
Abstract
Upon mitogen sensitization, lymphocytes undergo proliferation by oxyradical-based mechanisms. Through continuous resting–restimulation cycles, lymphocytes accumulate auto-induced oxidative lesions which lead to cell dysfunction and limit their viability. Astaxanthin (ASTA) is a nutritional carotenoid that shows notable antioxidant properties. This study aims to evaluate whether the in vitro ASTA treatment can limit oxyradical production and auto-oxidative injury in human lymphocytes. Activated lymphocytes treated with 5 µM ASTA showed immediate lower rates of O •−2 /H2O2 production whilst NO• and intracellular Ca2+ levels were concomitantly enhanced (≤4 h). In long-term treatments (>24 h), the cytotoxicity test for ASTA showed a sigmoidal dose–response curve (LC50 = 11.67 ± 0.42 µM), whereas higher activities of superoxide dismutase and catalase in 5 µM ASTA-treated lymphocytes were associated to significant lower indexes of oxidative injury. On the other hand, lower proliferative scores of ASTA lymphocytes might be a result of diminished intracellular levels of pivotal redox signaling molecules, such as H2O2. Further studies are necessary to establish the ASTA-dose compensation point between minimizing oxidative damages and allowing efficient redox-mediated immune functions, such as proliferation, adhesion, and oxidative burst.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lymphocytes are highly active immune cells that constantly generate reactive oxygen/nitrogen species (ROS/RNS) as part of their normal cellular activity (Chew and Park 2004). Lymphocytes undergo proliferation and clonal expansion upon stimulation with antigens/mitogens through redox-signaling molecules, such as H2O2 and NO• (Pahlavani et al. 2001). The immune system is highly reliant on accurate cell–cell communication for optimal function, and any damage to the signaling systems involved (e.g., membrane components) will result in impaired immune responsiveness (Victor et al. 2004). However, when lymphocytes pass through continuous resting–restimulation cycles, these cells accumulate oxidative lesions that factually culminate in cell dysfunction and, thus, limit their viability. Ultimately, oxidized lymphocytes trigger intrinsic apoptotic cascades to inactivate and decompose/recycle the nonviable immune cells, also known as activation-induced cell death (Curtin et al. 2002). Apoptosis undoubtedly serves an important immunoregulatory and homeostatic function by downregulating the proliferation of the less-viable lymphocytes after repetitive stimulation, though uncontrolled oxidative stress has been pointed out as the molecular basis of several immune-impaired pathologies such as diabetes, chronic inflammation, autoimmune disorders, sepsis, and HIV infection (Dobmeyer et al. 1997).
Since abundant circumstantial evidence links oxidative reactions to many consequences of immune diseases processes, some authors have currently described antioxidant therapies as auxiliary interventions to reduce the risk of such complications (Hussein et al. 2006b; Vinson 2006). However, the efficiency of antioxidant therapy drastically depends on the selected antioxidants, in situ concentrations, and timepoints in the disease course (Nunomura et al. 2006). Among several phytochemicals with confirmed antioxidant properties, carotenoids are of major interest due to their widespread occurrence in foodstuffs and particular ROS/RNS-scavenging mechanisms (Young and Lowe 2001). Apart from the inherent antioxidant properties of general food carotenoids, other mechanisms can actually account for their positive effects in the immune system (Elliott 2005). Beta-carotene possesses anti-inflammatory activity by functioning as a potential inhibitor of NF-кB activation with putative participation of retinoic acid metabolites (Bai et al. 2005). Not rare, non-provitamin A carotenoids such as lutein, lycopene, and astaxanthin are as active, and at times even more efficient, than β-carotene in enhancing cell-mediated and humoral immune responses in animals and humans (Chew and Park 2004). However, uptakes of lutein, β-carotene, and lycopene by MOLT-17 lymphocytes were proven to be both carotenoid-specific and dose-dependent (Astley et al. 2004). Dietary lutein consistently inhibits the growth of mammary tumors in mice and also prevents tumor-induced changes in subclasses of T-cell population, including Th, Tc, and IL-2R+T cells (Chew et al. 1996). Moreover, lutein-related proapoptotic effects are selectively triggered against tumors, but not in normal cells (Chew et al. 2003).
The carotenoid astaxanthin (3,3′-dihydroxy-β,β′-carotene-4,4′-dione; ASTA) has regained attention in the scientific media due to recent proposed antiapoptotic effects (even in immune cells) (Kim et al. 2009). ASTA is an orange-pinkish carotenoid essentially produced by micro- and macroalgal species, which accumulates in many marine organisms, such as shrimps, crabs, troutes, and salmons. It is a potential functional food and pharmaceutical supplement because of its excellent antioxidant activity (Barros et al. 2001; Cantrell et al. 2003; Young and Lowe 2001). Recent studies continue to evidence the multiple possibilities of ASTA application in providing benefits to human health (Hussein et al. 2006b). Furthermore, ASTA has also demonstrated cardioprotective (Fassett and Coombes 2009; Pashkow et al. 2008), neuroprotective (Liu et al. 2009), anti-inflammatory (Ohgami et al. 2003), antihypertensive (Hussein et al. 2006a), and anti-tumorogenic properties, especially against breast, prostate, and liver cancer (Kurihara et al. 2002). It has also been suggested as a protecting agent for bacterial infection of Helicobacter pylori in stomach ulcerations (Wang et al. 2000).
Thus, this study aims to evaluate whether the in vitro ASTA treatment can restrain ROS/RNS production and subsequent auto-oxidative injury in isolated human lymphocytes with putative improvement of cell proliferation. For this purpose, cytotoxic effects of ASTA and the proliferative capacity of T and B lymphocytes were compared to superoxide (O •−2 ), H2O2, and NO• production, intracellular Ca2+ mobilization, antioxidant enzyme activities (superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase), and indexes of oxidative damage in lipids (thiobarbituric acid reactive substances (TBARS) assay) and in proteins (carbonyl and thiol content).
Methods and materials
Reagents
Purified astaxanthin (ASTA) and most of other chemicals were purchased from Sigma–Aldrich Chemical Company (St. Louis, MO, USA), excepting the RPMI-1640 culture medium, lucigenin, pluronic acid, Vybrant 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation kit, and acetoxymethylester (Fura-2 AM) which were from Invitrogen (CA, USA). Common reagents for buffers (e.g., PBS) and regular laboratory solutions were obtained from Labsynth (Diadema, SP, Brazil).
Subjects
The Ethical Committee of the Universidade Cruzeiro do Sul approved the experimental procedure of this study. Around 30 healthy adult women and men (15 men and 15 women, mean age 27.0 ± 9.0) were included in the present study. All subjects did not present systemic or topical therapeutic regimen at least for the last 2 months. Subjects with a smoking history, alcohol habits, obesity, or any other systemic disease were excluded of the study (based on an anamnesis protocol).
Cell isolation and culture condition
Lymphocytes were obtained through the collection of human peripheral blood by venipuncture procedure in vacuum/siliconized tubes containing 0.1 mM EDTA. Peripheral blood lymphocytes were isolated under sterile conditions by using a density gradient present in the reagent Histopaque 1077 (Sigma–Aldrich) according to the manufacturer’s instruction. After centrifugation, the lymphocytes were counted in a neubauer chamber using Trypan blue (1%). Lymphocytes (1 × 106/mL) were cultured in 5 mL of RPMI 1640 supplemented as described above. The cells were treated with 5 µM of ASTA and cultured at 95% O2 for 24 h at 37°C. After this period, the cells were collected, centrifuged, and stored at −80°C to assays of enzymes activities and oxidative damage in biomolecules.
Effect of ASTA and DMSO on cell viability
A 150-µM stock solution of purified ASTA was prepared by dissolving carotenoid crystals in DMSO. Microcrystal/aggregates were disrupted by ultrasonication in a Vibra Cell apparatus (Connecticut, USA) for 5 min in ice-water bath. Tests were performed to establish the highest percentage of DMSO (v/v) in lymphocyte cultures that avoided significant cytotoxic effects. Then, the toxicity assay of ASTA in human lymphocytes was performed using cell density of 1 × 106 cell/mL exposed to different ASTA concentrations (0, 0.1, 1, 2, 5, 10, 20, and 30 μM in 1% DMSO) for 24 h in the culture medium. Immediately after being isolated and at the end of the incubation period, 5 × 105 cells were used to test cell viability, based on scores of membrane integrity. This assay was carried out in a FACScalibur flow cytometer (Becton Dickinson, Mountain View, CA) using 50 µg/mL propidium iodide dissolved in PBS pH 7.4. Propidium iodide (PI) is a membrane impermeant dye that preferably intercalates in nuclear DNA and, thus, indicates disruption of cellular membrane. Fluorescence of PI was determined in the FL2 channel (orange-red fluorescence at 585/542 nm).
Determination of lymphocyte proliferation capacity
The proliferation response of lymphocytes was determined using the Vybrant MTT cell proliferation (InVitrogen) according to the manufacturer’s instructions. Briefly, the MTT assay involves the conversion of the water soluble compound MTT to the insoluble formazan. The formazan is then solubilized, and the concentration determined by optical density at 570 nm. The cells (5 × 105 cell/well) were treated with ASTA (5 µM) and stimulated with concavalin A (20 µg Con A/mL) or lipopolysaccharide (100 µg LPS/mL) to evaluated T and B proliferation capacity, respectively. Absorbance was measured in 570 nm, and the results were expressed as optical density.
Nitric oxide production
Nitric oxide production was performed according to Ding et al. (1988) through nitrite (NO −2 ) determination. Nitric oxide (NO•) is rapidly converted to NO −2 in aqueous solutions; therefore, the total NO −2 concentration can be used as a stoichiometric indicator of NO• production in culture. Lymphocytes (5 × 105/well) were cultured with ASTA (5 µM) and LPS (10 μg/well) for 4 h. Afterwards, 100 µL of Griess reagent was added to culture containing both cells and supernatant. The absorbance was measured at 550 nm to estimate NO −2 concentrations based on a standard NaNO2 solution.
Hydrogen peroxide production
Hydrogen peroxide (H2O2) production was measured according to Pick and Mizel (1981) which is based on the horseradish peroxidase catalysis of the phenol red oxidation by H2O2. The production of H2O2 was measured in lymphocytes (5 × 105/well) in the absence and presence of phorbol myristate acetate (PMA; 20 ng/well) a promoter of respiratory burst in leucocytes. The absorbance was measured at 620 nm to evaluate H2O2 concentration (compared to a standard curve).
Measurement of intracellular superoxide anion production—dihydroethidium assay
Dihydroethidium (DHE) is a florescence probe and was used to measure the intracellular O •−2 production. Once inside the cell, DHE is rapidly oxidized to ethidium (a red fluorescent compound) by O •−2 with minor collaboration of other ROS (Hatanaka et al. 2006). Immediately after being obtained, lymphocytes (5 × 105) were incubated with 5 μM DHE for 15 min at room temperature in the dark. After incubation, cells were treated with ASTA (5 µM) and then promptly stimulated with PMA (20 ng/well) in Tyrode’s buffer (137 mM NaCl, 2.68 mM KCl, 0.49 mM MgCl2, 12 mM NaHCO3, 0.36 mM NaH2PO4, 5.6 mM d-glucose, and 5 mM acid HEPES, pH 7.4). Fluorescence of DHE emission was analyzed in a microplate reader (Tecan, Salzburg, Austria) at 590 nm (excitation wavelength at 396 nm).
Measurement of extracellular superoxide anion production—lucigenin assay
Extracellular O •−2 production (mainly produced by activated NADPH-oxidase) was measured by lucigenin chemiluminescence as previously described by (Hatanaka et al. 2006). Freshly prepared lymphocytes were incubated (5 × 105/well) with lucigenin (5 µM) and ASTA (5 µM), to a final volume of 300 µL. Superoxide production was triggered by addition of PMA (20 ng/well) in Tyrode’s buffer. Chemiluminescence of the excited product of lucigenin oxidation by O •−2 was monitored for 60 min in a luminometer (Tecan, Salzburg, Austria) and expressed as chemiluminescence relative units.
Intracellular Ca2+ concentration
Changes in cytosolic Ca2+ levels were monitored by fluorescence using the calcium-sensitive probe Fura 2-AM (Otton et al. 2007). The cells (1 × 106/mL) were washed and treated with 5 µM ASTA. Total intracellular release of [Ca2+] was monitored for 60 min in a microplate reader (Tecan, Salzburg, Austria). Transformation of the fluorescent signal to [Ca2+] (in nmol Ca2+ per minute) was performed by calibration with ionomycin (100 µM, maximum concentration) followed by EGTA addition (60 µM, minimum concentration) according to the Grynkiewicz equation (Grynkiewicz et al. 1985).
Preparation of homogenates for measurement of antioxidant enzymes and oxidative lesions
After the culture period, lymphocytes (5 × 106/mL) were harvested, and the cells were rupted by ultrasonication in a Vibra Cell apparatus (Connecticut, USA) using 500 µL of the assay-specific extraction solution/buffer, then centrifuged for 10 min, 10,000×g at 4°C. The supernatant was used for further analysis.
Lymphocyte antioxidant enzyme activities
Catalase (CAT), superoxide dismutase (SOD), glutathione peroxidase (GPX), and glutathione reductase (GR) activities were determined in lymphocytes using a spectrophotometer (Amersham Biosciences, UK). CAT activity was measured as described by Aebi (1984) based on the direct decomposition of hydrogen peroxide (H2O2). SOD activity was measured using the method described by Ewing and Janero (1995) which involves the reduction of O •−2 radicals by NBT following a linear first-order kinetic during 3 min. Glutatione peroxidase (Mannervik 1985) and glutathione reductase (Carlberg and Mannervik 1985) activities were measured based on the oxidation of β-NADPH in the presence of tert-butyl hydroperoxide used as substrate.
TBARS
The measurement of TBARS was described by Fraga et al. (1988) through the formation of a colored adduct after the stoichiometric reaction between TBA and several lipid derived aldehydes, including malondialdehyde (MDA). The absorbance at 535 nm was measured after the mixture reaches room temperature, and the TBARS content was estimated by a standard curve of 10 µM 1,1,3,3-tetraethoxypropane.
Thiol group and protein carbonyl formation
Thiol and carbonyl groups were evaluated as biomarkers of amino acid oxidation in total protein fractions, which were isolated from crude homogenate of cells (5 × 106) by precipitation with 20% trichloracetic acid solution in ice. Reduced thiol groups were detected by the formation of colored adducts after reaction with 4 mM 5.5′-dithio-bis (2-nitrobenzoic acid) solution (DTNB). The absorbance of DTNB-treated samples at 412 nm was calculated using GSH as a standard (Biteau et al. 2003). The same procedure was used to estimate protein carbonyls. The protein carbonyls were identified by the hydrazones formed with 10 mM DNPH in 0.25 M HCl. Absorbance of the peak detected within the range of 340–380 nm was measured, and the carbonyl group concentration was calculated based on the molar coefficient of ε = 2.2 × 104 M−1 cm−1 (Murphy and Kehrer 1989).
Protein determination
The total protein content of lymphocytes was measured by the method of Bradford (1976), using BSA as standard.
Statistical analysis
The results are expressed as mean ± SEM, and (n) is the number of experiments performed with lymphocyte cultures isolated from 30 volunteers upon ASTA effect (n ≥ 3). ANOVA was employed to detect significant differences among groups followed by the Tukey’s post-test (*p < 0.05).
Results
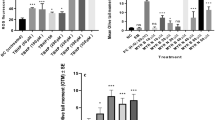
Preliminary toxicological tests were conducted to evaluate the cytotoxicity of either ASTA or its lipophilic vehicle DMSO on the isolated human lymphocytes. The viability scores from flow cytometry analysis are presented in Fig. 1a. The cytotoxicity of DMSO was assayed in terms of (v/v) percentage in order to estimate the maximum DMSO volumes allowed in cultures to avoid undesirable solvent lethality. In fact, DMSO over 4% (v/v) proved to be dramatically cytotoxic to lymphocytes after 24 h of incubation (data not shown). Based on these data, all ASTA-added systems were carried out using 1% of DMSO (v/v), in which desired ASTA concentrations could be efficiently solubilized. Cytotoxicity assays performed with different ASTA concentrations (Fig. 1a) demonstrated an abrupt decrease in lymphocyte viability above 5 µM: 64% at 10 µM ASTA and ca. 90% with either 20 or 30 μM ASTA. As shown in Fig. 1b, the LD50 value for ASTA in lymphocytes was 11.67 ± 0.42 µM. The well-fitted sigmoidal curve (R 2 = 0.998) also showed a high-slope value (p = 5.48 ± 1.08) suggesting magnified harmful effects of ASTA aggregation in the membranes of lymphocytes when carotenoid concentration reaches approximately 10 µM. Taking these calculations into account, all ASTA treatments in lymphocyte cultures herewith were carried out at 5 μM ASTA in 1% DMSO (v/v).

Membrane integrity scores (PI fluorescence) as indexes of ASTA cytotoxicity (a) in lymphocytes cultured for 24 h. The cells (1 × 106/mL) were cultured in different ASTA (0, 0.1, 1, 2, 5, 10, 20, and 30 μM) concentrations. b Dose–response curve of micromolar concentrations of ASTA to human lymphocytes in culture. The results are presented as mean ± SEM (n = 5). * p < 0.05 as compared to control group (0.0 μM ASTA)
Lymphocyte functionality was accurately evaluated by measurement of the proliferation capacity after stimulation with a specific mitogen. Figure 2 shows the MTT assay results after stimulation by Con A (a specific mitogen to T lymphocytes) or LPS (specific stimulus to B lymphocytes) for 48 h. Both stimuli caused a significant increase in the proliferation capacity of control: 25% and 35% for ConA- and LPS-stimulation systems, respectively. However, in vitro treatment of lymphocytes with 5 µM ASTA caused a similar decrease in cell proliferation either by Con A or LPS stimulation (∼30%).

Proliferation response of human lymphocytes to a Con A and b LPS stimulation. Cells (5 × 105/well) were incubated for 48 h in the absence or presence of 5 µM ASTA. The results are presented as mean ± SEM (n = 12, four different experiments) * p < 0.05 compared to control group without stimulation. # p < 0.05 compared to stimulated control group
The immediate/short-term effects of 5 µM ASTA on the isolated human lymphocytes were assessed by measuring key redox signaling components O •−2 , H2O2, NO•, and intracellular Ca2+. As shown in Fig. 3a, the NO• production in experimental groups treated with LPS was significant higher than in nontreated groups. The LPS-triggered NO• formation showed a significant increment of 90% in ASTA-treated groups compared to control group. On the other hand, H2O2 production showed a significant decrease by 24% after ASTA treatment and PMA stimulation as compared to control-stimulated group (Fig. 3b). Noteworthy, isolated human lymphocytes were distinctly stimulated with LPS or PMA in order to prioritize the activation of NOS and NADPH oxidase, respectively, thereby bursting NO• and O •−2 /H2O2 production.

Nitric oxide (a) and hydrogen peroxide production (b) in human lymphocytes treated with 5 µM ASTA. Cells (2 × 106/mL) were stimulated with LPS (40 µg/mL; a) or PMA (80 ng/mL; b) for detection by the Griess or phenol red reagents, respectively. The results are presented as mean ± SEM (n = 12; at least four different experiments). # p < 0.05 compared to stimulated-control group
Regarding DHE and lucigenin protocols, appropriate controls were carried out to exclude possible quenching effects of ASTA in those fluorescence/chemiluminescence assays. In cell-depleted systems, ASTA did not directly affect the ROS-detected fluorescences (data not shown). Nevertheless, our results demonstrated a robust decrease in extracellular O •−2 production (90%, as assessed by lucigenin assay) when PMA-stimulated lymphocytes were pretreated with 5 μM ASTA (compared to control group, Fig. 4a, b). A slight but significant decrease of 26% in DHE fluorescence was observed in activated lymphocytes treated with 5 μM ASTA, revealing diminished intracellular O •−2 production (Fig. 4c).

Superoxide anion production in human lymphocytes (5 × 105/well) freshly treated with ASTA (5 µM) and PMA-activated (20 ng/well). a Kinetic of extracellular superoxide anion production by the 5 μM lucigenin fluorescence assay. b The kinetic results are presented as mean ± SEM of eight assays from at least eight different experiments performed in triplicate. c Intracellular superoxide anion production by measurement of 5 μM dihydroethidium fluorescence (DHE). Negative controls were performed in cell-free systems with lucigenin + PMA or lucigenin + ASTA; and DHE + PMA or DHE + ASTA (data not shown). The values are presented as mean ± SEM from ten subjects in at least five experiments. * p < 0.05 compared to control group # p < 0.05 compared to control-stimulated group
Intracellular Ca2+ mobilization was significantly enhanced by 5 μM ASTA treatment in human lymphocytes (∼15%) when compared to the control group (Fig. 5). The increase in Ca2+ levels was sustained during 60 min of kinetic monitoring.

Total intracellular Ca2+ mobilization (nM) in ASTA-treated human lymphocytes. Cells (1 × 106/well) were previously loaded with 5 µM Fura 2-AM during 1 h and then incubated with 5 µM ASTA for extra 60 min. The results are presented as mean ± SEM (n ≥ 5). * p < 0.05 compared to control group
Upon the observed short-term variations in the ROS/RNS production of ASTA-treated lymphocytes, many questions arose regarding antioxidant responses of human lymphocytes upon long-term exposure to the carotenoid. Therefore, the activities of major antioxidant enzymes in experimental cells were analyzed after 24 h (Table 1). Among all front line antioxidant enzymes tested, only total SOD and CAT activities were significantly increased (89% and 72%, respectively), after 5 µM ASTA treatment, compared to the control. Interestingly, the enzyme activity of the mitochondrial MnSOD isoform was unaltered suggesting that major variations in long-term production of O •−2 preferably occurred at other cellular compartments than mitochondria. No significant changes were observed in both glutathione-related enzymes GPX and GR after 24 h of 5 µM ASTA treatment.
Table 1 also shows the extension of oxidative lesions in lipids (by TBARS test) and proteins (by carbonyl and thiol contents) of isolated lymphocytes. A significant decrease by 40% in TBARS content in parallel to a 38% increment in thiol groups (but not protein carbonyls) suggest the beneficial effects of 5 µM ASTA in preventing lipid and protein oxidation in human lymphocytes after 24 h.
Discussion
The orchestration of the immune response relies heavily on cell–cell communication processes. Thus, the oxidant–antioxidant balance is a key factor in preserving the immune cell function, not only regarding the integrity of their polyunsaturated fatty acid-rich membrane but also for control of signal transduction and gene expression (Meydani and Azzi 2009). Immune cells are frequently exposed to oxidizing injury provoked by antigen stimulation of ROS/RNS as part of their normal function (Meydani and Azzi 2009). Accordingly, there is relative consensus reinforcing the pivotal role of oxyradicals in survival, function, and/or proliferation ability of lymphocytes. Some immunological studies have shown that high antioxidant concentrations were actually detrimental to lymphocyte functions, since they inhibited H2O2-mediated proliferation and production of IL-2, both required for optimal T-cell expansion (Schmielau and Finn 2001).
In this study, 5 µM ASTA significantly inhibited O •−2 and H2O2 production in activated lymphocytes within 60 min, while total NO• formed after 4 h was exacerbated. Choi et al. (2008) also observed augmented production of NO• in ASTA-treated BV2 microglial cells stimulated with LPS. Through a hydrogen–atom abstraction mechanism, ASTA has the capacity to scavenge several relevant ROS in vivo, including O •−2 (Jackson et al. 2004), and the propagating intermediates of the lipid peroxidation chain reaction, peroxyl (ROO•), and alkoxyl (RO•) radicals (Barros et al. 2001). On the other hand, pretreatment of lymphocytes with ASTA for 24 h could impose mild oxidative insults to those cells in culture based on the described pro-oxidative properties of carotenoids under some circumstances (Young and Lowe 2001). Regarding the O2 tension in flask cultures of lymphocytes (pO2 different from venous/arterial blood) and the relatively high ASTA concentration applied, this hypothesis is very acceptable. Therefore, in response to the slight oxidative challenge imposed by ASTA treatment in cultures, lymphocytes showed higher activities of the antioxidant enzymes SOD and CAT (Table 1), lower indexes of oxidative lesions (TBARS and protein thiols, Table 1), and lower rates of O •−2 and H2O2 production after PMA activation (Figs. 3b and 4a–c). Noteworthy, the simultaneous decreases of O •−2 and H2O2 production are congruent to the fact that both ROS levels are tightly regulated in several cellular compartments by the ubiquitous SOD-catalyzed dismutation of O •−2 to H2O2.
Our results on ConA and LPS stimulation demonstrated a significant decrease in lymphocyte proliferation with 5 µM ASTA treatment. Lymphocyte proliferation and clonal expansion are known immune functions triggered by antigen/mitogen activation through redox-signaling molecules, especially H2O2 (Pahlavani et al. 2001). Thus, due to the powerful scavenging activity of ASTA, intracellular levels of O •−2 and its dismutation product, H2O2, were severely diminished to hinder efficient lymphocyte proliferation. Wojcik et al. (2008) demonstrated that the addition of β-carotene as well as ASTA delayed the proliferation of oval cells obtained from 2/3 in partial hepatectomised and diethylnitrosamine-treated rats. Accordingly, Tanaka et al. (1995) related that ASTA and its precursor canthaxanthin have anticarcinogenic chemopreventive properties partly due to inhibition of cell proliferation.
Increased intracellular Ca2+ mobilization is in agreement with higher NO• production, as Ca2+ ions (not necessarily bound to calmodulin) is a recognized activation factor for several nitric oxide synthase (NOS) isoforms (Feske 2007). In lymphocytes, constitutive endothelial and neuronal NOS (eNOS and nNOS, respectively) are the main cytoplasmic sources of NO•, whereas inducible NOS isoform (iNOS) is apparently absent in these immune cells (Ibiza et al. 2006). Whether primary T or B lymphocytes express any of the NOS isoforms remains questionable. Furthermore, the existence of a constitutive mitochondrial NOS isoenzyme (mtNOS) that also generates NO• according to cytosolic/mitochondrial Ca2+ ratio and mitochondrial membrane potential is now clear (Valdez et al. 2006). Despite the fact that ASTA intracellular targets remain unknown, the carotenoid was recently linked to the maintenance of high mitochondrial membrane potential and stimulated respiration efficiency but controlled mitochondrial ROS production in cultured cells, probably due to its inherent scavenging activity (Wolf et al. 2009). Thus, the participation of ASTA in limiting mitochondrial production of ROS, controlling high membrane potential (as described by Wolf et al. 2009), and, therefore, sustaining elevated levels of NO• by Ca2+-mediated activation of mtNOS should not be discarded. Appropriately, mitochondrial MnSOD activity was unaltered after 24 h of 5 µM ASTA treatment in lymphocytes (Table 1). Since the Griess reaction actually detects end products of NO• metabolism, the involvement of other RNS can be suggested, e.g., the powerful oxidizing agent ONOO− (Ferrer-Sueta and Radi 2009).
Independent to their inherent scavenging properties, three major topics of carotenoid healthy benefits have always been debated in the literature: (a) the synergistic effect with other antioxidants such as ascorbate and tocopherols (Schroeder et al. 2006); (b) the need of constant provision of carotenoids from diet in order to replace oxidized/metabolized derivatives and sustain steady-state levels in tissues (Yeum et al. 1996); and (c) putative participation of carotenoids and/or their metabolites in gene expression of antioxidant enzymes via NF-кB pathways (Kim et al. 2008). In our study, it is possible that the single dose of 5 µM ASTA efficiently afforded antioxidant protection to lymphocytes for the short-term assays. Afterwards, the induction of antioxidant frontline enzymes SOD (mainly the cytosolic isoform CuZn SOD) and peroxisomal CAT was apparently required to reestablish the intracellular redox balance in cultured lymphocytes. We cannot discard the participation of ASTA metabolites in this gene expression function (Suzuki et al. 2006).
In summary, our data suggest that ASTA displays interesting anti-inflammatory effects by preserving redox-sensitive (and essential) structures of human lymphocytes, although the applied dose apparently hindered lymphocyte proliferation. This can be mainly deduced by the increased NO• formation, the observed reduced O •−2 /H2O2 production, and induced SOD and CAT activities in parallel to lower indexes of oxidative injury in lipids and proteins. Despite that such relatively high ASTA doses are improbable to be achieved in vivo, the search for an optimal ASTA concentration that could compensate lower membrane/protein lesions with suitable levels of vital redox signals for sustaining immune cell functions is still open for further studies. This information is crucial in order to properly address the indication of ASTA as a nutritional therapy for prevention/prophylaxy of immune-impaired diseases, such as type 2 diabetes, sepsis, and cardiovascular disorders.
Abbreviations
- ANOVA:
-
Analysis of variance
- ASTA:
-
Astaxanthin
- BHT:
-
Butylated hydroxytoluene
- BSA:
-
Albumin
- [Ca2+]i :
-
Intracellular calcium
- CAT:
-
Catalase
- Con A:
-
Concanavalin A
- DHE:
-
Dihydroethidium
- DMSO:
-
Dimethyl sulfoxide
- DNPH:
-
2,4-Dinitrophenylhydrazine
- DTNB:
-
5,5′-Dithiobis(2-nitrobenzoic acid)
- EDTA:
-
Ethylenediaminetetraacetic acid
- EGTA:
-
Ethylene glycol tetracetic acid
- GSH:
-
Reduced glutathione
- GSSG:
-
Oxidized glutathione
- H2O2 :
-
Hydrogen peroxide
- LPS:
-
Lipopolysaccharide
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NADH:
-
Nicotinamide adenine dinucleotide
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NBT:
-
Nitroblue tetrazolium
- NEM:
-
N-ethylmalemide
- NO:
-
Nitric oxide
- PMA:
-
Phorbol-12-myristate 13-acetate
- PMS:
-
Phenazine methosulfate
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- TBA:
-
Thiobarbituric acid
References
Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–6.
Astley SB, Hughes DA, Wright AJ, Elliott RM, Southon S. DNA damage and susceptibility to oxidative damage in lymphocytes: effects of carotenoids in vitro and in vivo. Br J Nutr. 2004;91:53–61.
Bai SK, Lee SJ, Na HJ, Ha KS, Han JA, Lee H, et al. Beta-Carotene inhibits inflammatory gene expression in lipopolysaccharide-stimulated macrophages by suppressing redox-based NF-kappaB activation. Exp Mol Med. 2005;37:323–34.
Barros MP, Pinto E, Colepicolo P, Pedersen M. Astaxanthin and peridinin inhibit oxidative damage in Fe(2+)-loaded liposomes: scavenging oxyradicals or changing membrane permeability? Biochem Biophys Res Commun. 2001;288:225–32.
Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–4.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Cantrell A, McGarvey DJ, Truscott TG, Rancan F, Bohm F. Singlet oxygen quenching by dietary carotenoids in a model membrane environment. Arch Biochem Biophys. 2003;412:47–54.
Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol. 1985;113:484–90.
Chew BP, Park JS. Carotenoid action on the immune response. J Nutr. 2004;134:257S–61S.
Chew BP, Wong MW, Wong TS. Effects of lutein from marigold extract on immunity and growth of mammary tumors in mice. Anticancer Res. 1996;16:3689–94.
Chew BP, Brown CM, Park JS, Mixter PF. Dietary lutein inhibits mouse mammary tumor growth by regulating angiogenesis and apoptosis. Anticancer Res. 2003;23:3333–9.
Choi SK, Park YS, Choi DK, Chang HI. Effects of astaxanthin on the production of NO and the expression of COX-2 and iNOS in LPS-stimulated BV2 microglial cells. J Microbiol Biotechnol. 2008;18:1990–6.
Curtin JF, Donovan M, Cotter TG. Regulation and measurement of oxidative stress in apoptosis. J Immunol Methods. 2002;265:49–72.
Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–12.
Dobmeyer TS, Findhammer S, Dobmeyer JM, Klein SA, Raffel B, Hoelzer D, et al. Ex vivo induction of apoptosis in lymphocytes is mediated by oxidative stress: role for lymphocyte loss in HIV infection. Free Radic Biol Med. 1997;22:775–85.
Elliott R. Mechanisms of genomic and non-genomic actions of carotenoids. Biochim Biophys Acta. 2005;1740:147–54.
Ewing JF, Janero DR. Microplate superoxide dismutase assay employing a nonenzymatic superoxide generator. Anal Biochem. 1995;232:243–8.
Fassett RG, Coombes JS. Astaxanthin, oxidative stress, inflammation and cardiovascular disease. Future Cardiol. 2009;5:333–42.
Ferrer-Sueta G, Radi R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol. 2009;4:161–77.
Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702.
Fraga CG, Leibovitz BE, Tappel AL. Lipid peroxidation measured as thiobarbituric acid-reactive substances in tissue slices: characterization and comparison with homogenates and microsomes. Free Radic Biol Med. 1988;4:155–61.
Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50.
Hatanaka E, Levada-Pires AC, Pithon-Curi TC, Curi R. Systematic study on ROS production induced by oleic, linoleic, and gamma-linolenic acids in human and rat neutrophils. Free Radic Biol Med. 2006;41:1124–32.
Hussein G, Goto H, Oda S, Sankawa U, Matsumoto K, Watanabe H. Antihypertensive potential and mechanism of action of astaxanthin: III. Antioxidant and histopathological effects in spontaneously hypertensive rats. Biol Pharm Bull. 2006a;29:684–8.
Hussein G, Sankawa U, Goto H, Matsumoto K, Watanabe H. Astaxanthin, a carotenoid with potential in human health and nutrition. J Nat Prod. 2006b;69:443–9.
Ibiza S, Victor VM, Bosca I, Ortega A, Urzainqui A, O’Connor JE, et al. Endothelial nitric oxide synthase regulates T cell receptor signaling at the immunological synapse. Immunity. 2006;24:753–65.
Jackson HL, Cardounel AJ, Zweier JL, Lockwood SF. Synthesis, characterization, and direct aqueous superoxide anion scavenging of a highly water-dispersible astaxanthin-amino acid conjugate. Bioorg Med Chem Lett. 2004;14:3985–91.
Kim JH, Na HJ, Kim CK, Kim JY, Ha KS, Lee H, et al. The non-provitamin A carotenoid, lutein, inhibits NF-kappaB-dependent gene expression through redox-based regulation of the phosphatidylinositol 3-kinase/PTEN/Akt and NF-kappaB-inducing kinase pathways: role of H(2)O(2) in NF-kappaB activation. Free Radic Biol Med. 2008;45:885–96.
Kim YJ, Kim YA, Yokozawa T. Protection against oxidative stress, inflammation, and apoptosis of high-glucose-exposed proximal tubular epithelial cells by astaxanthin. J Agric Food Chem. 2009;57:8793–7.
Kurihara A, Nakazaki H, Watanabe M, Hasebe Y, Takita W, Seo A, et al. Hepatectomy and intraarterial infusion chemotherapy for liver metastasis from colorectal cancer. Gan To Kagaku Ryoho. 2002;29:2104–7.
Liu X, Shibata T, Hisaka S, Osawa T. Astaxanthin inhibits reactive oxygen species-mediated cellular toxicity in dopaminergic SH-SY5Y cells via mitochondria-targeted protective mechanism. Brain Res. 2009;1254:18–27.
Mannervik B. Glutathione peroxidase. Methods Enzymol. 1985;113:490–5.
Meydani M, Azzi A. Diabetes risk: antioxidants or lifestyle? Am J Clin Nutr. 2009;90:253–4.
Murphy ME, Kehrer JP. Oxidation state of tissue thiol groups and content of protein carbonyl groups in chickens with inherited muscular dystrophy. Biochem J. 1989;260:359–64.
Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–41.
Ohgami K, Shiratori K, Kotake S, Nishida T, Mizuki N, Yazawa K, et al. Effects of astaxanthin on lipopolysaccharide-induced inflammation in vitro and in vivo. Invest Ophthalmol Vis Sci. 2003;44:2694–701.
Otton R, da Silva DO, Campoio TR, Silveira LR, de Souza MO, Hatanaka E, et al. Non-esterified fatty acids and human lymphocyte death: a mechanism that involves calcium release and oxidative stress. J Endocrinol. 2007;195:133–43.
Pahlavani MA, Mele JF, Richardson A. Effect of overexpression of human Cu/Zn-SOD on activation-induced lymphocyte proliferation and apoptosis. Free Radic Biol Med. 2001;30:1319–27.
Pashkow FJ, Watumull DG, Campbell CL. Astaxanthin: a novel potential treatment for oxidative stress and inflammation in cardiovascular disease. Am J Cardiol. 2008;101:58D–68D.
Pick E, Mizel D. Rapid microassays for the measurement of superoxide and hydrogen peroxide production by macrophages in culture using an automatic enzyme immunoassay reader. J Immunol Methods. 1981;46:211–26.
Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–60.
Schroeder MT, Becker EM, Skibsted LH. Molecular mechanism of antioxidant synergism of tocotrienols and carotenoids in palm oil. J Agric Food Chem. 2006;54:3445–53.
Suzuki Y, Ohgami K, Shiratori K, Jin XH, Ilieva I, Koyama Y, et al. Suppressive effects of astaxanthin against rat endotoxin-induced uveitis by inhibiting the NF-kappaB signaling pathway. Exp Eye Res. 2006;82:275–81.
Tanaka T, Makita H, Ohnishi M, Mori H, Satoh K, Hara A. Chemoprevention of rat oral carcinogenesis by naturally occurring xanthophylls, astaxanthin and canthaxanthin. Cancer Res. 1995;55:4059–64.
Valdez LB, Zaobornyj T, Boveris A. Mitochondrial metabolic states and membrane potential modulate mtNOS activity. Biochim Biophys Acta. 2006;1757:166–72.
Victor VM, Rocha M, De la Fuente M. Immune cells: free radicals and antioxidants in sepsis. Int Immunopharmacol. 2004;4:327–47.
Vinson JA. Oxidative stress in cataracts. Pathophysiology. 2006;13:151–62.
Wang X, Willen R, Wadstrom T. Astaxanthin-rich algal meal and vitamin C inhibit Helicobacter pylori infection in BALB/cA mice. Antimicrob Agents Chemother. 2000;44:2452–7.
Wojcik M, Bobowiec R, Martelli F. Effect of carotenoids on in vitro proliferation and differentiation of oval cells during neoplastic and non-neoplastic liver injuries in rats. J Physiol Pharmacol. 2008;59 Suppl 2:203–13.
Wolf AM, Asoh S, Hiranuma H, Ohsawa I, Iio K, Satou A, et al. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J Nutr Biochem. 2009 (in press).
Yeum KJ, Booth SL, Sadowski JA, Liu C, Tang G, Krinsky NI, et al. Human plasma carotenoid response to the ingestion of controlled diets high in fruits and vegetables. Am J Clin Nutr. 1996;64:594–602.
Young AJ, Lowe GM. Antioxidant and prooxidant properties of carotenoids. Arch Biochem Biophys. 2001;385:20–7.
Acknowledgements
The authors are indebted to the constant assistance of Buttignol MHP, Campoio TR. This research is supported by Fundação do Amparo a Pesquisa do Estado de São Paulo (FAPESP 07/03334-6; 02/09405-9), PIBIC-CNPq (119256/2009-6; 109713/2008-7), and Universidade Cruzeiro do Sul.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bolin, A.P., Macedo, R.C., Marin, D.P. et al. Astaxanthin prevents in vitro auto-oxidative injury in human lymphocytes. Cell Biol Toxicol 26, 457–467 (2010). https://doi.org/10.1007/s10565-010-9156-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-010-9156-4