
Abstract
Boron (B) is a developmental and reproductive toxin. It is also essential for some organisms. Plants use uptake and efflux transport proteins to maintain homeostasis, and in humans, boron has been reported to reduce prostate cancer. Ca2+ signaling is one of the primary mechanisms used by cells to respond to their environment. In this paper, we report that boric acid (BA) inhibits NAD+ and NADP+ as well as mechanically induced release of stored Ca2+ in growing DU-145 prostate cancer cells. Cell proliferation was inhibited by 30% at 100μM, 60% at 250μM, and 97% at 1,000μM BA. NAD+-induced Ca2+ transients were partly inhibited at 250μM BA and completely at 1,000μM BA, whereas both NADP+ and mechanically induced transients were inhibited by 1,000μM BA. Expression of CD38 protein increased in proportion to BA exposure (0–1,000μM). In vitro mass spectrometry analysis showed that BA formed adducts with the CD38 products and Ca2+ channel agonists cyclic adenosine diphosphate ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP). Vesicles positive for the Ca2+ fluorophore fluo-3 acetoxymethyl ester accumulated in cells exposed to 250 and 1,000μM BA. The BA analog, methylboronic acid (MBA; 250 and 1,000μM), did not inhibit cell proliferation or NAD+, NADP+, or mechanically stimulated Ca2+ store release. Nor did MBA increase CD38 expression or cause the formation of intracellular vesicles. Thus, mammalian cells can distinguish between BA and its synthetic analog MBA and exhibit graded concentration-dependent responses. Based on these observations, we hypothesize that toxicity of BA stems from the ability of high concentrations to impair Ca2+ signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Boron is a ubiquitous element, the ninth most abundant dissolved element in seawater (414μM B), and exhibits beneficial and toxic effects in plants and animals. Boric acid (BA) and borates are natural constituents of dietary plant products and drinking water and used as ingredients in many consumer products. Plants regulate BA/borate homeostasis using uptake and efflux transporters (Miwa et al. 2007). In vertebrates, BA toxicity exhibits a U-shaped form typical of an essential nutrient (Rowe et al. 1998). The No observable adverse effect level (NOAEL) for BA is based on normal rat pup birth weight and corresponds to a maternal blood concentration of 123μM BA (Price et al. 1997; IPCS 1998). Concentrations greater than 275μM BA in rats are associated with fetal stunting and levels of 1,000μM BA with atrophy of the seminiferous tubules (Fail et al. 1998). By contrast, low levels of exposure have beneficial effects (IOM 2001).
Studies in chicks, rats, and pigs have shown that BA is important for normal growth and may improve bone strength (Hunt and Nielsen 1981; Chapin et al. 1997; Armstrong et al. 2000). Supplemental B was shown to improve brain function in an in-patient metabolic ward study designed to evaluate the effects of B depletion (Penland 1994). Two epidemiological studies have reported that the risk of prostate cancer in US men is reduced in proportion to dietary and groundwater B (Cui et al. 2004; Barranco et al. 2007). The biological plausibility of this is supported by several lines of investigation. In immunocompromised mice, BA supplementation reduced the growth of transplanted human prostate tumors, decreased insulin-like growth factor 1 tissue concentrations and lowered serum prostate-specific antigen levels (Gallardo-Williams et al. 2004). In cell cultures, BA reduced the proliferation of human cancer prostate cell lines in a dose-dependent manner over a range from 60 to 1,000μM BA and inhibited cell migration and invasion (Barranco and Eckhert 2004; Barranco and Eckhert 2006).
It has been known for more than 20years that intracellular Ca2+ signaling serves a vital role in regulating the proliferation of eukaryotic cells (Poenie et al. 1985). The relationship between BA and Ca2+ signaling has not been studied, but sufficient reason exists to do so. The major BA-binding molecules in biology are the five-carbon furanose sugars, ribose in animals and apiose in plants. In animals, BA binds NAD+, which has two ribose units, and functions both as a coenzyme in intermediary metabolism and intercellular paracrine-signaling mediator (Kim et al. 2003). As a paracrine, NAD+ is released from cells and interacts with the CD38 receptor on the plasma membrane of neighboring cells (Bruzzone et al. 2001). CD38 regulates cell proliferation by modulating stored Ca2+ release from the endoplasmic reticulum into the cytoplasm (Lee 2006). CD38 is a multifunctional enzyme that converts NAD+ into cyclic adenosine diphosphate ribose (cADPr) and catalyzes exchange of the nicotinamide group of NADP+ with nicotinic acid to produce nicotinic acid adenine dinucleotide phosphate (NAADP+). Both cADPr and NAADP mediate the release of Ca2+ from different intracellular stores using different calcium channels (Lee 2006; Zhang and Li 2007).
Ca2+ signaling and phosphorylation are the principal means by which cells respond to their environment. The release of stored calcium ions (Ca2+) from the endoplasmic reticulum into the cytoplasm is a transitory event since Ca2+ rapidly binds to and induces changes in the shape of a class of regulatory proteins referred to as calcium-binding proteins. Our objective was to test the hypothesis that BA inhibits Ca2+ store release in human prostate cancer cells. DU-145 prostate cancer cells are well suited for investigating cellular processes modulated by BA because their proliferation is sensitive to BA over a wide concentration range. We used three approaches to stimulate the release of intracellular Ca2+ stores. These included: NAD+, which is converted by CD38 to cADPr, an agonist of the Ca2+ ryanodine channel; NADP+, which is converted by CD38 to NAADP, an agonist of the NAADP Ca2+ channels (Lee 2006; Zhang and Li 2007); and mechanical stimulation, a CD38-independent mechanism that activates the inositol 1,4,5-triphosphate (IP3) calcium channel (Moerenhout et al. 2001).
Materials and methods
Cell culture and media preparations
The prostate cancer cell line DU-145 was acquired from American Type Culture Collection (ATCC, Manassas, VA, USA) and LNCaP was a gift from Dr. Allen Pantuck at the University of California – Los Angeles. Cells were cultured at 37°C, 5% CO2 in Roswell Park Memorial Institute (RPMI) 1640 media (Invitrogen), supplemented with 10% fetal bovine serum, penicillin/streptomysin (100U/mL; 100μg/mL), and l-glutamine (200mM; Gemini Bioproducts, West Sacramento, CA, USA). Nontumorigenic lines, RWPE-1 and PWR-1E, were acquired from ATCC and cultured as recommended in keratinocyte serum-free medium supplemented with 50μg/mL bovine pituitary extract, 5ng/mL human recombinant epidermal growth factor, and 1× antibiotic/antimycotic mixture (100U/mL penicillin, 100μg/mL streptomycin, 0.25μg/mL Fungizone). The boron-depleted medium was prepared by the addition of the boron-specific ionic exchange resin Amberlite IRA-743 (Sigma-Aldrich, USA) to the supplemented media, followed by overnight agitation on a Koala-Ty rotator (80rpm at 4°C). The resin-treated medium was transferred to sterile containers for boron supplementation. BA and methylboronic acid (MBA) stock solutions were prepared in ion exchange treated water with resistivity greater than 18MΩ.
Cell proliferation
Cells were plated onto six-well culture plates, at a density of 3 × 105 cells per well, and treated daily with serum-free media supplemented with BA or MBA (0–1000μM) as previously described (Barranco and Eckhert 2004). To provide sufficient material for the study, cells were allowed to grow until control plates (0μM BA) were near but not at confluence (7 or 8days). Cells were removed by trypsinization and counted using a hemacytometer, with the aid of Trypan Blue to exclude nonviable cells (Life Technologies, Rockville, MD, USA).
Stored calcium ion [Ca2+] release in proliferating inhibited cells
DU-145 cells were plated on 100 × 20-mm culture plates and cultured in 0, 250, and 1,000μM BA- or MBA-supplemented media. On day7, cells were trypsinized and replated onto15-mm glass coverslips (16,000 cells per square centimeter), covered with their corresponding BA- or MBA-supplemented media, and allowed to settle overnight. The following day, cells were incubated for 30min in media supplemented with Fluo-4-acetoxymethyl ester (AM; 4–6μM) (Sigma-Aldrich; Fluka cat no. 93596), a cell-permeable Ca2+ indicator dye, and then washed once with mammalian Ringer’s solution (0.14M NaCl, 0.005M KCl, 0.002M CaCl2, 0.001M MgCl2, 0.01M glucose, 0.01M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES] at pH7.4). Coverslip-adherent cells were placed in a perfusion chamber (1mL/min flow rate) and exposed to Ringer’s solution containing NAD+ or NADP+ to stimulate the release of stored Ca2+. The protocol was: 0–1min baseline Ringer’s solution followed by exposure to NAD+ or NADP+ in the presence or absence of BA or MBA. Fluo-4-AM was excited with a 488-nm argon laser line, and emissions were collected with a 505 LP filter using a Zeiss LSM 5 Pascal confocal microscope (Zeiss, Thornwood, NY, USA) coupled to an upright fixed-stage microscope (Axioskop 2 FS mot) and equipped with a Axoplan 63X (NA 0.95) water-immersion objective. Additional magnification, time series, and background subtraction were controlled by Zeiss LSM acquisition software. All images were acquired as 12-bit images. NAD+ (10mM) and NADP+ (5mM) were solubilized in mammalian Ringer’s solution.
pH dependence of NAD+- and NADP+-induced Ca2+ release
The pH of mammalian Ringer’s solution containing NAD+ (10mM) and NADP+ (5mM) is 3.96 and 3.59, respectively. To evaluate the importance of the acidifying characteristics, the NAD+- and NADP+-supplemented Ringer’s solutions was adjusted to pH7.4, with 5.8 and 8.76mM NaOH, respectively. The effect of pH on Ca2+ release was examined using Ringer’s solution acidified to pH3.96 and 3.59, with 4.04 and 5.55mM HCl, respectively.
Mechanical stimulation of stored [Ca2+] release
Flow cytometry measurements of basal intracellular Ca2+ levels required a loading buffer (RPMI 1640 without phenol red) supplemented with 1μM indo-1-AM (Sigma-Aldrich cat no. 13261), a cell-permeable fluorescent probe for Ca2+ (excitation 330nm, emission 398nm). Trypsinized cultures of DU-145 cells exposed to BA (0, 250, and 1,000μM) for either 24h or 8days were resuspended in indo-1 loading buffer (106 cells per milliliter) and allowed to incubate for 30min at 37°C, 5% CO2, in 12 × 75-mm polystyrene test tubes. Following incubation, [Ca2+]i baseline fluorescence was recorded on a Becton Dickinson BD-LSR Analytic Flow Cytometer with 400 ± 40 (FL5) and 510 ± 20nm (FL4) band-pass filters. Results were presented as ratios of fluorescence (FL5/FL4) at 325nm. Steps prior to instrument recording included vortexing test tubes (5s at speed setting 2.5 on Fisher Scientific model 232) to induce cellular suspension and attaching the sample to the inlet port (5s), followed by 5s of intake before the initiation of data collection. Fluorescence was recorded for 90s, with average indo-1-AM emission documented over the time intervals: 1–30, 31–60, and 61–90s. Data were analyzed using FlowJo Software (Treestar Software, San Carlos, CA, USA).
Analysis of cADPr and NAADP adducts
The boration of cADPr and NAADP was evaluated in vitro using mass spectrometry. All solutions were prepared in fresh ultrapure water and then diluted to the desired concentration in water/acetonitrile/triethylamine (WAT, 50:50:0.2, v/v/v, pH10.3). Borate complexes were prepared by mixing equal volumes of BA and cADPr or NAADP solutions, yielding final concentrations of 500 and 100μM, respectively. To determine the K A, a minimum of 12 different measurements (made on three separate occasions, each with different fresh sample preparations) were made and analyzed. A Perkin-Elmer Sciex (Thornhill, Canada) API III triple quadrupole mass spectrometer fitted with an Ionspray™ source was tuned and calibrated in the positive ion mode, as previously described (Kim et al. 2004). Instrument resolution allowed for a 15–20% valley between the 13C-containing satellites of the polypropylene glycol/NH4 + singly charged calibrant ion, at m/z 906. For the analysis of borate esterification to the nucleotides, the instrument polarity was reversed, and the ion spray voltage was lowered to −3.5kV. Samples dissolved in WAT were introduced (10μL per injection) into a stream of similar solvent entering the ion source (10μL/min). Spectra were collected (profile mode) while the instrument was scanning from m/z 520 to 620 (0.1Da step size, 6ms dwell time, 6.66s/scan, orifice −60V). Representative spectra were computed as the average of all the spectra accrued from each injection, using instrument-supplied software (MacSpec, version 3.3 PE Sciex, Ontario, Canada). The equilibrium constant equations were:
Identification of CD38 mRNA expression
Ribonucleic acid (RNA) was isolated from DU-145 cells using Trizol (Invitrogen) and purified via the RNeasy Mini Kit (Qiagen), according to manufacturer’s instructions. Isolated RNA was treated with 2U deoxyribonucleic acid (DNA)-free (Ambion) to remove DNA contamination. Five micrograms of total RNA was utilized during synthesis of first-strand complementary DNA with Superscript II reverse transcriptase (Invitrogen), according to the manufacturer’s protocol. Primer sequences for CD38, as follows, were designed from accession number NM_001775: 5′-CAACTCTG TCTTGGCGTCAG-3′, forward and 5′-GTTGCTGCAGTCCTTTCTCC-3′, reverse, leading to a 480-bp product. Cycler conditions for CD38 were: 1min at 94°C, followed by 35 cycles of 30s at 94°C, 30s at 59°C, and 1.5min at 70°C. The polymerase chain reaction (PCR) master mix consisted of: 11μL water, 2μL MgCl2 (25mM), 2μL 10× buffer, 0.4μL (0.43mM) deoxyribonucleotide triphosphate, 0.4μL (2U) Taq DNA polymerase (Promega), 0.087μL template DNA, and 1μL (55nM) of either primer (Invitrogen). All PCR reactions were carried out in a PTC-200 Peltier Thermal Cycler (MJ Research). Product confirmation was performed by BglII restriction enzyme digestion (New England Biolabs) with a 1-h incubation at 37°C. Products were loaded onto 1% agarose gels containing ethidium bromide (1μg/mL), and run at 100V for 1h, prior to visualization under UV light and photographing.
Identification of CD38 protein expression
Protein expression was evaluated using immunoaffinity analysis. DU-145 cells were exposed to BA for 1–7days, and then culture plates were washed with phosphate-buffered saline (PBS), and cells scraped off with a rubber policeman and centrifuged at 1,200rpm for 5min. The protein was extracted from cell pellets using a lysis buffer (250mM NaCl, 1% NP40, 50mM HEPES [pH7.0], 5mM ethylenediamine tetraacetic acid, 1mM dithiothreitol, 10% protease inhibitor mixture [Sigma no. P8340]), sonication, and incubation at 4°C for 40min. Samples containing 30μg of protein from the treated cells were loaded and separated on a 10% stacking, 12% separating (sodium dodecyl sulfate polyacrylamide gel electrophoresis) gels for 30min at 100V, followed by 1h (200V), transferred to nitrocellulose membranes for 4h (40V), and shaken overnight in blocking solution (nonfat dry milk 4g, 38mM Tris base, 125mM NaCl, 100μL Tween 20, double-distilled H2O 100mL). Two-hour primary CD38 antibody (Santa Cruz Biotechnology) exposure (1/200 dilution) was followed by a 10min wash in PBS/Tween 20 (0.1%). One-hour secondary antibody exposure (1/1,000 dilution) was followed by 3 × 10min wash in PBS/Tween 20 (0.1%). Probed membranes were submerged in electrochemiluminescence detection reagent (Amersham), wrapped in cellophane, and exposed to X-ray film (Fuji).
Intracellular Ca2+ distribution in resting BA and MBA exposed cells
Live-cell Ca2+ images were obtained from DU-145 cells plated onto glass coverslips (3,000–45,000 cells per square centimeter) and cultured for 8days in media supplemented with BA and MBA (0–1,000μM). On day8, adherent cells were incubated for 30min in culture media supplemented with Fluo-4-AM (4μM) and then washed once with mammalian Ringer’s solution prior to viewing under confocal microscopy.
Statistical analysis
SigmaStat 3.1 statistical software (Systat Software, Point Richmond, CA, USA) was used to determine the most appropriate test based on the data comparisons. Analysis of variance was followed by Bonferroni and Holme tests for the determination of statistical differences on cell proliferation.
Results
Inhibition of cell proliferation by BA and not MBA
A dose-dependent reduction in cell proliferation occurred in cells exposed to 0–1,000μM BA. By contrast, its analog MBA was inactive (Fig. 1). The structure of MBA differs from that of BA in that one of the three hydroxyl groups of BA is substituted with a methyl group. Each point represented the mean of six independent measurements ± sem.

Differential effects of BA and MBA on DU-145 cell proliferation. A dose-dependent reduction in cell proliferation occurred in cells exposed to BA (filled bars) for 8days (100–1,000μM) but not MBA (empty bars). Values are presented as the mean of six independent measurements ± sem. Asterisk represents statistically significant means compared to the 0μM BA control (p < 0.01); double asterisk represents significant differences between BA and MBA at a similar dose (p < 0.01)
Inhibition of NAD+ and NADP+ Ca2+ store release by BA and not MBA
Measurements of Fluo-4-AM showed NAD+ and NADP+ both induced Ca2+ release in DU-145 prostate cancer cells. The Ca2+ transients were inhibited by BA. Lower concentrations of BA were required to inhibit NAD+- than NADP+-stimulated release (Fig. 2a,c). The analog, MBA (1,000μM), did not inhibit either NAD+- or NADP+-induced Ca2+ cytoplasmic transients (Fig. 2b,d).

NAD+- and NADP+-stimulated Ca2+ release in DU-145 cells grown for 8days in the presence of BA or MBA (250 and 1,000μM). a BA treatment (250 and 1,000μM) inhibited NAD+ (10mM) induced Ca2+ release, whereas b MBA (1,000μM) treatment did not. c BA (1,000μM) treatment reduced NADP+ (5mM)-induced Ca2+ release, whereas d MBA (1,000μM) did not. Traces represent mean fluorescence (Fluo-4 AM) of four individual cell responses
Essentiality of pH during NAD+- and NADP+-induced Ca2+ release
The solubilization of NAD+ and NADP+ lowers the pH of Ringer’s solution. When the pH was adjusted to 7.4, neither NAD+ nor NADP+ stimulated Ca2+ release. To assess if the observed Ca2+ release was a result of low pH, Ringer’s solution was acidified to 3.96 and 3.59, the experimentally determined pH of NAD+ (10mM) and NADP+ (5mM) in Ringer’s solution, respectively, and applied to cells. Exposure to low pH alone did not induce intracellular Ca2+ release (data not shown).
Inhibition of mechanical stimulation of Ca2+ Store Release by BA and not MBA
Mechanical stimulation of stored Ca2+ release was induced by vortexing suspended DU-145 cells. Stored Ca2+ release was measured by flow cytometry using the cell-permeable Ca2+ probe indo-1-AM. Ca2+ transients decayed over 90s (Fig. 3a). Exposure to 1,000μM BA blocked the Ca2+ transient, whereas MBA (1,000μM) did not (Fig. 3b). Values represent the mean of three independent fluorescent measurements ± sem.

Release of stored Ca2+ by mechanical stimulation. Flow cytometry analysis of indo-1-AM fluorescence showing cytoplasmic Ca2+ concentrations following the stimulation of release by vortexing (5s) and attaching the sample to the inlet port (10s). Time lapses indicate subanalyses of fluorescent values recorded for 90s. a Exposure to 1,000μM BA prevented Ca2+ release so that cytoplasmic Ca2+ did not rise above basal levels. b MBA was inactive and did not prevent mechanical stimulation from activating Ca2+ release into the cytoplasm. Bars represent the mean of three independent fluorescent measurements ± sem. Asterisk, significantly different from 0μM BA (p < 0.01)
ESI-MS analysis of borate complexed with NAADP and cADPr
In the WAT solvent (pH10.3), the negative ion electrospray ionisation mass spectrometry (ESI-MS) spectra of the 100-μM solutions of cADPr occurred as a prominent singly charged, alkali metal-free molecule [(M–H)−] at m/z 539.9 (calculated 540.0Da; Fig. 4a). With the addition of BA, the spectra revealed additional signals at m/z 565.8, assigned as the singly charged BA–cADPr complex (calculated 566.0Da for (M–H)−) and m/z 583.8 assigned as a borate–cADPr complex (calculated 584.0Da for (M–H)−; Fig. 4b). Since the concentrations of all the different species in the equilibrium (Eq. 1) were either known or could be estimated, the equilibrium constant was calculated (Eq. 2). Using the relative peak areas of the free cADPr, BA–cADPr complex, and borate–cADPr complexes, the calculated affinity constant (K A) was 655 ± 99L/mol.

Negative ion ESI-MS spectra of NAADP, NAADP–BA complexes, cADPr, and cADPr–BA complexes in WAT mixtures at pH10.3. a 100μM cADPr showing an intense (M–H)− signal at m/z 539.9 (calculated 540.0Da). The signals at m/z 549.0 and 557.8 are assigned as unknown impurities in the cADPr sample. b A mixture of 100μM cADPr and 500μM 11B(OH)3 produced signals corresponding to the cADPr–BA complex at m/z 565.8 (calculated 566.0Da) and the cADPr–borate complex at m/z 583.7 (calculated 584.0). K A for the complex formation was calculated as 655 ± 99L/mol. c 100μM NAADP showing an intense (M–H)− signal at m/z 742.7 (calculated 743.1Da). The signal at m/z 764.7 (calculated 765.1) is assigned to a NAADP–Na+ adduct used as a reference standard. d A mixture of 100μM NAADP and 500μM 11B(OH)3 produced signals corresponding to the NAADP–BA complex at m/z 768.7 (calculated 769.1Da). K A for the complex formation was calculated as 82 ± 3.8L/mol
We used NAADP–Na+ as a reference mass. The negative ion ESI-MS spectra of the 100-μM solutions of NAADP revealed a prominent molecular anion for the singly charged, (M–H)− at m/z 742.7 (calculated 743.1Da) and the NAADP–Na+ adduct at m/z 764.7 (calculated 765.1Da; Fig. 4c). Following the addition of 11B(OH)3 (500μM final concentration), the spectra revealed additional signals at m/z 768.7, assigned as the singly charged BA–NAADP complex (calculated 769.1Da for (M–H)−; Fig. 4d). The sum areas of the m/z 742.7, 764.7, and 768.7 peaks correspond to the mass spectrometric response from the NAADP injected (1 × 10−9mol). From the known concentrations of NAADP and BA and the measured relative areas of the free nucleotide and borate-complexed nucleotide, the concentration of the BA–NAADP complex was calculated. The calculated K A for the BA–NAADP complex was 82 ± 3.8L/mol.
BA adducts of cADPr and NAADP formed in the cell probably occur in picomolar concentrations. This was below our level of detection, and so the present results do not provide evidence to conclude boration occurs under in vivo conditions (Macgregor et al. 2007).
Expression of CD38 mRNA and protein in BA-treated DU-145 cells
PCR analysis demonstrated that the CD38 (480bp) transcript was present in untreated DU-145 cells (Fig. 5a, lane 2). Product confirmation, via digestion with the restriction enzyme BglII (lane 3), showed the predicted fragments 159 and 321bp. Western blot analysis showed that growing cells in the presence of BA induced a dose-dependent increase in CD38 protein, whereas the exposure to MBA did not (Fig. 5b). We examined three prostate tumor cell lines (DU-145, LNCaP, and PC-3) and showed they all expressed CD38, whereas our two nontumorigenic prostate cancer cell lines (RWPE-1 and PWR-1E) did not (Fig. 5b).

a CD38 expression in BA-treated DU-145 cells and alternative prostate cell lines. a CD38 mRNA in nontreated DU-145 cells; lanes: marker [M], CD38 PCR product [CD38] and restriction enzyme digest of CD38 PCR product with BglII (BglII). b Immunoaffinity bands (Westerns) to CD38 protein. Top to bottom, DU-145 cells exposed to BA (0–1,000μM) for 1–3days; dose-dependent increase in CD38 protein in cells exposed to BA for 7days; CD38 protein levels were unchanged by exposure to MBA for 7days; CD38 expression evident in all tumorigenic (DU:DU-145; LN:LNCaP; PC:PC-3) prostate cells lines but not in nontumorigenic (RW:RWPE-1; PW:PWR-1E) prostate cell lines
Intracellular Ca2+ distribution in BA and MBA exposed DU-145 cells
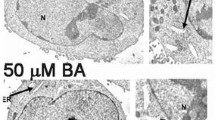
Fluorescent imaging of intracellular Ca2+ revealed that the exposure of growing DU-145 cells in 250 and 1,000μM BA resulted in the redistribution of Ca2+ into small vesicles that accumulated at the periphery (Fig. 6). This did not occur when cells were exposed to MBA (Fig. 6).

Fluo-4-AM was used to visualize intracellular Ca2+ distribution and concentration in DU-145 cells exposed for 8days to BA and MBA. a Green fluorescent Ca2+ indicator was uniformly distributed within DU-145 cells at 0mM BA. b, c Fluo-4 became localized within vesicles when exposure was increased to 250 and 1,000μM BA. d–f The distribution of fluo-4 was unchanged by incubating DU-145 cells in MBA (250 and 1,000μM)
Discussion
Coordinated cell growth regulation is a major theme in B biology. Autoinducer 2 is a quorum-sensing signaling molecule released by bacteria that requires a single B atom for activity (Chen et al. 2002). Autoinducer 2 binds to the surface receptor Lux P, and when a population reaches a “quorum,” it coordinates gene expression and behavior among cells of different species to enable biofilm formation. Plants also require B for growth, flowering, and seed formation (Blevins and Lukaszewski 1994). In animals, concentrations less than 10μm B impair embryonic growth in rainbow trout embryos and the initial cleavages of Zebrafish embryos (Eckhert 1998; Rowe and Eckhert 1999). Low maternal dietary B levels lead to malformations in Xenopus embryos and impaired development of cultured mouse trophoblasts (Fort et al. 1999; Lanoue et al. 1998). In this paper, we show that concentrations of 100μM BA and greater inhibit proliferation of DU-145 cells in a dose-dependent manner achieving near-complete inhibition at 1,000μM BA (Fig. 1). These results in DU-145 cells are in concert with whole-animal toxicology studies. The critical effect used for calculating the tolerable intake and reference dose is the amount of BA ingested by pregnant rats that causes a 5% reduction in pup birth weight (Allen et al. 1996). The NOAEL for BA is based on normal rat pup birth weight and corresponds to a maternal blood concentration of 123μM BA, whereas the lowest observed adverse effect level corresponds to 275μM BA (Price et al. 1997). Using the IPCS regression curve to calculate blood concentrations, we estimate that atrophy of the seminiferous tubules of rats occurs at 1,000μM BA (Fail et al. 1998; IPCS 1998).
Calcium ion waves formed by the release of Ca2+ from endoplasmic reticulum stores into the cytoplasm are important modulators of eukaryotic cell proliferation (Whitfield 1995; Kao et al. 1990; Steinhardt and Alderton 1988; Xu et al. 2003). Several cytoplasmic Ca2+-binding proteins, including troponin C, calmodulin, annexin II, and calpain, serve as intermediaries that regulate progress through the cell cycle (Choi et al. 2006). For example, in vascular smooth muscle cells, an elevation of intracellular Ca2+ concentration is required for G1- to S-phase cell cycle progression. The presence of a highly conserved 22 amino acid N-terminal calmodulin-binding motif in mammalian cyclin E is responsive to physiological changes in cytoplasmic Ca2+ and activates the kinase of mouse and human cyclin E/CDK2 (Choi et al. 2006).
The inhibition of Ca2+ store pumps in human prostate LNCaP cells inhibits the proliferation as well as the transition to androgen independence (Abeele et al. 2003). In the present study, the exposure of DU-145 cells to 250μM BA significantly reduced NAD+-induced cytoplasmic Ca2+ transients, and 1,000μM BA completely abolished them (Fig. 2a). BA was less effective in inhibiting NADP+-stimulated Ca2+ release showing partial inhibition at 1,000μM (Fig. 2c). This could in part be due to the lower affinity constant (K A) of BA for NAADP. Ca2+ transients induced by mechanical stimulation were also blocked by BA (1,000μM). In calf pulmonary artery endothelial cells, mechanical stimulation is mediated by phospholipase C and the IP3 calcium channel (Moerenhout et al. 2001). DU-145 cells express the IP3 receptor so the effect of BA on the Ca2+ release apparatus is not limited to the CD38 Ca2+ release pathway. Further studies are necessary to determine if BA interacts directly with the ryanodine and IP3 Ca2+ channels or their accessory proteins.
Mammalian cells secrete NAD+ for transport on connectin 43 to adjacent cells where it binds to CD38 and is converted into cADPr (Bruzzone et al. 2001). CD38 is a multifunctional enzyme present on the surface and internal membranes of cells that regulates intracellular function through Ca2+ messengers (Lee 2006). At neutral pH, ADP-ribosyl cyclase catalyzes the conversion of NAD+ to cADPr, which then binds to the ryanodine receptor complex releasing the accessory protein, FKBP12. This results in a conformational change that opens a channel in the ryanodine receptor allowing stored Ca2+ in the endoplasmic reticulum to enter the cytoplasm (Li et al. 2001). At acidic pH, ADP-ribosyl cyclase catalyzes the conversion of NADP+ to NAADP with the maximum rate occurring at pH4.0 (Lee 2006). In the present study, acidic conditions were necessary for NAD+ and NADP+ to stimulate Ca2+ release but not sufficient on their own. The acidic dependence of NAADP is in agreement with its reported ability to control Ca2+ release from acidic stores (Macgregor et al. 2007; Zhang and Li 2007). We also show that BA formed complexes with cADPr and NAADP (Fig. 4). Our instrumentation was not able to resolve structures in the nanomolar range to determine if these complexes occur in prostate epithelial cell extracts. Achieving this sensitivity would make it possible to determine if cADPr–BA and NAADP–BA complexes are formed in cells. We previously reported that NAD–BA is a noncompetitive inhibitor of ADP-ribosyl cyclase activity in Aplysia, a sea hare that lives in a high boron environment (Kim et al. 2006). The K i for this was 40.5mM BA, a level 1,000-fold higher than human plasma. The K i of human ADP-ribosyl cyclase for BA has not been reported.
The exposure of growing DU-145 cells to BA induced a dose-dependent increase in CD38 protein expression that coincided with reduced proliferation (Fig. 5b). The data do not permit us to discern if this increase represented a feedback response to BA’s dampening of CD38 Ca2+-mediated signals. We previously reported that the nontumorigenic prostate cell lines (RWPE-1 and PWR-1E) were fourfold less sensitive to BA-induced proliferative inhibition (Barranco and Eckhert 2004). In this paper, we show that all three tumor cell lines expressed CD38, whereas the two nontumorigenic cell lines tested did not (Fig. 5b). This may explain the difference.
Exposure to BA (250 and 1,000μM BA) resulted in the formation of vesicles containing Ca2+ that accumulated at the cell periphery (Fig. 6). We previously reported these vesicles stain positive for the acidic dye Lyso Tracker Green (Molecular Probes), and their appearance is associated with acidification of the culture media (Barranco and Eckhert 2006). They resemble the punctate staining pattern of NAADP-responsive acidic Ca2+ stores in the heart and the endosomes that degrade BA uptake transporter BOR1 after Arabidosis thaliana is exposed to 100μM BA, a level that exceeds its nutritional requirement (Macgregor et al. 2007; Tankano et al. 2005).
Boron is ubiquitous in the environment, and both bacteria and plants have exploited the element to regulate cell proliferation, growth, differentiation, and intercellular behavior. We therefore examined the ability of DU-145 cells to discriminate between BA, the form present in biological fluids, and a synthetic analog. We selected MBA since arylboronic acids undergo biological catalyase by oxygenases (Negret-Raymond et al. 2003). In contrast to BA, MBA did not bind to cADPr or NAADP or inhibit Ca2+ store release, cell proliferation, or CD38 expression. MBA’s lack of activity cannot be explained by differences in permeability. Animal cell plasma membranes have been shown, along with plant membranes, to be permeable to BA via passive diffusion allowing immediate access to intracellular sites (Dordas and Brown 2001). Size and solubility govern their capacity to diffuse passively through plasma membranes and both BA (water solubility = 0.75M; molecular weight = 61.83) and MBA (water solubility = 5.4M; molecular weight = 59.86) are water soluble and of similar size. The failure of MBA to mimic BA together with observed concentration-dependent effects of BA on prostate epithelial cell proliferation and embryonic growth in fish and amphibians provides strong evidence that vertebrate cells have retained the ability to recognize and respond to environmental BA concentrations.
In conclusion, we hypothesize that the toxicity of BA/borates stems from the ability of high concentrations to inhibit stored Ca2+ release. Plant cells express uptake and efflux BA transport proteins that allow them to regulate BA homeostasis and protect against toxicity, but this mechanism can be overloaded and has not been shown to exist in animals (Sutton et al. 2007). Defining the low and high concentrations of BA that trigger the activation of BA uptake and efflux transporters in plants would provide a glimpse of the BA concentration range biology considers optimum for health.
References
Allen BC, Strong PL, Price CJ, Hubbard SA, Daston GP. Benchmark dose analysis of developmental toxicity in rats exposed to boric acid. Fundam Appl Toxicol. 1996;232:194–204.
Abeele FV, Shuba Y, Roudbaraki M, Lemonnier L, Vanoverberghe K, Mariot P, et al. Store-operated Ca2+ channels in prostate cancer epithelial cells: function, regulation, and role in carcinogenesis. Cell Calcium. 2003;33:357–73.
Armstrong TA, Spears JW, Crenshaw TD, Nielsen FH. Boron supplementation of a semipurified diet for weanling pigs improves feed efficiency and bone strength characteristics and alters plasma lipid metabolites. J Nutr. 2000;139:2575–81.
Barranco WT, Eckhert CD. Boric acid inhibits prostate cancer cell proliferation. Cancer Lett. 2004;216:21–9.
Barranco WT, Eckhert CD. Cellular changes in boric acid-treated DU-145 prostate cancer cells. Br J Cancer. 2006;94:884–90.
Barranco WT, Hudak PF, Eckhert CD. Evaluation of ecological and in vitro effects of boron on prostate cancer risk. Cancer Causes Control. 2007;18:71–77 (Publisher Erratum. Cancer Causes Control. 2007; 18: 583–4).
Blevins DG, Lukaszewski KM. Proposed physiologic functions of boron in plants pertinent to animal and human metabolism. Environ Health Perspect. 1994;102(Suppl 7):31–3.
Bruzzone S, Franco L, Guida L, Zochi E, Contini P, Bisso A, et al. A self-restricted CD38-connexin 43 cross-talk affects NAD+ and cyclic ADP-ribose metabolism and regulates intracellular calcium in 3T3 fibroblasts. J Biol Chem. 2001;276:48300–8.
Chapin RE, Ku WW, Kenney MA, McCoy H, Gladen B, Wine RN, et al. The effects of dietary boron on bone strength in rats. Fundam Appl Toxicol. 1997;35:205–15.
Chen X, Schauder S, Potier N, Dorsselaer AV, Pelczer I, Bassler BL, et al. Structural Identification of a Bacterial Quorum Sensing Signal Containing Boron. Nature. 2002;415:545–9.
Choi J, Chiang A, Tauler N, Gros R, Pirani A, Husain MA. Calmodulin-binding site on cyclin E mediates Ca2+-sensitive G1/S transitions in vascular smooth muscle cells. Circ Res. 2006;98:1273–81.
Cui Y, Winton MI, Zhang Z-F, Rainey C, Marshall J, De Kernion JB, et al. Dietary boron intake and prostate cancer risk. Oncol Rep. 2004;11:887–92.
Dordas C, Brown PH. Permeability and the mechanism of transport of boric acid across the plasma membrane of Xenopus laevis oocytes. Biol Trace Elem Res. 2001;81:127–39.
Eckhert CD. Boron stimulates embryonic trout growth. J Nutr. 1998;128:2488–93.
Fail PA, Chapin RE, Price CJ, Heindel JJ. General, reproductive, developmental, and endocrine toxicity of boronated compounds. Reprod Toxicol. 1998;12:1–18.
Fort DJ, Propst TL, Stover EL, Murray FJ, Strong PL. Adverse effects from low dietary and environmental boron exposure on reproduction, development, and maturation in Xenopus laevis. J Trace Elem Exp Med 1999;12:175–86.
Gallardo-Williams MT, Chapin RE, King PE, Moser GJ, Goldsworthy TL, Morrison JP, et al. Boron supplementation inhibits the growth and local expression of IGF-1 in human prostate adenocarcinoma (LNCaP) tumors in nude mice. Toxicol Pathol 2004;32:73–8.
Hunt CD, Nielsen FH. Interaction between boron and cholecalciferol in the chick. In: McHowell J, Gawthorne JH, White CL, editors. Trace elements in man and animals. vol 4. Canberra, Australia: Australian Academy of Sciences; 1981. p. 597–600.
IOM. Dietary reference intakes for vitamin A, vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. A report of the Panel on Micronutrients, Subcommittees on Upper Reference Levels of Nutrients and of Interpretation and Use of Dietary Reference Intakes, and the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. Food and Nutrition Board, Institute of Medicine. Washington, DC: National Academy; 2001. p. 13-16–14.
IPCS. Boron, environmental health criteria 204. Geneva, Switzerland: WHO; 1998. p. 61–5.
Kao JP, Alderton JM, Tsien RY, Steinhardt RA. Active involvement of Ca2+ in mitotic progression of Swiss 3T3 fibroblasts. J Cell Biol 1990;111:183–96.
Kim DH, Faull KF, Norris AJ, Eckhert CD. Borate–nucleotide complex formation depends on charge and phosphorylation state. J Mass Spectrometry 2004;39:743–51.
Kim DH, Marbois BN, Faull KF, Eckhert CD. Esterification of borate with NAD+ and NADH as studied by electrospray ionization mass spectrometry and 11B NMR spectroscopy. J Mass Spectrom 2003;38:632–40.
Kim DH, Que Hee S, Norris A, Faull KF, Eckhert CD. Boric acid inhibits ADP-ribosyl cyclase non-competitively. J Chromatogr A 2006;1115:246–52.
Lanoue L, Taubeneck MW, Muniz J, Hanna LA, Strong PL, Murray FJ, et al. Assessing the effects of low boron diets on embryonic and fetal development in rodents using in vitro and in vivo model systems. Biol Trace Elem Res 1998;66:271–98.
Lee HC. Structure and enzymatic functions of human CD38. Mol Med 2006;12:317–23.
Li PL, Tang WX, Valdivia HH, Zou AP, Campbell WB. cADP-ribose activates reconstituted ryanodine receptors from coronary arterial smooth muscle. Am J Physiol Heart Circ Physiol 2001;280:H208–15.
Macgregor A, Yamasaki M, Rakovic S, Sanders L, Parkesh R, Churchill GC, et al. NAADP controls cross-talk between distinct Ca2+ stores in the heart. J Biol Chem 2007;282:15302–11.
Miwa K, Takano J, Omori H, Seki S, Shinozaki K, Fujiwarai T. Plants tolerate of high boron levels. Science 2007;318:1417.
Moerenhout M, Vereecke J, Himpens B. Mechanism of intracellular Ca2+ wave propagation elicited by mechanical stimulation in cultured endothelial CPAE cells. Cell Calcium 2001;29:117–23.
Negret-Raymond AC, Weder B, Wacket LP. Catabolism of arylboronic acids by Arthrobacter nicotinovorans strain PBA. Appl Environ Microbiol 2003;69:4263–7.
Penland JG. Dietary boron, brain function, and cognitive performance. Envir Health Perspect 1994;102(Suppl 7):65–72.
Poenie M, Alderton J, Tsien RY, Steinhardt RA. Changes of free calcium levels with stages of the cell division cycle. Nature 1985;315:147–9.
Price CJ, Strong PL, Murray FJ, Goldberg MM. Blood boron concentrations in pregnant rats fed boric acid throughout gestation. Reprod Toxicol 1997;11:833–42.
Rowe IR, Bouzan C, Nabili S, Eckhert CD. The response of trout and zebrafish embryos to low and high boron concentrations is U-shaped. Biol Trace Elem Res 1998;66:261–70.
Rowe RI, Eckhert CD. Boron is required for zebrafish embryogenesis. J Exp Biol 1999;202:1649–54.
Steinhardt RA, Alderton J. Intracellular free calcium rise triggers nuclear envelope breakdown in the sea urchin embryo. Nature 1988;332:364–6.
Sutton T, Bauman U, Hayes J, Collins NC, Shi B-J, Schnurbusch T, et al. Boron-toxicity tolerance in barley arising from efflux transporter amplification. Science 2007;318:1446–9.
Tankano J, Kyoko Miwa K, Yuan L, von Wiren N, Fujiwara T. Endocytosis and degradation of BOR1, a boron transporter of Arabidopsis thaliana, regulated by boron availability. Proc Natl Acad Sci USA 2005;102:12276–81.
Whitfield JF. Calcium in cell cycles and cancer. 2nd ed. Boca Raton, FL: CRC; 1995. p. 1–214.
Xu N, Luo KQ, Chang DC. Ca2+ signal blockers can inhibit M/A transition in mammalian cells by interfering with the spindle checkpoint. Biochem Biophys Res Commun 2003;306:737–45.
Zhang F, Li P-L. Reconstitution and characterization of a nicotinic acid adenine dinucleotide phosphate (NAADP)-sensitive Ca2+ release channel from liver lysosomes of rats. J Biol Chem 2007;282:25259–69.
Acknowledgments
We wish to thank Michael Gulrajani in charge of the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility. This facility is supported by the National Institutes of Health awards CA-16042 and AI-28697 and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA.
Funding
The work for this grant was funded in part by University of California Toxic Substances Research and Training Program and the US Army Medical Research and Material Command Prostate Cancer Research Program Idea Grant to C.E., DAMD 17-03-0067.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barranco, W.T., Kim, D.H., Stella Jr., S.L. et al. Boric acid inhibits stored Ca2+ release in DU-145 prostate cancer cells. Cell Biol Toxicol 25, 309–320 (2009). https://doi.org/10.1007/s10565-008-9085-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-008-9085-7