse neurodegeneration model (Sterky et al. 2011), Hsu and colleagues found that α-synuclein aggregates in sporadic PD promote mitochondrial dysfunction (Hsu et al. 2000). A more recent study indicated that overexpression of Parkin in a rat model for PD contributed to α-synuclein clearance suggesting that PARK2 may have further roles is the pathogenesis of disease (Lonskaya et al. 2013). Despite the fact that α-synuclein is an autophagy substrate, its elevated levels inhibited autophagosome formation both in fruit fly and in cell culture, due to mislocalization of ATG9 (Winslow et al. 2010). A recent study reported similar autophagy defects owing to mutation of the retromer member VPS35, which is associated with familiar forms of PD (Zavodszky et al. 2014). Furthermore, leucine-rich repeat kinase 2 (LRKK2)—whose mutations was revealed in autosomal-dominant PD patients—was recently described also as an autophagy regulator; however, evidence on the exact autophagic role of this protein is controversial (Bravo-San Pedro et al. 2013; Gómez-Suaga et al. 2012; Manzoni et al. 2013).
Proteins with well-known autophagic roles were found to be affected in other neurodegenerative disorders as well. For instance, mutations of the selective autophagy receptor SQSTM1 and optineurin were observed in ALS patients; moreover, optineurin was also detected in cytosolic aggregates in HD or other polyQ diseases (Hirano et al. 2013; Mori et al. 2012; Teyssou et al. 2013). Mutations of dynactin 1—which has an essential role in dynein-mediated autophagosome trafficking in neurons—were found to cause neurodegenerative diseases, as ALS and Perry syndrome (Farrer et al. 2009). Recent studies showed that loss of WDR45/WIPI4 protein, one of the mammalian homologs of ATG18 results in impaired autophagy in parallel with SENDA (static encephalopathy of childhood with neurodegeneration in adulthood), a subtype of neurodegeneration with brain iron accumulation (Saitsu et al. 2013).
Autophagy in cardiovascular disease
Autophagy contributes to the maintenance of normal cardiovascular function and thus functions as a protective mechanism under physiological conditions (reviewed in Lavandero et al. 2015). Impairment of this process was observed in the pathogenesis of various heart and vascular diseases. Several early studies revealed increased autophagic activity in human atherosclerotic plaques, which probably occurs due to the simultaneous presence of hypoxia, inflammation, increased ROS and oxidized lipoprotein level (Muller et al. 2011). Elevated autophagy, by removal of damaged organelles, might serve as a defending strategy during the pathogenesis of atherosclerosis. Supporting this hypothesis, recent study showed that upregulation of autophagy via resveratrol reduces inflammatory response of endothelial cells (ECs) (Chen et al. 2013). Furthermore, autophagy induction protected vascular smooth muscle cells (VSMCs) against cell death induced by 7-ketocholesterol, a major component of oxidized lipoproteins (He et al. 2013c). Autophagy in macrophages has a beneficial effect on the cardiovascular system as well: its inhibition in them promotes apoptosis and NADPH oxidase-mediated oxidative stress and increases plaque formation in an inflammasome-dependent manner (Liao et al. 2012). However, other reports demonstrated that excessive autophagy may also contribute to cell death in ECs and VSMCs; for instance, increased autophagic activity via long-term activation of XBP-1 eventually leads to apoptosis of ECs (Margariti et al. 2013). Moreover, even phagocytosis of cells dying by autophagy promoted pro-inflammatory response in macrophages (Petrovski et al. 2011).
Autophagy is also involved in the pathogenesis of cardiomyopathies. Several studies described impaired autophagy in lamin A/C gene mutation related dilated cardiomyopathy, probably caused by increased mTORC1 signaling (Choi et al. 2012). In humans, dilated cardiomyopathy was also found to be connected to the Mst1-mediated phosphorylation of BECN1 facilitating its interaction with Bcl-2, thereby leading to autophagy inhibition (Maejima et al. 2013). Upon desmin-related cardiomyopathy, autophagy may function as a protective mechanism through the removal of misfolded alphaB-crystallin protein; it is supported by a study demonstrating that loss of BECN1 promoted heart failure and early mortality in a mouse model system (Tannous et al. 2008). Furthermore, sustained expression of ATG7 was able to rescue not only impaired autophagy but also decreased cardiac hypertrophy (Pattison et al. 2011). Danon disease, a glycogen storage disease-related form of cardiomyopathy, is primarily caused by impaired autophagosome-lysosome fusion owing to LAMP2 mutations (for a recent review see Rowland et al. 2016). Similar to cardiovascular diseases, excessive autophagy may also contribute to cardiomyopathies. For instance, histone deacetylases (HDACs) were shown to decrease cardiac hypertrophy through suppression of elevated autophagic activity in mice (Cao et al. 2011). In addition, ceramide synthase-5 was recently described as a positive regulator of both lipid-induced autophagy and lipotoxic cardiomyopathy (Russo et al. 2012).
Autophagy plays a crucial role during ischemia/reperfusion. During ischemia, autophagy protects tissues via removal of damaged mitochondria and also provides molecules for the metabolic cellular processes upon low nutrient levels. In line with this, AMPK-activated autophagy functions as a major cytoprotective process and reduces apoptotic cell death in this condition (Troncoso et al. 2012). Furthermore, Huang and colleagues demonstrated that preconditioning by repeated short ischemic periods facilitates cardioprotection through autophagy induction (Huang et al. 2010b). Following reperfusion, transient elevation in autophagy level mediating a beneficial effect on cell survival was observed in various model systems (Huang et al. 2010a; Xie et al. 2014). Interestingly, it seems that upregulation of autophagy during reperfusion occurs through a different pathway, as it was independent of AMPK activation while BECN1 abundance had a central role in the process (Matsui et al. 2007). However, Valentim and colleagues showed that autophagy may also be detrimental during reperfusion (Valentim et al. 2006). Later reports indicated that ischemia/reperfusion induced a protective autophagic response, but cell death eventual occurs due to defective autophagosome clearance (Ma et al. 2012a; Ma et al. 2012b).
Autophagy plays a similarly controversial role in heart failure. In mice, autophagy induced by prolonged ATG7 expression was found to be beneficial by antagonizing ventricular hypertrophy through increased protein degradation (Bhuiyan et al. 2013). Supporting the protective role of autophagy, a significant decrease in the levels of core autophagic proteins was detected upon heart failure in human biopsy samples (Kassiotis et al. 2009). Moreover, high fat diet, a well-known risk factor underlying cardiac hypertrophy and dysfunction also resulted in impaired autophagosome maturation by disrupting autophagosome-lysosome fusion (Xu et al. 2013). miR-212 and miR-132, two regulator microRNAs which play a key role in hypertrophic growth of cardiomyocytes leading to heart failure in mice, caused impaired starvation-induced autophagy in parallel, suggesting that autophagic response might protect cells from pathological conditions (Ucar et al. 2012). By contrast, autophagy inhibition by 3-methyladenine or depletion of ATG genes preserved cardiomyocytes from injury caused by hyperglycemia-like conditions (Kobayashi et al. 2012). Furthermore, loss of myocardial mTORC1 activity in mice promoted both autophagy and apoptosis and led to heart failure (Shende et al. 2011). Thus, despite its beneficial effects, excessive autophagy may exacerbate the symptoms of cardiac hypertrophy and heart failure.
Autophagy in diabetes and obesity
Autophagy, as a surveillance mechanism, plays a crucial role in the maintenance of cellular homeostasis under normal conditions in pancreatic β-cells. Recent studies demonstrated that ATG7 deletion in mice lead to diabetes-like symptoms such as low insulin levels, hyperglycemia, and reduced β-cell mass (Quan et al. 2012). Consistently, induction of autophagy via mTOR inhibitors reduced ER stress in β-cells and protected cells against apoptosis (Bachar-Wikstrom et al. 2013). Diabetes is often associated with cardiomyopathy increasing the incidence of heart failure. In a mouse model of diabetic cardiomyopathy, Xie and colleagues recently found significantly reduced AMPK activity, which in turn resulted in decreased autophagy and cardiac dysfunction (Xie et al. 2011). In line with this, enhanced cardiac autophagy stimulated via the AMPK pathway protected cells from apoptosis in diabetic mice and also in high glucose-treated cardiac myoblasts (He et al. 2013a). Regarding kidney functions, high glucose level was found to disrupt autophagy in podocytes leading to diabetic glomerular dysfunction (Fang et al. 2013). Moreover, in the epithelial cells of proximal tubules in both mouse and human kidney, Yamahara and colleagues found that obesity may lead to impaired autophagy exposing these cells to more severe damage under diabetic conditions (Yamahara et al. 2013).
Obesity is caused by the chronic imbalance of energy homeostasis and nutrient storage. Autophagy plays an essential role in lipid metabolism via the selective degradation of lipid droplets (LDs)—a process called lipophagy (for a recent review see Wang 2016). Among others, it is supported by the fact that hepatocyte-specific deletion of core ATG genes facilitated triglyceride storage into LDs (Singh et al. 2009a). In line with this, in both genetic and dietary models of obesity, downregulation of autophagy resulted in ER stress and insulin resistance, which could be rescued by restoration of ATG7 levels (Yang et al. 2010). Moreover, supporting the protective role of autophagy against obesity, a recent study reported that acute exercise induced autophagy in skeletal and cardiac muscle, liver, pancreas, adipose tissue, and cerebral cortex and it was necessary for altered glucose metabolism and endurance during physical exertion (He et al. 2012). In addition, autophagy in agouti-related peptide (AgRP) neurons of hypothalamus reduced food intake upon starvation and promoted the production of α-melanocyte-stimulating hormone, which is usually associated with lean phenotype (Kaushik et al. 2011). However, other findings showed that autophagy does not possess a definite role either in obesity. Skeletal muscle-specific inhibition of this process by ATG7 deletion just protected mice from increased adipogenesis, insulin resistance, and obesity induced by diet (Kim et al. 2013b). Similarly, adipose-specific ATG7 knockout and depletion of ATG5 and ATG7 reduced lipid accumulation in white adipose tissue, increased brown adipose tissue mass, and increased insulin sensitivity in mice (Zhang et al. 2009).
Autophagy in cancer
Perhaps not surprisingly, autophagy has a dual role in cancer as well: in normal cells, it acts as housekeeping and protective process, which prevents from tumorigenesis; however, it may also contribute to the survival of already existing tumor cells, thus promoting tumor progression.
BECN1 was the first autophagy gene identified as a haploid-insufficient tumor suppressor present only in one copy in 40–75% of breast, ovarian, and prostate cancers (Aita et al. 1999). Further studies also supported that BECN1 may function as a tumor suppressor, since loss of BECN1 function facilitated tumorigenesis in mice (Qu et al. 2003). Consistently, AKT-mediated inhibitory phosphorylation of BECN1 resulted in autophagy inhibition and subsequent tumorigenesis (Wang et al. 2012). More recently, other ATG genes were found to be relevant in cancer development. Mutations in ATG2B, ATG5, ATG9B, and ATG12 genes were detected in human samples of patients with various types of gastrointestinal cancers (An et al. 2011; Kang et al. 2009). In accordance with it, mosaic ATG5 and liver-specific ATG7 deletion leads to the development of benign liver tumors in a mouse model system (Takamura et al. 2011). In addition, Sato and colleagues identified various point mutations of mTOR resulting in its constitutive kinase activity and, as a consequence, inhibition of autophagy in human cancers (Sato et al. 2010). Furthermore, several well-known tumor suppressor proteins, i.e., PTEN, TSC1/2, and LKB1 function as positive regulators of autophagy (reviewed in Cheng et al. 2013).
Together, these data suggest a tumor suppressor role for autophagy, although the exact mechanism through which it may inhibit tumorigenesis remains unclear. A study suggested that loss of autophagy results in tumorigenesis due to the impaired removal of the autophagy receptor SQSTM1, which in turn accumulates and leads to oxidative stress and tumor development (Mathew et al. 2009). Supporting this, Li and colleagues demonstrated that overexpression of SQSTM1 contributes to the pathogenesis of clear cell renal cell carcinoma (Li et al. 2013). Interestingly, SQSTM1 was also found to be required for RAS-induced tumorigenesis: SQSTM1 deficiency prevented the tumorigenic activity of RAS (Duran et al. 2008). However, based on the study of Rosenfeldt and colleagues, it seems that, in mouse pancreas, impaired autophagy only promotes pre-malignant tumor development and loss of other tumor suppressor factors, such as p53 is required for further tumor progression (Rosenfeldt et al. 2013).
On the other hand, increased autophagic activity can facilitate cancer development and progression by promoting the survival of existing tumor cells upon low oxygen levels or other stress conditions (Guo et al. 2011; Lock et al. 2011). For example, a recent work of Sabatini’s group demonstrated that autophagy inhibition increases the vulnerability of melanoma cells to leucine deprivation, suggesting that autophagy has an important role in tumor growth and survival during periods of nutrient starvation (Sheen et al. 2011). In a mouse model of breast cancer, loss of RB1CC1/FIP200 leads to a suppression in both initiation and progression of mammary tumors, suggesting that autophagy is necessary for tumor development (Wei et al. 2011). Interestingly, its cancer promoting effect can be resulted from a positive impact on metastasis and drug resistance as well. A recent study showed that, by facilitating the secretion of promigratory factors, autophagy might contribute to the invasion of epithelial cells transformed with oncogenic RAS (Lock et al. 2014). Furthermore, pancreatic tumor cells are highly dependent on autophagy, as it increases their drug resistance (Yang et al. 2011b). Consistently, other studies showed that high levels of autophagy in biopsy samples from metastatic melanoma patients can be used as a prognostic factor for invasiveness, chemotherapy resistance, and the duration of survival (Ma et al. 2011). According to a more recent study of Ma and colleagues, therapy-induced autophagy promoted tumor cell resistance and decreased the patients’ response to BRAFi treatment (Ma et al. 2014). Interestingly, in mouse models of various types of lung tumors, while autophagy prevents cells from malignant transformation, it also promotes cell growth, probably due to the degradation of damaged mitochondria and thereby reducing ROS levels (Guo et al. 2013; Rao et al. 2014; Strohecker et al. 2013). Thus, in tumor cells, autophagy contributes to growth and survival by multiple mechanisms: it supplies the cells with sufficient amounts of nutrients, inhibits apoptotic cell death by removal of damaged mitochondria and by reduction of oxidative stress, and promotes drug resistance during chemotherapy.
Concluding remarks
Based on the numerous studies of the last 50 years, it is apparent that autophagy is an essential stress-response mechanism and it is required for the maintenance of cellular homeostasis under physiological conditions. Mutations in ATG genes and impaired function of core autophagic proteins were associated with different types of infections, neurodegenerative, cardiovascular and metabolic diseases, and cancer (Table 1), suggesting that autophagy plays a crucial role during the pathogenesis of severe human diseases. Although investigation of human samples or invertebrate and vertebrate disease models provides a deeper insight into the pathological roles of autophagy, observations are often controversial. One underlying factor may be that in spite of several studies, many questions remained open concerning the molecular mechanisms involved in autophagy and the exact pathomechanisms of these disorders (Lindqvist et al. 2015).
Although our incomplete understanding on autophagy and its contribution to diseases limits therapeutic targeting, recent studies, seeking for pharmacological approaches able to enhance or inhibit autophagy, has yielded promising results in animal models for human diseases (for a detailed review see Levine et al. 2015). For instance, Ravikumar and colleagues demonstrated that activation of autophagy via mTORC1 inhibitors (such as rapamycin and its analog CCI-779) prevented neurodegeneration in Drosophila and mouse models of Huntington’s disease (Ravikumar et al. 2004). Other rapamycin analogs (e.g., RAD001 and AP23573) also induced autophagy and in parallel, successfully attenuated medical symptoms in animal models of atherosclerosis or in patients with different tumor types including sarcoma (Martinet et al. 2007; Mita et al. 2008). Interestingly, some other inhibitors of mTORC1 kinase activity (e.g. Torin 1 and PP242) may be even more effective than rapamycin (Feldman et al. 2009; Thoreen et al. 2009). Trehalose, an mTORC1-independent autophagy activator, had a beneficial effect on the clearance of mutant α-synuclein and also protected cells against apoptosis in a HD model (Sarkar et al. 2007). Autophagy-inducing drugs were also successfully deployed against hepatic fibrosis (Hidvegi et al. 2010) and viral infections (Shoji-Kawata et al. 2013).
In other cases, inhibition of autophagy seems to be needed: it induced apoptotic death of tumor cells in a mouse model of lymphoma (Amaravadi et al. 2007) and in various types of tumors (McAfee et al. 2012). Currently, further autophagy regulator agents targeting core autophagic proteins, such as ATG4B, ATG7, or VPS34, are under different phases of testing (reviewed in Jiang and Mizushima 2014).
However, pharmacological targeting of autophagy is still at its beginnings; and, unfortunately, it is likely that most approved autophagy-modulating drugs also affect other pathways—and hence processes side effects. Furthermore, different studies often reported contradictory role for autophagy in human diseases suggesting that it has remained unclear whether this process should be enhanced or inhibited in order to mitigate symptoms. Thus, further investigations are necessary, which would reveal the yet unknown molecular mechanisms of autophagy, especially under pathological conditions. These future advances might pave the way for the development of more specific autophagy inhibitors and enhancers, which could eventually help us to harness autophagy in favor of efficient therapeutic interventions against various human diseases.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- ATG:
-
Autophagy related
- CM:
-
Cardiomyopathy
- GAP:
-
GTPase activating protein
- GAS:
-
Group A Streptococcus
- GDP:
-
Guanosine diphosphate
- GEF:
-
Guanosine nucleotide exchange factor
- GTP:
-
Guanosine triphosphate
- HD:
-
Huntington’s disease
- HF:
-
Heart failure
- HIV:
-
Human immunodeficiency virus
- HOPS:
-
Homotypic fusion and vacuole protein sorting
- KSHV:
-
Kaposi sarcoma-associated herpesvirus
- (m)TOR:
-
(Mechanistic/mammalian) target of rapamycin
- (MAP1)LC3:
-
(Microtubule-associated protein 1) light chain 3
- (m)TORC1:
-
(Mechanistic/mammalian) TOR complex 1
- PAS:
-
Pre-autophagosomal structure or phagophore assembly site
- PD:
-
Parkinson’s disease
- PI3P:
-
Phosphatidylinositol 3-phosphate
- SENDA:
-
Static encephalopathy of childhood with neurodegeneration in adulthood
- SNARE:
-
Soluble NSF Attachment Protein (SNAP) receptor
- ULK:
-
unc51-like kinase
- UVRAG:
-
Ultra violet radiation resistance associated gene
- VPS:
-
Vacuolar protein sorting
References
Aichinger M, Wu C, Nedjic J, Klein L. Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J Exp Med. 2013;210(2):287–300. doi:10.1084/jem.20122149.
Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59(1):59–65. doi:10.1006/geno.1999.5851.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117(2):326–36. doi:10.1172/JCI28833.
An CH, Kim MS, Yoo NJ, Park SW, Lee SH. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol Res Pract. 2011;207(7):433–7. doi:10.1016/j.prp.2011.05.002.
Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276(38):35243–6. doi:10.1074/jbc.C100319200.
Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. 2011;440(2):283–91. doi:10.1042/BJ20101894.
Bachar-Wikstrom E, Wikstrom JD, Ariav Y, Tirosh B, Kaiser N, Cerasi E, et al. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes. 2013;62(4):1227–37. doi:10.2337/db12-1474.
Bains M, Zaegel V, Mize-Berge J, Heidenreich KA. IGF-I stimulates Rab7-RILP interaction during neuronal autophagy. Neurosci Lett. 2011;488(2):112–7. doi:10.1016/j.neulet.2010.09.018.
Balderhaar HJK, Ungermann C. CORVET and HOPS tethering complexes—coordinators of endosome and lysosome fusion. J Cell Sci. 2013;126(6):1307–16. doi:10.1242/jcs.107805.
Beale R, Wise H, Stuart A, Ravenhill BJ, Digard P, Randow F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe. 2014;15(2):239–47. doi:10.1016/j.chom.2014.01.006.
Beitz JM. Parkinson’s disease: a review. Front Biosci (Schol Ed). 2014;6:65–74. doi:10.2741/S415.
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–81. doi:10.1128/MCB.00166-09.
Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123(12):5284–97. doi:10.1172/JCI70877.
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28(27):6926–37. doi:10.1523/JNEUROSCI.0800-08.2008.
Borel S, Robert-Hebmann V, Alfaisal J, Jain A, Faure M, Espert L, et al. HIV-1 viral infectivity factor interacts with microtubule-associated protein light chain 3 and inhibits autophagy. AIDS. 2015;29(3):275–86. doi:10.1097/QAD.0000000000000554.
Bravo-San Pedro JM, Niso-Santano M, Gómez-Sánchez R, Pizarro-Estrella E, Aiastui-Pujana A, Gorostidi A, et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci. 2013;70(1):121–36. doi:10.1007/s00018-012-1061-y.
Bury JJ, Highley JR, Cooper-Knock J, Goodall EF, Higginbottom A, McDermott CJ, et al. Oligogenic inheritance of optineurin (OPTN) and C9ORF72 mutations in ALS highlights localisation of OPTN in the TDP-43-negative inclusions of C9ORF72-ALS. Neuropathology. 2016;36(2):125–34. doi:10.1111/neup.12240.
Campbell GR, Bruckman RS, Chu Y-L, Spector SA. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J Biol Chem. 2015;290(8):5028–40. doi:10.1074/jbc.M114.605428.
Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108(10):4123–8. doi:10.1073/pnas.1015081108.
Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109(46):E3168–76. doi:10.1073/pnas.1210500109.
Cervantes S, Bunnik EM, Saraf A, Conner CM, Escalante A, Sardiu ME, et al. The multifunctional autophagy pathway in the human malaria parasite, Plasmodium falciparum. Autophagy. 2014;10(1):80–92. doi:10.4161/auto.26743.
Chang Y, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. 2009;20(7):2004–14. doi:10.1091/mbc.E08-12-1250.
Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. 2014;20(5):503–10. doi:10.1038/nm.3521.
Chen M-L, Yi L, Jin X, Liang X-Y, Zhou Y, Zhang T, et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013;9(12):2033–45. doi:10.4161/auto.26336.
Cheng Y, Ren X, Hait WN, Yang J-M. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev. 2013;65(4):1162–97. doi:10.1124/pr.112.007120.
Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity. 2014;40(6):924–35. doi:10.1016/j.immuni.2014.05.006.
Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. 2012;4(144):144ra102. doi:10.1126/scitranslmed.3003875.
Choy A, Dancourt J, Mugo B, O’Connor TJ, Isberg RR, Melia TJ, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012;338(6110):1072–6. doi:10.1126/science.1227026.
Darsow T, Rieder SE, Emr SD. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J Cell Biol. 1997;138(3):517–29. doi:10.1083/jcb.138.3.517.
Dash S, Chava S, Aydin Y, Chandra PK, Ferraris P, Chen W, et al. Hepatitis C virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic ER-stress response. Viruses. 2016;8(5) doi:10.3390/v8050150.
Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27(7):1110–21. doi:10.1038/emboj.2008.31.
Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2012; doi:10.1242/jcs.114819.
Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–37. doi:10.1038/nri3532.
Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–54. doi:10.1016/j.ccr.2008.02.001.
Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012;19(5):788–97. doi:10.1038/cdd.2011.149.
Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS One. 2013;8(4):e60546. doi:10.1371/journal.pone.0060546.
Farrer MJ, Hulihan MM, Kachergus JM, Dächsel JC, Stoessl AJ, Grantier LL, et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41(2):163–5. doi:10.1038/ng.293.
Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38. doi:10.1371/journal.pbio.1000038.
Funakoshi T, Matsuura A, Noda T, Ohsumi Y. Analyses of APG13 gene involved in autophagy in yeast, Saccharomyces cerevisiae. Gene. 1997;192(2):207–13.
Furuta N, Fujita N, Noda T, Yoshimori T, Amano A. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell. 2010;21(6):1001–10. doi:10.1091/mbc.E09-08-0693.
Furuya N, Yu J, Byfield MP, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1(1):46–52.
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–31. doi:10.1038/ncb2012.
Gómez-Suaga P, Luzón-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S, et al. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet. 2012;21(3):511–25. doi:10.1093/hmg/ddr481.
Guo JY, Chen H-Y, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–70. doi:10.1101/gad.2016311.
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447–61. doi:10.1101/gad.219642.113.
Gutierrez MG, Munafó DB, Berón W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117(Pt 13):2687–97. doi:10.1242/jcs.01114.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26. doi:10.1016/j.molcel.2008.03.003.
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell Elsevier Ltd. 2010;141(4):656–67. doi:10.1016/j.cell.2010.04.009.
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER–mitochondria contact sites. Nature. 2013;495(7441):389–93. doi:10.1038/nature11910.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–9. doi:10.1038/nature04724.
Hara T, Takamura A, Kishi C, Iemura S-I, Natsume T, Guan J-L, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510. doi:10.1083/jcb.200712064.
Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol Nat Publ Group. 2009;11(12):1433–7. doi:10.1038/ncb1991.
He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–5. doi:10.1038/nature10758.
He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013a;62(4):1270–81. doi:10.2337/db12-0533.
He C, Zhu H, Zhang W, Okon I, Wang Q, Li H, et al. 7-Ketocholesterol induces autophagy in vascular smooth muscle cells through Nox4 and Atg4B. Am J Pathol. 2013b;183(2):626–37. doi:10.1016/j.ajpath.2013.04.028.
Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–97. doi:10.1016/j.immuni.2012.09.014.
Heng MY, Duong DK, Albin RL, Tallaksen-Greene SJ, Hunter JM, Lesort MJ, et al. Early autophagic response in a novel knock-in model of Huntington disease. Hum Mol Genet. 2010;19(19):3702–20. doi:10.1093/hmg/ddq285.
Heo J-M, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60(1):7–20. doi:10.1016/j.molcel.2015.08.016.
Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329(5988):229–32. doi:10.1126/science.1190354.
Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C, et al. Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology. 2013;80(5):458–63. doi:10.1212/WNL.0b013e31827f0fe5.
Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5(7):973–9. doi:10.4161/auto.5.7.9296.
Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, et al. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000;157(2):401–10.
Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, et al. Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am J Physiol Heart Circ Physiol. 2010a;298(2):H570–9. doi:10.1152/ajpheart.00716.2009.
Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, et al. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res. 2010b;3(4):365–73. doi:10.1007/s12265-010-9189-3.
Huynh KK, Eskelinen E-L, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26(2):313–24. doi:10.1038/sj.emboj.7601511.
Hyttinen JMT, Niittykoski M, Salminen A, Kaarniranta K. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta. 2013;1833(3):503–10. doi:10.1016/j.bbamcr.2012.11.018.
Isakson P, Holland P, Simonsen A. The role of ALFY in selective autophagy. Cell Death Differ Nat Publ Group. 2013;20(1):12–20. doi:10.1038/cdd.2012.66.
Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell Elsevier Inc. 2012;151(6):1256–69. doi:10.1016/j.cell.2012.11.001.
Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6):764–76.
Jewell JL, Russell RC, Guan K-L. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol Nat Publ Group. 2013;14(3):133–9. doi:10.1038/nrm3522.
Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24(1):69–79. doi:10.1038/cr.2013.161.
Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014;25(8):1327–37. doi:10.1091/mbc.E13-08-0447.
Juhász G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21(23):3061–6. doi:10.1101/gad.1600707.
Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8(4):318–24. doi:10.1016/j.cmet.2008.08.013.
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009;217(5):702–6. doi:10.1002/path.2509.
Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, et al. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120(11 Suppl):S191–7. doi:10.1161/CIRCULATIONAHA.108.842252.
Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, et al. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011;14(2):173–83. doi:10.1016/j.cmet.2011.06.008.
Khweek AA, Caution K, Akhter A, Abdulrahman BA, Tazi M, Hassan H, et al. A bacterial protein promotes the recognition of the Legionella pneumophila vacuole by autophagy. Eur J Immunol. 2013;43(5):1333–44. doi:10.1002/eji.201242835.
Kim JJH, Kim YC, Fang C, Russell RC, Kim JJH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell Elsevier Inc. 2013a;152(1–2):290–303. doi:10.1016/j.cell.2012.12.016.
Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim Y-N, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013b;19(1):83–92. doi:10.1038/nm.3014.
Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33(1):109–22.
Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell Elsevier Inc. 2009;34(3):259–69. doi:10.1016/j.molcel.2009.04.026.
Klionsky D, Agholme L, Agnello M, Agostinis P, Aguirre-ghiso JA, Ahn HJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222. doi:10.1080/15548627.2015.1100356.
Kobayashi S, Xu X, Chen K, Liang Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy. 2012;8(4):577–92. doi:10.4161/auto.18980.
Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126(Pt 2):580–92. doi:10.1242/jcs.114926.
Krüger U, Wang Y, Kumar S, Mandelkow E-M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012;33(10):2291–305. doi:10.1016/j.neurobiolaging.2011.11.009.
Kumar A, Singh A, Ekavali. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol. Rep. 2015;67(2):195–203. doi:10.1016/j.pharep.2014.09.004.
Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186(2):255–68. doi:10.1083/jcb.200903070.
Labbadia J, Morimoto RI. Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci. 2013;38(8):378–85. doi:10.1016/j.tibs.2013.05.003.
Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest. 2015;125(1):55–64. doi:10.1172/JCI73943.
Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010a;32(2):227–39. doi:10.1016/j.immuni.2009.12.006.
Lee J-H, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010b;141(7):1146–58. doi:10.1016/j.cell.2010.05.008.
Lee J-SJ-Y, Li Q, Lee J-SJ-Y, Lee S-H, Jeong JH, Lee H-R, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol Nat Publ Group. 2009;11(11):1355–62. doi:10.1038/ncb1980.
Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125(1):14–24. doi:10.1172/JCI73938.
Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell. 2013;24(6):738–50. doi:10.1016/j.ccr.2013.10.025.
Li Y, Wang Y, Kim E, Beemiller P, Wang C-Y, Swanson J, et al. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem. 2007;282(49):35803–13. doi:10.1074/jbc.M705231200.
Liang Q, Chang B, Brulois KF, Castro K, Min C-K, Rodgers MA, et al. Kaposi’s sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J Virol. 2013;87(22):12499–503. doi:10.1128/JVI.01898-13.
Liang XH, Jackson S, Seaman MNJ, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi:10.1038/scibx.2008.460.
Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15(4):545–53. doi:10.1016/j.cmet.2012.01.022.
Lindmo K, Brech A, Finley KD, Gaumer S, Contamine D, Rusten TE, et al. The PI 3-kinase regulator Vps15 is required for autophagic clearance of protein aggregates. Autophagy. 2008;4(4):500–6.
Lindqvist LM, Simon AK, Baehrecke EH. Current questions and possible controversies in autophagy. Cell death Discov. 2015;1:15036. doi:10.1038/cddiscovery.2015.36.
Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4(4):466–79. doi:10.1158/2159-8290.CD-13-0841.
Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22(2):165–78. doi:10.1091/mbc.E10-06-0500.
Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15(8):702–13. doi:10.1016/j.cub.2005.02.053.
Longatti A, Lamb CA, Razi M, Yoshimura S, Barr FA, Tooze SA. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol. 2012;197(5):659–75. doi:10.1083/jcb.201111079.
Lonskaya I, Hebron ML, Algarzae NK, Desforges N, Moussa CE-H. Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson’s disease. Neuroscience. 2013;232:90–105. doi:10.1016/j.neuroscience.2012.12.018.
Lőrincz P, Lakatos Z, Maruzs T, Szatmári Z, Kis V, Sass M. Atg6/UVRAG/Vps34-containing lipid kinase complex is required for receptor downregulation through endolysosomal degradation and epithelial polarity during Drosophila wing development. Biomed Res Int. 2014;2014:851349. doi:10.1155/2014/851349.
Lucin KM, O’Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79(5):873–86. doi:10.1016/j.neuron.2013.06.046.
Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Diwan A. Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy. 2012a;8(9):1394–6. doi:10.4161/auto.21036.
Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012b;125(25):3170–81. doi:10.1161/CIRCULATIONAHA.111.041814.
Ma X-H, Piao S, Wang D, McAfee QW, Nathanson KL, Lum JJ, et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res. 2011;17(10):3478–89. doi:10.1158/1078-0432.CCR-10-2372.
Ma X-H, Piao S-F, Dey S, McAfee Q, Karakousis G, Villanueva J, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124(3):1406–17. doi:10.1172/JCI70454.
Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19(11):1478–88. doi:10.1038/nm.3322.
Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy. 2007;3(4):374–6.
Manzoni C, Mamais A, Dihanich S, Abeti R, Soutar MPM, Plun-Favreau H, et al. Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta. 2013;1833(12):2900–10. doi:10.1016/j.bbamcr.2013.07.020.
Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem. 2013;288(2):859–72. doi:10.1074/jbc.M112.412783.
Martinet W, Verheye S, De Meyer GRY. Everolimus-induced mTOR inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy. 2007;3(3):241–4.
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13(5):567–76. doi:10.1038/nn.2528.
Maruzs T, Lőrincz P, Szatmári Z, Széplaki S, Sándor Z, Lakatos Z, et al. Retromer ensures the degradation of autophagic cargo by maintaining lysosome function in Drosophila. Traffic. 2015;16(10):1088–107. doi:10.1111/tra.12309.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen H-Y, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–75. doi:10.1016/j.cell.2009.03.048.
Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–22. doi:10.1161/01.RES.0000261924.76669.36.
Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190(4):511–21. doi:10.1083/jcb.200911141.
Maurer K, Reyes-Robles T, Alonzo F, Durbin J, Torres VJ, Cadwell K. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe. 2015;17(4):429–40. doi:10.1016/j.chom.2015.03.001.
Mauvezin C, Nagy P, Juhász G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6:7007. doi:10.1038/ncomms8007.
McAfee Q, Zhang Z, Samanta A, Levi SM, Ma X-H, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109(21):8253–8. doi:10.1073/pnas.1118193109.
Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J Biol Chem. 2014;289(6):3547–54. doi:10.1074/jbc.M113.536912.
Meijer AJ, Lorin S, Blommaart EF, Codogno P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids. 2015;47(10):2037–63. doi:10.1007/s00726-014-1765-4.
Metzger S, Walter C, Riess O, Roos RAC, Nielsen JE, Craufurd D, et al. The V471A polymorphism in autophagy-related gene ATG7 modifies age at onset specifically in Italian Huntington disease patients. PLoS One. 2013;8(7):e68951. doi:10.1371/journal.pone.0068951.
Mita M, Sankhala K, Abdel-Karim I, Mita A, Giles F. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin Investig Drugs. 2008;17(12):1947–54. doi:10.1517/13543780802556485.
Moreau K, Lacas-Gervais S, Fujita N, Sebbane F, Yoshimori T, Simonet M, et al. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell Microbiol. 2010;12(8):1108–23. doi:10.1111/j.1462-5822.2010.01456.x.
Mori F, Tanji K, Toyoshima Y, Yoshida M, Kakita A, Takahashi H, et al. Optineurin immunoreactivity in neuronal nuclear inclusions of polyglutamine diseases (Huntington’s, DRPLA, SCA2, SCA3) and intranuclear inclusion body disease. Acta Neuropathol. 2012;123(5):747–9. doi:10.1007/s00401-012-0956-x.
Muller C, Salvayre R, Nègre-Salvayre A, Vindis C. Oxidized LDLs trigger endoplasmic reticulum stress and autophagy: prevention by HDLs. Autophagy. 2011;7(5):541–3.
Nagy P, Hegedűs K, Pircs K, Varga Á, Juhász G, Varga A, et al. Different effects of Atg2 and Atg18 mutations on Atg8a and Atg9 trafficking during starvation in Drosophila. FEBS Lett. 2013;588(3):408–13. doi:10.1016/j.febslet.2013.12.012.
Nagy P, Kárpáti M, Varga A, Pircs K, Venkei Z, Takáts S, et al. Atg17/FIP200 localizes to perilysosomal Ref(2)P aggregates and promotes autophagy by activation of Atg1 in Drosophila. Autophagy. 2014;10(3):453–67. doi:10.4161/auto.27442.
Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013;55:39–50. doi:10.1042/bse0550039.
Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5(1):61–9. doi:10.1016/j.celrep.2013.08.042.
Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000;406(6798):906–10. doi:10.1038/35022604.
Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc Natl Acad Sci U S A. 2012;109(51):20800–7. doi:10.1073/pnas.1218674109.
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. doi:10.1038/embor.2009.256.
Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307(5710):727–31. doi:10.1126/science.1106036.
Orvedahl A, MacPherson S, Sumpter R, Tallóczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7(2):115–27. doi:10.1016/j.chom.2010.01.007.
Padmaja MV, Jayaraman M, Srinivasan AV, Srisailapathy CRS, Ramesh A. PARK2 gene mutations in early onset Parkinson’s disease patients of South India. Neurosci Lett. 2012;523(2):145–7. doi:10.1016/j.neulet.2012.06.062.
Pankiv S, Alemu EA, Brech A, Bruun J-A, Lamark T, Overvatn A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188(2):253–69. doi:10.1083/jcb.200907015.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45. doi:10.1074/jbc.M702824200.
Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109(2):151–60. doi:10.1161/CIRCRESAHA.110.237339.
Petrovski G, Ayna G, Majai G, Hodrea J, Benko S, Mádi A, et al. Phagocytosis of cells dying through autophagy induces inflammasome activation and IL-1β release in human macrophages. Autophagy. 2011;7(3):321–30.
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118(6):2190–9. doi:10.1172/JCI33585.
Pilli M, Arko-Mensah J, Ponpuak M, Roberts EA, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37(2):223–34. doi:10.1016/j.immuni.2012.04.015.
Pryor WM, Biagioli M, Shahani N, Swarnkar S, Huang W-C, Page DT, et al. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci Signal. 2014;7(349):ra103. doi:10.1126/scisignal.2005633.
Pua HH, Dzhagalov I, Chuck M, Mizushima N, He Y-W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204(1):25–31. doi:10.1084/jem.20061303.
Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell Elsevier. 2013;154(6):1285–99. doi:10.1016/j.cell.2013.08.044.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20. doi:10.1172/JCI20039.
Quan W, Lim YM, Lee MS. Role of autophagy in diabetes and endoplasmic reticulum stress of pancreatic β-cells. Exp Mol Med. 2012;44(2):81–8. doi:10.3858/emm.2012.44.2.030.
Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi:10.1038/ncomms4056.
Ravikumar B, Imarisio S, Sarkar S, O’Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci. 2008;121(Pt 10):1649–60. doi:10.1242/jcs.025726.
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12(8):747–57. doi:10.1038/ncb2078.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–95. doi:10.1038/ng1362.
Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol Dis. 2011;43(1):68–78. doi:10.1016/j.nbd.2010.11.003.
Rolando M, Escoll P, Nora T, Botti J, Boitez V, Bedia C, et al. Legionella pneumophila S1P-lyase targets host sphingolipid metabolism and restrains autophagy. Proc Natl Acad Sci U S A. 2016;113(7):1901–6. doi:10.1073/pnas.1522067113.
Romanov J, Walczak M, Ibiricu I, Schüchner S, Ogris E, Kraft C, et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012;31(22):4304–17. doi:10.1038/emboj.2012.278.
Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. doi:10.1038/nature12865.
Rowland TJ, Sweet ME, Mestroni L, Taylor MRG. Danon disease—dysregulation of autophagy in a multisystem disorder with cardiomyopathy. J Cell Sci. 2016;129(11):2135–43. doi:10.1242/jcs.184770.
Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, et al. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest. 2012;122(11):3919–30. doi:10.1172/JCI63888.
Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45(4):445–9. doi:10.1038/ng.2562.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell Elsevier Ltd. 2010;141(2):290–303. doi:10.1016/j.cell.2010.02.024.
Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282(8):5641–52. doi:10.1074/jbc.M609532200.
Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010;29(18):2746–52. doi:10.1038/onc.2010.28.
Sato TK, Darsow T, Emr SD. Vam7p, a SNAP-25-like molecule, and Vam3p, a syntaxin homolog, function together in yeast vacuolar protein trafficking. Mol Cell Biol. 1998;18(9):5308–19.
Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J Nat Publ Group. 2009;28(9):1341–50. doi:10.1038/emboj.2009.80.
Sekito T, Kawamata T, Ichikawa R, Suzuki K, Ohsumi Y. Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells. 2009;14(5):525–38. doi:10.1111/j.1365-2443.2009.01299.x.
Sheen J-H, Zoncu R, Kim D, Sabatini DM. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell. 2011;19(5):613–28. doi:10.1016/j.ccr.2011.03.012.
Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123(10):1073–82. doi:10.1161/CIRCULATIONAHA.110.977066.
Shi C-S, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3(123):ra42. doi:10.1126/scisignal.2000751.
Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, et al. Regulation of intracellular accumulation of mutant huntingtin by Beclin 1. J Biol Chem. 2006;281(20):14474–85. doi:10.1074/jbc.M600364200.
Shin D-M, Jeon B-Y, Lee H-M, Jin HS, Yuk J-M, Song C-H, et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010;6(12):e1001230. doi:10.1371/journal.ppat.1001230.
Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–6. doi:10.1038/nature11866.
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. Nat Publ Group. 2009a;458(7242):1131–5. doi:10.1038/nature07976.
Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, et al. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009b;119(11):3329–39. doi:10.1172/JCI39228.
Sinha S, Colbert CL, Becker N, Wei Y, Levine B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy. 2008;4(8):989–97.
Sterky FH, Lee S, Wibom R, Olson L, Larsson N-G. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A. 2011;108(31):12937–42. doi:10.1073/pnas.1103295108.
Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3(11):1272–85. doi:10.1158/2159-8290.CD-13-0397.
Su M, Mei Y, Sanishvili R, Levine B, Colbert CL, Sinha S. Targeting γ-herpesvirus 68 Bcl-2-mediated down-regulation of autophagy. J Biol Chem. 2014;289(12):8029–40. doi:10.1074/jbc.M113.515361.
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi:10.1101/gad.2016211.
Takáts S, Nagy P, Varga Á, Pircs K, Kárpáti M, Varga K, et al. Autophagosomal syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol. 2013;201(4):531–9. doi:10.1083/jcb.201211160.
Takáts S, Pircs K, Nagy P, Varga Á, Kárpáti M, Hegedűs K, et al. Interaction of the HOPS complex with syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell. 2014;25(8):1338–54. doi:10.1091/mbc.E13-08-0449.
Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406(6798):902–6. doi:10.1038/35022595.
Tang H-W, Wang Y-B, Wang S-L, Wu M-H, Lin S-Y, Chen G-C. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J Nat Publ Group. 2011;30(4):636–51. doi:10.1038/emboj.2010.338.
Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105(28):9745–50. doi:10.1073/pnas.0706802105.
Tatti M, Motta M, Di Bartolomeo S, Scarpa S, Cianfanelli V, Cecconi F, et al. Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Hum Mol Genet. 2012;21(23):5159–73. doi:10.1093/hmg/dds367.
Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LAM, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. 2012;11(6):563–75. doi:10.1016/j.chom.2012.04.012.
Teyssou E, Takeda T, Lebon V, Boillée S, Doukouré B, Bataillon G, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125(4):511–22. doi:10.1007/s00401-013-1090-0.
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–32. doi:10.1074/jbc.M900301200.
Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011;25(6):1934–42. doi:10.1096/fj.10-175158.
Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2012;93(2):320–9. doi:10.1093/cvr/cvr321.
Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–74.
Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078. doi:10.1038/ncomms2090.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–60. doi:10.1126/science.1096284.
Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, et al. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006;40(6):846–52. doi:10.1016/j.yjmcc.2006.03.428.
Vázquez CL, Colombo MI. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 2010;17(3):421–38. doi:10.1038/cdd.2009.129.
Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009;16(7):1006–17. doi:10.1038/cdd.2009.34.
Vingtdeux V, Chandakkar P, Zhao H, D’Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-β peptide degradation. FASEB J. 2011;25(1):219–31. doi:10.1096/fj.10-167361.
von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein Á, Bloor S, Rutherford TJ, et al. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell. 2012;48(3):329–42. doi:10.1016/j.molcel.2012.08.024.
Wang C-W. Lipid droplets, lipophagy, and beyond. Biochim. Biophys Acta. 2016;1861(8 Pt B):793–805. doi:10.1016/j.bbalip.2015.12.010.
Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338(6109):956–9. doi:10.1126/science.1225967.
Wang T, Ming Z, Xiaochun W, Hong W. Rab7: role of its protein interaction cascades in endo-lysosomal traffic. Cell Signal Elsevier Inc. 2011;23(3):516–21. doi:10.1016/j.cellsig.2010.09.012.
Wartosch L, Günesdogan U, Graham SC, Luzio JP. Recruitment of VPS33A to HOPS by VPS16 is required for lysosome fusion with endosomes and autophagosomes. Traffic. 2015;16(7):727–42. doi:10.1111/tra.12283.
Wei H, Wei S, Gan B, Peng X, Zou W, Guan J-L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25(14):1510–27. doi:10.1101/gad.2051011.
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–88. doi:10.1016/j.molcel.2008.06.001.
Winslow AR, Chen C, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. α-synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190(6):1023–37. doi:10.1083/jcb.201003122.
Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129(10):1139–51. doi:10.1161/CIRCULATIONAHA.113.002416.
Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60(6):1770–8. doi:10.2337/db10-0351.
Xu X, Hua Y, Nair S, Sreejayan N, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol. 2013;5(1):61–3. doi:10.1093/jmcb/mjs055.
Yamahara K, Kume S, Koya D, Tanaka Y, Morita Y, Chin-Kanasaki M, et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol. 2013;24(11):1769–81. doi:10.1681/ASN.2012111080.
Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198(2):219–33. doi:10.1083/jcb.201202061.
Yan J, Kuroyanagi H, Kuroiwa A, Matsuda Y, Tokumitsu H, Tomoda T, et al. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. Biochem Biophys Res Commun. 1998;246(1):222–7.
Yang D-S, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain. 2011a;134(Pt 1):258–77. doi:10.1093/brain/awq341.
Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–78. doi:10.1016/j.cmet.2010.04.005.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011b;25(7):717–29. doi:10.1101/gad.2016111.
Yang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17(12):5094–104. doi:10.1091/mbc.E06-06-0479.
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–6. doi:10.1038/nature09076.
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee J-H, et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171(1):87–98. doi:10.1083/jcb.200505082.
Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–82. doi:10.1073/pnas.2436255100.
Yuk JM, Yoshimori T, Jo EK. Autophagy and bacterial infectious diseases. Exp Mol Med. 2012;44(2):99–108. doi:10.3858/emm.2012.44.2.032.
Zaffagnini G, Martens S. Mechanisms of selective autophagy. J Mol Biol. 2016;428(9 Pt A):1714–24. doi:10.1016/j.jmb.2016.02.004.
Zavodszky E, Seaman MNJ, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, et al. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun. 2014;5:3828. doi:10.1038/ncomms4828.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–903. doi:10.1074/jbc.M800102200.
Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar B, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003;5(6):578–81. doi:10.1038/ncb999.
Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A. 2009;106(47):19860–5. doi:10.1073/pnas.0906048106.
Zheng S, Clabough EBD, Sarkar S, Futter M, Rubinsztein DC, Zeitlin SO. Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet. 2010;6(2):e1000838. doi:10.1371/journal.pgen.1000838.
Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–76. doi:10.1038/ncb1854.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lippai, M., Szatmári, Z. Autophagy—from molecular mechanisms to clinical relevance. Cell Biol Toxicol 33, 145–168 (2017). https://doi.org/10.1007/s10565-016-9374-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-016-9374-5