
Abstract
Autophagy is a highly regulated process in eukaryotes to maintain homeostasis and manage stress responses. Understanding the regulatory mechanisms and key players involved in autophagy will provide critical insights into disease-related pathogenesis and potential clinical treatments. In this review, we describe the hallmark events involved in autophagy, from its initiation, to the final destruction of engulfed targets. Furthermore, based on structural and biochemical data, we evaluate the roles of key players in these processes and provide rationale as to how they control autophagic events in a highly ordered manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy is a highly regulated self-clearance process in eukaryotes that delivers long-lived proteins, lipids, glycogens, and organelles from the cytosol to lysosomes for destruction [1,2,3]. It also functions as an adaptive cellular mechanism to deal with stress stimuli such as starvation, oxidation, and pathogen invasion [4, 5]. Malfunctions in autophagy are associated with a wide array of human diseases including cancer and neurodegeneration [6, 7]. Thus, studies of autophagy will provide the basis for the development of potential therapeutic strategies that target these diseases.
Autophagy is divided into roughly three types, according to the pathways that deliver cargo to the lysosomes: macroautophagy, microautophagy, and chaperone-mediated autophagy [4]. Macroautophagy, which is the focus of this review, and referred to as simply autophagy hereafter, is characterized by the formation of a double-membrane vesicular structure named autophagosome that sequesters bulk cytosol and fuses with lysosomes (or vacuole in yeasts and plants [8, 9].
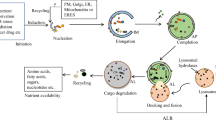
As a hallmark event in autophagy, autophagosome formation is dynamically regulated by a group of structurally and functionally conserved proteins, which are encoded by autophagy-related genes (ATG) in yeast (Table 1) [10]. In response to autophagic signals, Atg proteins of the core autophagic machinery are mostly clustered in the vicinity of the vacuole/lysosome/and assembled into a structure named phagophore assembly site (or preautophagosomal structure, PAS) that also contains the forming vesicle (phagophore or isolation membrane) [4]. The phagophore undergoes an expansion process and eventually develops into a mature autophagosome in which a sealed double membrane enwraps the cargo. Then the outer membrane of the autophagosome quickly fuses with the lysosome membrane, exposing its inner membrane and enwrapped contents to degradation by lysosomal hydrolases. At last, the degradation products are transported back to the cytosol and used for protein synthesis and other cellular functions [4, 8, 9, 11]. Figure 1 illustrates a simplified autophagic process that is directed by green arrowheads.

Schematic depiction of the autophagic process. The autophagic process is directed by green arrowheads. The whole process can be roughly divided into six steps: (1) initiation of autophagy; (2) biogenesis of phagophore; (3) expansion of phagophore; (4) formation of autophagosome; (5) fusion with lysosome, and (6) reformation of lysosome. The red arrows indicate the potential membrane sources for autophagosomes, including cytoplasm, ER (endoplasmic reticulum), mitochondria, and Golgi. The blue arrow indicates that degraded materials are transported back to the cytosol for reuse via lysosomal transporters. PAS phagophore assembly site
In this review, we dissect the autophagic process into several steps for ease of discussion, which includes (1) initiation of autophagy; (2) biogenesis of the phagophore; (3) expansion of the phagophore; (4) formation of the autophagosome; (5) fusion with the lysosome, and (6) reformation of the lysosome (also refer to Fig. 1). As we describe these steps in detail, we also summarize the biochemical and structural properties of key Atg proteins that participate in each respective step (Table 1). We use the yeast nomenclature Atg1–31 to represent autophagic proteins and the names of their mammalian homologues are shown in parentheses in Table 1. Not only we focus on the yeast Atg proteins for structural and functional analysis, but discuss important autophagic functions or events relating to their mammalian homologues. Of note, most yeast Atg proteins have obvious mammalian homologues (based on amino acid sequence), with the exception of a few specific proteins. For example, Atg17, Atg31, and Atg29 are found in yeast, but have no mammalian homologues, suggesting in these cases, homology may be based on function rather than sequence. In addition, the mammalian protein Atg101 does not have an apparent yeast counterpart [12, 13], thus demonstrating that yeast and mammalian cells may use different regulatory mechanisms in certain autophagic scenarios [5].
Initiation of autophagy: the Atg1 complex
Autophagy is induced in response to several stress signals, such as amino acid deprivation, DNA damage, low energy, and hypoxia. These signals trigger the activation of distinct pathways that primarily converge on the nutrient sensor, Tor kinase complex 1 (TORC1) (Fig. 2a). Lack of amino acids inhibits the translocation of TORC1 to lysosomal membranes, where TORC1 is typically activated by RAS-homologue enriched in brain (RHEB). This translocation process is mediated by RAG GTPases and their interacting protein complex, Ragulator [14]. Low oxygen levels induce the hypoxia-inducible gene (REDD1), which in turn promotes the activity of the TSC1–TSC2 complex, a GTPase-activating protein for RHEB [15, 16]. As a result, GDP-loaded RHEB fails to activate TORC1. Low energy levels are sensed by a high cellular AMP-to-ATP ratio and this signal promotes phosphorylation and activation of AMPK by LKB1. Activated AMPK can regulate TORC1 activity positively or negatively through several signaling pathways. In one scenario, AMPK can inhibit TORC1 activation by phosphorylation of regulator-associated protein of TOR (RAPTOR) [17]. Alternatively, AMPK can also be activated by the sestrin family members (SESN1 or SESN2), which are induced by the p53 tumor suppressor protein in response to DNA damage [18].

Structural analysis of the Atg1 complex. a Signaling pathways that transmit stress signals to regulate TORC1 and autophagy. b The Atg1/Atg13 complex structure. The domain organization schemes of Atg1 and Atg13 are shown above. Below left is the kinase domain structure of Atg1 in complex with its inhibitor (yellow) (4WNP), where the activation loop is highlighted in red. Below middle is the domain complex structure of Atg1 MIT (green)/Atg13 MIM (cyan) (4P1N). Below right is the HORMA domain complex structure of Atg13 (cyan)/Atg101 (grey) (4YK8), where Atg13 is folded in the closed state, compared to Atg101’s open state conformation. c The Atg13–Atg17–Atg31–Atg29 complex structure. Atg17 (yellow) forms a dimer with a curvature radius close to 10 nm and appears as a letter “S”. Atg29 (blue) and Atg31 (magenta) are located at the concave face of Atg17. Atg13 interacts with the N-terminus and C-terminus via 17BR (red) and 17LR (orange), respectively. The ability of Atg13 to bind to different Atg17 molecules simultaneously allows the supramolecular molecular assembly. 5JHF is used as a reference here but 4HPQ and 4P1W should also be noted
Initiation of autophagy in yeast is characterized by assembly of the Atg1–Atg13–Atg17–Atg31–Atg29 complex (Atg1 complex) and its localization to the PAS. In contrast, a similar Atg1 complex (ULK1/2–Atg13–Atg101–FIP200, refer to abbreviations in the legend of Table 1) is constitutively formed in mammalian cells and translocated to the PAS in response to autophagic signals [5, 19,20,21]. Assembly of the Atg1 complex is determined by the phosphorylation status of Atg13, which serves as an adaptor protein bridging Atg1 and the Atg17 subcomplex (Atg17–Atg31–Atg29) [19, 22]. Under nutrient-rich conditions, TORC1 heavily phosphorylates the C-terminal domain of Atg13 [21, 23, 24], which in turn disrupts the interaction between Atg13 and Atg1 or Atg17, and the Atg1 complex fails to be formed [25]. In contrast, when TORC1 activity is inhibited (Fig. 2a), Atg13 is quickly dephosphorylated. Consequently, the interaction between Atg13 and Atg1 or Atg17 is increased, facilitating the formation of the Atg1 complex [21, 24]. Figure 2b indicates the interaction between Atg1 and Atg13, which is mediated by Atg1 microtubule interacting and transport (MIT) domains of Atg1 and MIT-interacting motif (MIM) domains of Atg13 [19]. When two MIT domains form two antiparallel three-helix bundles (like collars), two MIM domains fold into an extended helix–loop–helix, reminiscent of a short tie wrapping collars.
Atg13 contains a Hop1p, Rev1p, and Mad2 domain (HORMA) at its N-terminus (Fig. 2b), which corresponds to the “closed” state of Mad2 (the spindle checkpoint protein) and plays an essential role in recruiting the Vps34 complex to the PAS during autophagy via the interaction with Atg14 [26]. Additionally, it was reported that an LC3 interacting region (LIR) is present in human Atg13 and required for proper autophagosome formation [27]. As such, Atg13 is regarded as a key regulator and organizer during the early stage of autophagy.
Atg1 (ULK1/2) is a serine/threonine protein kinase whose activity is regulated by Atg13 binding and by TORC1 phosphorylation [21, 24]. The kinase activity of Atg1 is required for autophagy, whereas it is not required for localization of the Atg1 complex to the PAS [25, 28, 29]. Indeed, C-terminal early autophagy targeting/tethering (EAT) domain of Atg1 plays a rather important role in localization, due to its membrane functions including membrane targeting [29], membrane curvature sensing, and lipid vesicle tethering [30]. The structure of Atg1’s kinase domain was solved most recently, which reveals a canonical bilobal kinase fold (Fig. 2b) [31]. The mechanistic roles of Atg1’s kinase activity in autophagy have been intensely studied in recent years. A number of mammalian substrates have been identified including activating molecule in Beclin 1-regulated autophagy (AMBRA1) and Beclin 1 (Atg6 homolog) [32, 33]. Atg1 phosphorylates AMBRA1 and releases the core machinery from dynein [32]. In addition, Atg1 phosphorylates Beclin 1 and activates Vps34 lipid kinase activity [33]. These findings link Atg1 catalytic activity to earlier regulatory events in mammalian cells, including translocation of the Atg1 complex to the endoplasmic reticulum (ER) membrane and subsequent omegasome formation.
Atg17–Atg31–Atg29 is constitutively formed and serves as a platform for PAS assembly [30]. Crystallographic analysis of this subcomplex shows that Atg17 forms a dimer via its C-terminal helix (Fig. 2c) [30, 34]. Each Atg17 monomer appears in a crescent shape with a curvature radius close to 10 nm and two Atg17 molecules are arranged like a letter “S”. Dimerization of Atg17 is required for PAS assembly. Intriguingly, the curvature radius of Atg17 is similar to that of Atg9 containing vesicles, which earliest cluster at the PAS. It should be noted that Atg29 and Atg31 sterically hinder lipid vesicle binding at the concave face of the Atg17 crescent in the crystal structure (Fig. 2c). Furthermore, the ability of the Atg1 EAT domain to tether vesicles is lost in the presence of Atg13–Atg17–Atg31–Atg29 [30]. Considering the structural hindrance and the dynamic properties of Atg1, other regulatory factors are likely involved in PAS construction. Most importantly, Atg13 interacts with both the N-terminus and C-terminus of Atg17 via its 17BR and 17LR domains, thus promoting the supramolecular assembly of the Atg1 complex (Fig. 2c) [34].
Biogenesis of phagophore: the Vps34 complex
Phagophore biogenesis requires the lipid kinase activity of the Vps34 complex, in which Vps34 is the core enzyme [35,36,37]. As the only phosphatidylinositol 3-kinase (PI3K) in yeast (and the only class III PI3K in mammalian cells), Vps34 phosphorylates the membrane lipid phosphatidylinositol (PI) to produce phosphatidylinositol 3-phosphate (PI3P). Blockage of this reaction with PI3K inhibitors, such as wortmannin and LY294002, inhibits PI3P production and autophagy [37]. The structures of Vps34 alone or in complex with inhibitors provided an important model for its enzymatic activation and inhibition [38]. Interestingly, Vps34 has an inherent auto-repressive mechanism (Fig. 3a). When in solution, the last C-terminal helix of Vps34 (shown in magenta) covers and blocks the ATP-binding site so as to avoid futile ATP hydrolysis. When Vps34 is recruited to the membrane during autophagy, there is a conformational switch of this helix (shown in cyan), which binds to the membrane and sways from the top of the ATP-binding site. As a result, the membranous substrate gains access to the catalytic center. Noteworthily, the ATP-binding site of Vps34 is unique in the PI3K class members, because it is so constrained that only small adenine analogues (inhibitors) can fit [38].

Structural analysis of the Vps34 complex. a The helical and kinase domain structure of Vps34. The domain organization scheme of Vps34 is shown above. Below is the structure of Vps34 helical and catalytic domains (2X6H). The different colors of C-terminal helix indicate a potential conformational switch from its cytosolic state (magenta) to its membrane-associated state (cyan). The activation loop is highlighted in blue and the catalytic loop in black, when the inhibitor is shown in red. b The complex structure of Vps30–Vps38–Vps15–Vps34-nb. The domain organization schemes of individual subunits are shown above. Below is the complex structure (5DFZ) that appears as a letter “Y”. One arm of “Y” is formed by intertwined Vps15 (orange) and Vps34 (green), where Vps15 contacts the activation loop of Vps34 to regulate its activity. The other arm is formed by intertwined Vps30 (yellow) and Vps38 (blue), which embraces the WD40 domain of Vps15 and the C2 domain of Vps34. nb (nanobody) is used as a crystallization chaperone
The activity of Vps34 is regulated by interactions with its partners including Vps15 (regulatory subunit) and Atg6 (Beclin 1, Vps30) [39,40,41,42]. Vps15 is a protein kinase whose enzymatic activity is required for activation of Vps34 [41, 42]. Additionally, its N-terminally myristoylation plays a role in Vps34 membrane targeting [41, 43]. In general, Atg6 functions as a protein platform to recruit additional factors into the Vps34 complex, such as Atg14 and Vps38 (UVRAG), to form the autophagy-specific complex (complex I) and the endosome sorting complex (complex II), respectively [40, 44]. This notion is supported by a most recent structure of the Vps38–Vps30–Vps15–Vps34 complex (Fig. 3b) [45]. This exciting work reveals a unique path for the complex assembly and molecular basis of regulation of Vps34 activity by Vps15 [45]. It also sheds light on the differential activity of complex II and complex I on their specific phosphorylation targets.
While the Atg14–Atg6–Vps15–Vps34 complex structure has yet to be determined, like Vps38, Atg14 uses its N-terminal coiled-coil to assemble with Vps34–Vps15–Atg6 and its C-terminal Barkor/Atg14(L) autophagosome targeting sequence (BATS) domain for membrane functions such as binding to curved membranes enriched in PI3P [46]. Atg14 also serves as a regulatory nexus of autophagy by interacting directly or indirectly with autophagy activators such as Ambra1, Bif-1 (Bax interacting factor, also known as endophilin B1), and vacuole membrane protein 1 (VMP1) [47,48,49] or autophagy inhibitors such as RUN domain and cysteine-rich domain containing, Beclin 1-interacting (Rubicon) [5, 50]. A long-standing, unanswered question in the field of autophagy is where and how phagophore biogenesis occurs. The formation of omegasomes near the ER is one model to explain phagophore biogenesis in mammalian cells, which is supported by experimental evidence [51, 52]. Omegasomes are “Ω” like and PI3P-rich membrane subdomains that dynamically connect to the ER membrane and serve as a platform for phagophore biogenesis and expansion [53, 54].
At the very beginning, autophagic signals activate the Atg1 complex and stimulate its localization to a subdomain of the ER membrane. Immediately, ER exit sites (ERESs) play an essential role in the hierarchical assembly of the autophagy machinery [55]. ERESs have been shown to be core autophagosomal biogenesis components, because autophagosomes are spatially, physically, and functionally linked to them [55]. ERESs are specialized regions of ER membranes that generate COPII transport vesicles. Interestingly, COPII-coated vesicles are also required for autophagy [56], suggesting ERESs might contribute one membrane source to both phagophore biogenesis and expansion via COPII transport vesicles. Consistently, COPII transport vesicles deliver the ER membranes containing the soluble N-ethylamaleimide-sensitive factor attachment protein receptors (SNARE) protein Ufe1 to sites of autophagosome formation [57].
In subsequent steps, the Atg1 complex recruits the Vps34 complex, likely via the interaction between Atg13 and Atg14 [26]. Vps34 is activated to produce PI3P. As a result, the local membrane concentration of PI3P is elevated, which in turn increases the membrane curvature. Considering that Atg14 has a high binding affinity towards curved membranes, a feed-forward loop is thus formed, leading to the facilitated recruitment of the Vps34 complex to the targeted region. Additionally, mammalian studies show that activation of Vps34 also induces the recruitment of COPII to the ER–Golgi intermediate compartment (ERGIC) to generate small vesicle active in Atg8/LC3 lipidation, which suggests ERGIC as another membrane source for autophagosome formation [58]. Finally, omegasomes are formed, from which the phagophore is generated as an extended membrane.
Besides increasing the membrane curvature, PI3P also functions as membrane anchors to recruit PI3P-dependent membrane-associated proteins that might assist or direct omegasome formation. These proteins include double FYVE-containing protein 1 (DFCP1) and Atg18 (WIPI1/2/3/4). DFCP1 binds to PI3P via its FYVE motif and clusters on the PAS in response to autophagic signals [54]. Of note, FYVE/PX/PH are the typical PI3P-binding motifs [59]. DFCP1 serves as a marker of omegasomes, but it is not required for autophagy [54]. Atg18 binds to PI3P via the FRRG motif. Noteworthily, Atg18’s lipid binding activity is not required for the formation of the Atg18–Atg2 complex, whereas it is required for the recruitment of this complex to the PAS [60]. The precise location of DFCP1 and Atg18 is different in the PAS [61, 62]. DFCP1 is mainly localized to the omegasome, while Atg18 is concentrated on the phagophore protruding from omegasomes, suggesting that Atg18 might be mainly responsible for phagophore expansion. This notion is supported by the role of Atg18 role in regulating two key events in phagophore expansion: Atg9 shuttling and Atg8 (LC3) lipidation [63, 64]. Reportedly, the Atg18–Atg2 complex, together with the Atg1 complex, regulates the retrograde transport of Atg9 from PAS to peripheral organelles [65, 66]. Additionally, Atg18, together with its family member Atg21, facilitates the localization of Atg8 and Atg16 to the PAS and protects Atg8–PE from cleavage by Atg4 before the phagophore expansion is completed [67].
Transmembrane proteins VMP1 and Atg9 also play a critical role during omegasome formation. VMP1 is localized to the PAS during autophagy and promotes Vps34 complex activity via its interaction with Atg6 [49, 62, 68]. Atg9 contributes to omegasome formation and phagophore biogenesis by transporting lipids and/or critical autophagic factors from peripheral organelles (e.g., the trans-Golgi network and endosomes) to the PAS [64].
The phagophore membrane is connected to the omegasome/ER membrane, which suggests that the ER membrane (e.g., ERES) is indeed the membrane source autophagosome. However, we cannot exclude the possibility that the omegasome only functions as a membrane anchor, while the phagophore forms most of its membrane through Atg9-mediated vesicle fusion in the expanding step [69,70,71,72]. Several membrane origins have been indicated for the autophagosome (Fig. 1). In the mitochondria model, the phagophore is derived from the outer membrane of mitochondria and developed with the lipid transfer from mitochondria [73]. Interestingly, this model requires the connections between ER and mitochondria and is unique to starvation-induced autophagy. In the cytoplasm membrane model, the phagophore comes from clathrin-mediated budding of the cytoplasmic membrane, which is dependent on the interaction between Atg16 (Atg16L1/2) and the heavy chain of clathrin [74]. With all the models, it is very likely that the autophagosome has multiple mechanisms to obtain membranes, all of which are dynamically regulated in response to different stress signals.
Expansion of phagophore: Atg9 and Atg8 lipidation
Atg9 shuttling and Atg8 lipidation are two key events in the phagophore expansion [63, 64]. Atg9 is the only transmembrane protein in the core Atg machinery and strictly required for autophagosome formation [75]. It is recruited to the PAS during autophagy in an Atg17-dependent manner, which is based on the Atg9–Atg17 interaction and requires the presence of Atg1 [76]. Atg9 shuttles between the PAS and peripheral organelles, and mediates material exchange between them [1, 4, 64, 66, 77]. In doing so, Atg9 containing vesicles are budded from peripheral organelles, anterograde transport lipids and/or critical autophagic factors to PAS, and then fused with the existing phagophore. During this process, Atg9 self-interaction plays a critical role [78, 79]. On the other hand, Atg9-containing vesicles are retrograde transported to peripheral organelles to recycle Atg9 and/or retrieve PAS-resident factors [65]. In this way, Atg9 shuttling regulates phagophore expansion. Importantly, SNARE proteins control Atg9-mediated vesicle fusion [80].
Atg8 lipidation is dependent on two ubiquitin-like conjugating systems: Atg12 and Atg8 conjugating systems. (1) The Atg12 conjugating system comprises Atg12 (ubiquitin-like module), Atg5 (substrate), Atg7 (E1 enzyme), and Atg10 (E2 enzyme) (Table 1). This system does not require an E3 enzyme and Atg12–Atg5 conjugate is constantly formed. In comparison, Atg12–Atg5 further complexes with Atg16 to form an E3 enzyme for the Atg8 conjugating system. (2) The Atg8 conjugating system consists of Atg8 (ubiquitin-like module), Atg4 (a cysteine protease to process Atg8), phosphatidylethanolamine (PE, substrate), Atg7 (E1 enzyme), Atg3 (E2 enzyme), and Atg5–Atg12–Atg16 (E3 enzyme). This system has been well characterized by structural biology approaches. First, Atg4 processes Atg8 into an active form by cleaving its last residue (R) and exposing the second last residue (G). It shows the structure of Atg4 (yellow) in complex with Atg8 (blue) in Fig. 4a. In the structure, the C-terminal tail of Atg8 is inserted into the catalytic core of Atg4 containing the catalytic residue C74 (marked in red), and induces a considerable conformational change of W142 and the regulatory loop (G257-A263) at the entrance of Atg4’s active site, compared to Atg4’s apo structure [81]. This structure also reveals the structural basis for Atg8–PE deconjugation by Atg4, which functions to recycle Atg8 or sensitize the autophagosome for its fusion with lysosomes [82]. Next, the structure in Fig. 4b shows how Atg8 (blue) is conjugated to Atg7 (pink). In the structure, the C-terminal tail of Atg8 is inserted into the catalytic core of Atg7’s C-terminal domain (CTD) and positioned next to Atg7’s catalytic residue, C507 (marked in red) [83].

Structural analysis of the Atg8 conjugating system. a The complex structure of Atg8–Atg4. The tail of Atg8 (blue) is inserted into the catalytic center of Atg4 (yellow) (2ZOD). C74 (red) stands for the catalytic residue of Atg4. W142 (red) and the regulatory loop (G257-A263, magenta) undergo a large conformational change, compared to Atg4’s apo structure (grey, 2CY7). b The complex structure of Atg8–Atg7. The tail of Atg8 (blue) is inserted into the catalytic center of Atg7 (pink) (3RUI). C507 (red) stands for the catalytic residue of Atg7, which is located in the proximity of the last amino acid glycine of Atg8. c The complex structure of Atg7–Atg3. Two Atg7 molecules (Atg7′ and Atg7″ labeled here) form a dimer via their CTDs and coordinate in Atg3 binding and catalysis (4GSL). Atg7’ NTD (purple) (Atg7″ NTD is not shown) pushes the backside of Atg3 (orange) to force its front side (containing the catalytic residue C234 in black) facing the catalytic center of Atg7″ CTD (pink) (containing the catalytic residue C507 in red). The juxtaposition of active sites of Atg7″ and Atg3 facilitates the transfer of Atg8 (modeled here) between two enzymes, as indicated by the arrow. d The complex structure of Atg3–Atg12–Atg5–Atg16. The Atg12–Atg5–Atg16 complex is present as a tetramer due to Atg16 dimerization (3A7O), and anchored to the target membrane via the association of Atg16 and Atg5 with the membrane, as indicated by the arrows. From the interaction between Atg3 (orange) and Atg12 (green) (4NAW), it is speculated that Atg8-conjugated Atg3 is recruited to the target membrane by Atg12. Then, Atg8 is transferred from Atg3 to the membrane substrate PE, as indicated by the arrow. e The factors involved in the autophagosomal fusion. Stx17, SNAP29, and VAMP8 form a SNARE complex that mediates the membrane tethering and fusion (4WY4). The regulatory factors are listed, which play either a positive or negative role in this process
While the structures of Atg3–Atg8 and Atg7–Atg3–Atg8 are yet known, the structure of Atg7–Atg3 sheds lights on how Atg8 might be transferred from E1 to E2 (Fig. 4c). Two Atg7s form a dimer via their own CTDs and coordinate in Atg3 binding and catalysis. With this spatial arrangement, active sites of Atg7 (C507 in red) and Atg3 (C234 in black) are juxtaposed so as to facilitate the transfer of Atg8 (blue) between two enzymes (the curved arrowhead line). Also the structure of Atg8 in complex with Atg3–Atg12–Atg5–Atg16 has not been determined. Nevertheless, the structures of Atg3–Atg12–Atg5–Atg16 and Atg16–Atg16 provide helpful hints as to how Atg8 is finally conjugated to PE (Fig. 4d). The Atg12–Atg5–Atg16 complex is present as a tetramer due to Atg16 dimerization [84], and anchored to the target membrane via the association of Atg16 and Atg5 with the membrane [85]. Atg8-conjugated Atg3 is recruited to the target membrane by Atg12. Eventually, Atg8 is transferred from Atg3 to the membrane substrate PE, ending one round of Atg8 lipidation.
Maturation of autophagosome: Atg8 and Atg4
The expanding phagophore develops as a cup-shaped structure and surrounds the nearby cargo to sequester them from the cytosol. How cargoes are recruited to the expanding phagophores is a mystery. Recently, a number of cargo-specific receptors have been identified including Atg19, p62, neighbor of BRCA1 gene 1(NBR1) in degrading ubiquitinated protein aggregates, or Atg32, Nix (also called Bnip3L) in mitophagy, indicating that cargo recruitment is a selective process [86]. Interestingly, these receptors contain the LXXW motif/LIR that is specifically recognized by Atg8 [87], indicating Atg8 is involved in the selective cargo recruitment besides its role in phagophore expansion. It is intriguing to speculate that this Atg8-mediated interaction between expanding phagophores and encapsulating cargo is key to stabilization of the expanding membrane curvature [88]. In this regard, Atg8 serves as a sensor of membrane curvature during autophagosome formation. Consistent with this notion, the amount of Atg8 controls the size of autophagosomes [89]. Eventually, the expanding phagophore becomes a mature double-membrane vesicle with both ends sealed. While the exact underlying regulatory mechanisms of this process remain unknown, it is wellknown that Atg8–PE deconjugation by Atg4 is critical to the sealing step [90].
Fusion with lysosome: the SNARE complex
Mature autophagosomes dissociate from the PAS and fuse with lysosomes to form autolysosomes, during which process, the outer membrane of the autophagosome quickly fuses with the lysosome membrane (see Fig. 1) [4, 8, 9, 11]. The fusion step is controlled by a number of factors that play a positive or negative role.
A SNARE complex is key to the fusion step [91]. Syntaxin 17 (Stx17) specifically recognizes and localizes to the outer membrane of mature autophagosomes but not of expanding phagophores, where it forms a complex with SNAP29 and the lysosomal SNARE protein VAMP8 to facilitate membrane tethering and fusion (Fig. 4e). Atg14 has been shown to bind to Stx17 and SNAP29, promoting Stx17-mediated fusion events [92]. Interestingly, Atg14 is recruited by Stx17 to the ER–mitochondria contact site to participate in autophagosome formation [93]. O-GlcNAc-modification of SNAP29 negatively regulates fusion, which is mediated by the glycan transferase OGT [94]. Furthermore, the Vici syndrome protein EPG5 interacts with molecules on both lysosomes (Rab7, VAMP7/8) and autophagosomes (LC3, assembled Stx17–SNAP29 SNARE complex). As a result, it promotes the assembly of Stx17–SNAP29–VAMP7/8 complex and facilitates fusion [95].
Stx17 also associates with the homotypic fusion and protein sorting (HOPS) complex proteins, which participate in the regulation of the autophagosomal fusion. Additionally, the HOPS complex interacts with pleckstrin homology domain containing protein family member 1 (PLEKHM1), which functions as an adaptor protein that connects endocytic and autophagy pathways to lysosomes [96]. PLEKHM1 facilitates the fusion step through its binding to the HOPS complex and LC3 on the autophagosome membranes. Interestingly, the cholesterol-sensing Rab7 effector, ORP1L regulates the formation of ER–autophagosome contact sites, which inhibits the localization of PLEKHM1 to Rab7 and consequently suppresses the autophagosome–lysosome fusion [97]. Moreover, tectonin beta-propeller repeat-containing protein 1 (TECPR1), also plays a role in the fusion step [98].
Lastly, emerging evidence indicates that certain lysosomal lipids regulate autophagosome–lysosome fusion. Phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) antagonizes the fusion step by counteracting actin filament stabilization on the lysosomal surface. The enzyme inositol polyphosphate-5-phosphatase E (INPP5E), which catalyzes PI(3,5)P2, was shown to be involved in autophagosome–lysosome fusion process. Mutations in INPP5E are responsible for autophagy-related diseases such as Joubert syndrome [99].
After fusion, the inner membrane and inside cargo are attacked by a series of lysosome-resident acidic hydrolases, such as cathepsins, lipases, and glycosidases that break down the cargo into basic building blocks of proteins, lipids, and sugars, respectively [100, 101]. Disruption of hydrolases always causes autophagy defect and lysosome dysfunction. Atg15, a lysosomal lipase which is delivered to lysosome from ER through MVB pathway, is required for autophagic bodies breakdown, the first step of autophagic protein turnover in lysosome/vacuole [102, 103]. Since the role of Atg15 in membrane lytic events, depletion of Atg15 inhibits both lipophagy and pexophagy [104, 105].
Subsequently, the resultant monomeric units from autophagic degradation are transported back to the cytosol through lysosomal membrane transporters (permeases), and participate in the cellular maintenance of energy, metabolic, and organelle homeostasis under autophagic conditions [106]. Atg22, Avt3 and Avt4 serve as partially redundant vacuolar effluxes, which mediate the efflux of leucine and other amino acids resulting from autophagy [107,108,109]. As far, no fatty acid or sterol effluxer protein has been identified, though some evidence derived from electron microscopic investigation indicates Atg22 might be a candidate in this process [105].
Reformation of lysosome–clathrin
During autophagy, lysosomes are exhausted to form autolysosomes, which are hybrid vesicles with mixed membrane compositions and containing components. Lysosomes are regenerated through a cellular process named autophagic lysosomal reformation (ALR), which is characterized by the formation of tubular structures extended from the autolysosomes [110]. Clathrin plays a key role in this process by regulating budding off of the autolysosome membranes to form reformation tubules, and also by regulating pitching off of the reformation tubular membranes to generate proto-lysosomes (lysosomal vesicles containing no hydrolases) [111, 112]. Proto-lysosomes inherit the ancestral lysosomal membranes and membrane transporters via a sorting mechanism [113], and further mature into functional lysosomes by taking in newly synthesized hydrolases through the Golgi–endosome pathway [101] (Fig. 1). Noteworthily, ALR is strictly coupled to autophagy, because ALR is eliminated when activity of TORC1, lysosomal proteases, or permeases is inhibited [110, 114].
As a conserved degradation pathway, autophagy has been reported to relate to several critical human diseases, such as neuronal degeneration diseases and cancer [6, 7]. It is a highly dynamic and complex process coupled with many steps. Although tremendous progress was made for understanding of autophagy in the past 24 years, the mechanisms of autophagy remain far from clear.
So far, the structures of many Atg proteins (Atg8 homologues) and their complexes (ATG5–ATG12–ATG16, Class III PI3K and Atg17–Atg29–Atg31, et al.) have been solved which facilitates drug development. For example, the structure of ECD domain in Beclin 1 shed light on the autophagy induction mechanism of tat-Beclin 1 peptides. Most recently, the co-crystal structures of Vps34 with two high-specificity inhibitors, PIK-III and SAR405, provide better understanding of two novel action mechanisms for PI3K kinase inhibition. Since conjugation system and autophagic ClassIII PI3K complex were established, autophagosome–lysosome fusogenic system is also reconstituted in vitro recently. These in vitro biochemistry systems provide promising strategies which can be exploited to develop more autophagy-specific inhibitors or inducers. Most structural biology studies mainly focus on the early stage of autophagy. With the development of autophagy studies, the structural information of important autophagy complex is urgently needed, especially in the fusion and lysosome reformation steps.
In addition, research on several parts in autophagy is still missing, such as omegasome formation, the composition of autophagosome, the content between inner membrane and outer membrane. How is the curvature of the isolation membrane determined? How is isolation membrane stitched? How is autophagic SNARE recruited to complete autophagosome but not to isolation membrane?
Selective autophagy including mitophagy, xenophagy, lipophagy, pexophagy stands out due to their critical physiological and pathological function in vivo. A subset of special genes has been identified such as different receptors for different types of selective autophagy. However, whether there are non-overlapping genes with non-selective autophagy machinery remain largely unknown. And it is also not clear about the mechanical difference between non-selective autophagy and selective autophagy. Recently, noncanonical autophagy was discovered, which bypasses a subset of autophagy essential genes or complexes. Of note, it is still elusive that these processes are real autophagy or just autophagy genes-involved processes.
In the future, both structural biology and biochemistry studies on potential drug targets in the different autophagic pathways will provide more critical information which probably further facilitates drug design and ultimately benefit patients.
References
Mizushima N (2011) Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol 76:397–402. doi:10.1101/sqb.2011.76.011023
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20(3):460–473. doi:10.1089/ars.2013.5371
Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12(9):814–822. doi:10.1038/ncb0910-814
Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9(10):1102–1109
Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 11(4):468–476. doi:10.1038/ncb1854
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132(1):27–42. doi:10.1016/j.cell.2007.12.018
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306(5698):990–995
Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132. doi:10.1146/annurev-cellbio-092910-154005
Hurley JH, Schulman BA (2014) Atomistic autophagy: the structures of cellular self-digestion. Cell 157(2):300–311. doi:10.1016/j.cell.2014.01.070
Klionsky DJ, Codogno P, Cuervo AM, Deretic V, Elazar Z, Fueyo-Margareto J, Gewirtz DA, Kroemer G, Levine B, Mizushima N, Rubinsztein DC, Thumm M, Tooze SA (2010) A comprehensive glossary of autophagy-related molecules and processes. Autophagy 6(4):438–448. doi:10.4161/auto.6.4.12244
Chen Y, Klionsky DJ (2011) The regulation of autophagy—unanswered questions. J Cell Sci 124(Pt 2):161–170. doi:10.1242/jcs.064576
Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N (2009) Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5(7):973–979
Mercer CA, Kaliappan A, Dennis PB (2009) A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5(5):649–662
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141(2):290–303
Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG Jr (2004) Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18(23):2893–2904
DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW (2008) Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 22(2):239–251
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30(2):214–226
Budanov AV (2011) Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid Redox Signal 15(6):1679–1690. doi:10.1089/ars.2010.3530
Fujioka Y, Suzuki SW, Yamamoto H, Kondo-Kakuta C, Kimura Y, Hirano H, Akada R, Inagaki F, Ohsumi Y, Noda NN (2014) Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat Struct Mol Biol. doi:10.1038/nsmb.2822
Stanley RE, Ragusa MJ, Hurley JH (2014) The beginning of the end: how scaffolds nucleate autophagosome biogenesis. Trends Cell Biol 24(1):73–81. doi:10.1016/j.tcb.2013.07.008
Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y (2000) Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150(6):1507–1513
Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y (2005) Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 16(5):2544–2553
Dorsey FC, Rose KL, Coenen S, Prater SM, Cavett V, Cleveland JL, Caldwell-Busby J (2009) Mapping the phosphorylation sites of Ulk1. J Proteome Res 8(11):5253–5263. doi:10.1021/pr900583m
Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y (2010) Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol 30(4):1049–1058. doi:10.1128/MCB.01344-09
Cheong H, Nair U, Geng J, Klionsky DJ (2008) The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol Biol Cell 19(2):668–681
Jao CC, Ragusa MJ, Stanley RE, Hurley JH (2013) A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Natl Acad Sci USA 110(14):5486–5491. doi:10.1073/pnas.1220306110
Suzuki H, Tabata K, Morita E, Kawasaki M, Kato R, Dobson RC, Yoshimori T, Wakatsuki S (2014) Structural basis of the autophagy-related LC3/Atg13 LIR complex: recognition and interaction mechanism. Structure 22(1):47–58. doi:10.1016/j.str.2013.09.023
Cheong H, Klionsky DJ (2008) Dual role of Atg1 in regulation of autophagy-specific PAS assembly in Saccharomyces cerevisiae. Autophagy 4(5):724–726
Chan EY, Longatti A, McKnight NC, Tooze SA (2009) Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol 29(1):157–171. doi:10.1128/MCB.01082-08
Ragusa MJ, Stanley RE, Hurley JH (2012) Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell 151(7):1501–1512. doi:10.1016/j.cell.2012.11.028
Lazarus MB, Novotny CJ, Shokat KM (2015) Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol 10(1):257–261. doi:10.1021/cb500835z
Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, D’Amelio M, Nardacci R, Romagnoli A, Piacentini M, Cecconi F, Fimia GM (2010) The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 191(1):155–168. doi:10.1083/jcb.201002100
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15(7):741–750. doi:10.1038/ncb2757
Yamamoto H, Fujioka Y, Suzuki SW, Noshiro D, Suzuki H, Kondo-Kakuta C, Kimura Y, Hirano H, Ando T, Noda NN, Ohsumi Y (2016) The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev Cell 38(1):86–99
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. doi:10.1146/annurev-genet-102808-114910
Kihara A, Noda T, Ishihara N, Ohsumi Y (2001) Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 152(3):519–530
Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ (1997) The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 243(1–2):240–246
Miller S, Tavshanjian B, Oleksy A, Perisic O, Houseman BT, Shokat KM, Williams RL (2010) Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science 327(5973):1638–1642. doi:10.1126/science.1184429
Panaretou C, Domin J, Cockcroft S, Waterfield MD (1997) Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 3-kinase. Substrate presentation by phosphatidylinositol transfer protein to the p150.Ptdins 3-kinase complex. J Biol Chem 272(4):2477–2485
McKnight NC, Zhenyu Y (2013) Beclin 1, an essential component and master regulator of PI3K-III in health and disease. Curr Pathobiol Rep 1(4):231–238. doi:10.1007/s40139-013-0028-5
Stack JH, Herman PK, Schu PV, Emr SD (1993) A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3-kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO J 12(5):2195–2204
Yan Y, Flinn RJ, Wu H, Schnur RS, Backer JM (2009) hVps15, but not Ca2+/CaM, is required for the activity and regulation of hVps34 in mammalian cells. Biochem J 417(3):747–755. doi:10.1042/BJ20081865
Herman PK, Stack JH, DeModena JA, Emr SD (1991) A novel protein kinase homolog essential for protein sorting to the yeast lysosome-like vacuole. Cell 64(2):425–437
Fu LL, Cheng Y, Liu B (2013) Beclin-1: autophagic regulator and therapeutic target in cancer. Int J Biochem Cell Biol 45(5):921–924. doi:10.1016/j.biocel.2013.02.007
Rostislavleva K, Soler N, Ohashi Y, Zhang L, Pardon E, Burke JE, Masson GR, Johnson C, Steyaert J, Ktistakis NT, Williams RL (2015) Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science 350(6257):aac7365
Fan W, Nassiri A, Zhong Q (2011) Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc Natl Acad Sci USA 108(19):7769–7774. doi:10.1073/pnas.1016472108
Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447(7148):1121–1125
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9(10):1142–1151
Molejon MI, Ropolo A, Re AL, Boggio V, Vaccaro MI (2013) The VMP1–Beclin 1 interaction regulates autophagy induction. Sci Rep 3:1055. doi:10.1038/srep01055
Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11(4):385–396. doi:10.1038/ncb1846
Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A (2009) A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11(12):1433–1437. doi:10.1038/ncb1991
Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL (2009) 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 5(8):1180–1185
Walker S, Chandra P, Manifava M, Axe E, Ktistakis NT (2008) Making autophagosomes: localized synthesis of phosphatidylinositol 3-phosphate holds the clue. Autophagy 4(8):1093–1096
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182(4):685–701. doi:10.1083/jcb.200803137
Graef M, Friedman JR, Graham C, Babu M, Nunnari J (2013) ER exit sites are physical and functional core autophagosome biogenesis components. Mol Biol Cell 24(18):2918–2931
Tan D, Cai Y, Wang J, Zhang J, Menon S, Chou HT, Ferro-Novick S, Reinisch KM, Walz T (2013) The EM structure of the TRAPPIII complex leads to the identification of a requirement for COPII vesicles on the macroautophagy pathway. Proc Natl Acad Sci USA 110(48):19432–19437
Lemus L, Ribas JL, Sikorska N, Goder V (2016) An ER-localized SNARE protein is exported in specific COPII vesicles for autophagosome biogenesis. Cell Rep 14(7):1710–1722
Ge L, Zhang M, Schekman R (2014) Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. Elife 3:e04135. doi:10.7554/eLife.04135
Kutateladze TG (2010) Translation of the phosphoinositide code by PI effectors. Nat Chem Biol 6(7):507–513. doi:10.1038/nchembio.390
Obara K, Sekito T, Niimi K, Ohsumi Y (2008) The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem 283(35):23972–23980. doi:10.1074/jbc.M803180200
Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, Tooze SA (2010) Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6(4):506–522. doi:10.4161/auto.6.4.11863
Itakura E, Mizushima N (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6(6):764–776
Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y (2000) A ubiquitin-like system mediates protein lipidation. Nature 408(6811):488–492. doi:10.1038/35044114
Longatti A, Tooze SA (2012) Recycling endosomes contribute to autophagosome formation. Autophagy 8(11):1682–1683. doi:10.4161/auto.21486
Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ (2004) The Atg1–Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell 6(1):79–90
Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA (2006) Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 119(Pt 18):3888–3900
Nair U, Cao Y, Xie Z, Klionsky DJ (2010) Roles of the lipid-binding motifs of Atg18 and Atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J Biol Chem 285(15):11476–11488. doi:10.1074/jbc.M109.080374
Ropolo A, Grasso D, Pardo R, Sacchetti ML, Archange C, Lo Re A, Seux M, Nowak J, Gonzalez CD, Iovanna JL, Vaccaro MI (2007) The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem 282(51):37124–37133
Moreau K, Renna M, Rubinsztein DC (2013) Connections between SNAREs and autophagy. Trends Biochem Sci 38(2):57–63. doi:10.1016/j.tibs.2012.11.004
Reggiori F, Shintani T, Nair U, Klionsky DJ (2005) Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 1(2):101–109
Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F (2010) An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 190(6):1005–1022. doi:10.1083/jcb.200912089
Kovacs AL, Palfia Z, Rez G, Vellai T, Kovacs J (2007) Sequestration revisited: integrating traditional electron microscopy, de novo assembly and new results. Autophagy 3(6):655–662
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141(4):656–667. doi:10.1016/j.cell.2010.04.009
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12(8):747–757. doi:10.1038/ncb2078
Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y, Klionsky DJ (2000) Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol 148(3):465–480
Sekito T, Kawamata T, Ichikawa R, Suzuki K, Ohsumi Y (2009) Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells 14(5):525–538. doi:10.1111/j.1365-2443.2009.01299.x
Legakis JE, Yen WL, Klionsky DJ (2007) A cycling protein complex required for selective autophagy. Autophagy 3(5):422–432
He C, Baba M, Klionsky DJ (2009) Double duty of Atg9 self-association in autophagosome biogenesis. Autophagy 5(3):385–387
He C, Baba M, Cao Y, Klionsky DJ (2008) Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell 19(12):5506–5516. doi:10.1091/mbc.E08-05-0544
Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ (2011) SNARE proteins are required for macroautophagy. Cell 146(2):290–302. doi:10.1016/j.cell.2011.06.022
Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F (2009) The structure of Atg4B–LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J 28(9):1341–1350. doi:10.1038/emboj.2009.80
Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S, Chen Y, Zheng XL, Klionsky DJ, Liang Y, Xie Z (2012) Dual roles of Atg8–PE deconjugation by Atg4 in autophagy. Autophagy 8(6):883–892. doi:10.4161/auto.19652
Hong SB, Kim BW, Lee KE, Kim SW, Jeon H, Kim J, Song HK (2011) Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat Struct Mol Biol 18(12):1323–1330. doi:10.1038/nsmb.2165
Fujioka Y, Noda NN, Nakatogawa H, Ohsumi Y, Inagaki F (2010) Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem 285(2):1508–1515. doi:10.1074/jbc.M109.053520
Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19(5):2092–2100. doi:10.1091/mbc.E07-12-1257
Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7(3):279–296
Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif—crucial for selective autophagy. J Cell Sci 126(Pt 15):3237–3247. doi:10.1242/jcs.126128
Johansen T, Lamark T (2014) Selective autophagy goes exclusive. Nat Cell Biol 16(5):395–397. doi:10.1038/ncb2961
Xie Z, Nair U, Klionsky DJ (2008) Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 19(8):3290–3298. doi:10.1091/mbc.E07-12-1292
Nair U, Yen WL, Mari M, Cao Y, Xie Z, Baba M, Reggiori F, Klionsky DJ (2012) A role for Atg8–PE deconjugation in autophagosome biogenesis. Autophagy 8(5):780–793. doi:10.4161/auto.19385
Itakura E, Kishi-Itakura C, Mizushima N (2012) The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151(6):1256–1269. doi:10.1016/j.cell.2012.11.001
Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, Pfuetzner RA, Brunger AT, Zhong Q (2015) ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520(7548):563–566
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495(7441):389–393
Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovacs AL, Zhang Z, Feng D, Chen S, Zhang H (2014) O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol 16(12):1215–1226
Wang Z, Miao G, Xue X, Guo X, Yuan C, Zhang G, Chen Y, Feng D, Hu J, Zhang H (2016) The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell 63(5):781–795
McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I (2015) PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57(1):39–54
Wijdeven RH, Janssen H, Nahidiazar L, Janssen L, Jalink K, Berlin I, Neefjes J (2016) Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat Commun 7:11808
Chen D, Fan W, Lu Y, Ding X, Chen S, Zhong Q (2012) A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12–Atg5 conjugate. Mol Cell 45(5):629–641. doi:10.1016/j.molcel.2011.12.036
Hasegawa J, Iwamoto R, Otomo T, Nezu A, Hamasaki M, Yoshimori T (2016) Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J 35(17):1853–1867
Bonten EJ, Annunziata I, d’Azzo A (2014) Lysosomal multienzyme complex: pros and cons of working together. Cell Mol Life Sci 71(11):2017–2032. doi:10.1007/s00018-013-1538-3
Appelqvist H, Waster P, Kagedal K, Ollinger K (2013) The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 5(4):214–226. doi:10.1093/jmcb/mjt022
Teter SA, Eggerton KP, Scott SV, Kim J, Fischer AM, Klionsky DJ (2001) Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J Biol Chem 276(3):2083–2087. doi:10.1074/jbc.C000739200
Epple UD, Suriapranata I, Eskelinen EL, Thumm M (2001) Aut5/Cvt17p, a putative lipase essential for disintegration of autophagic bodies inside the vacuole. J Bacteriol 183(20):5942–5955. doi:10.1128/JB.183.20.5942-5955.2001
Epple UD, Eskelinen EL, Thumm M (2003) Intravacuolar membrane lysis in Saccharomyces cerevisiae. Does vacuolar targeting of Cvt17/Aut5p affect its function? J Biol Chem 278(10):7810–7821. doi:10.1074/jbc.M209309200
van Zutphen T, Todde V, de Boer R, Kreim M, Hofbauer HF, Wolinski H, Veenhuis M, van der Klei IJ, Kohlwein SD (2014) Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol Biol Cell 25(2):290–301. doi:10.1091/mbc.E13-08-0448
Mizushima N, Klionsky DJ (2007) Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 27:19–40. doi:10.1146/annurev.nutr.27.061406.093749
Suriapranata I, Epple UD, Bernreuther D, Bredschneider M, Sovarasteanu K, Thumm M (2000) The breakdown of autophagic vesicles inside the vacuole depends on Aut4p. J Cell Sci 113(Pt 22):4025–4033
Yang Z, Klionsky DJ (2007) Permeases recycle amino acids resulting from autophagy. Autophagy 3(2):149–150
Yang Z, Huang J, Geng J, Nair U, Klionsky DJ (2006) Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell 17(12):5094–5104. doi:10.1091/mbc.E06-06-0479
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465(7300):942–946. doi:10.1038/nature09076
Rong Y, Liu M, Ma L, Du W, Zhang H, Tian Y, Cao Z, Li Y, Ren H, Zhang C, Li L, Chen S, Xi J, Yu L (2012) Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol 14(9):924–934. doi:10.1038/ncb2557
Chen Y, Yu L (2013) Autophagic lysosome reformation. Exp Cell Res 319(2):142–146. doi:10.1016/j.yexcr.2012.09.004
Braulke T, Bonifacino JS (2009) Sorting of lysosomal proteins. Biochim Biophys Acta 1793(4):605–614. doi:10.1016/j.bbamcr.2008.10.016
Rong Y, McPhee CK, Deng S, Huang L, Chen L, Liu M, Tracy K, Baehrecke EH, Yu L, Lenardo MJ (2011) Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci USA 108(19):7826–7831. doi:10.1073/pnas.1013800108
Acknowledgements
We thank Jiangsu collaborative innovation center of meat production and processing for technical assistance and J.D. Hulleman from UT Southwestern Medical Center for reading the manuscript. The work was supported by grants to R.L. from the National Natural Science Foundation of China (Grant no. 31771532), the National Key Research and Development Program of China (Grant no. 2017YFD0400200), the Jiangsu Natural Science Funds for Distinguished Young Scholar (Grant no. SBK2017010325), the Jiangsu Natural Science Funds (Grant no. SBK2016043530), the fundamental research funds for the central universities (Grant no. 0806j0498), and the National Natural Science Foundation of China (Grant no. 31771529) to R.Y.G. Funding was provided by Natural Science Foundation of Jiangsu Province (Grant no. BK20160729).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhi, X., Feng, W., Rong, Y. et al. Anatomy of autophagy: from the beginning to the end. Cell. Mol. Life Sci. 75, 815–831 (2018). https://doi.org/10.1007/s00018-017-2657-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2657-z