
Abstract
The esterification of cyclohexene with formic acid is the first step for indirect hydration to cyclohexanol. In the work reported in this study, phosphotungstic acid (HPW) was incorporated into the metal–organic framework Cu3(BTC)2 through a one-step hydrothermal synthesis method to catalyze the esterification reaction. The crystal structure of composite HPW–Cu3(BTC)2 was modulated during the preparation process to convert the catalyst from the {111} crystal facets of the original regular octahedral structure to the {100} crystal facets of the cubic structure using p-toluic acid (pTA) as the regulator. The conversion of the crystal facets reduces the formation through holes while exposing more active sites of HPW. When the mass ratio of the modulator to the organic ligand terephthalic acid was 20, the crystal facets of the catalyst were completely covered by the {100} crystal facets. After the crystal transformation, the conversion of cyclohexene was increased to 86.8%, and the selectivity of cyclohexyl formate was 84.2%. Agglomeration occurred after the catalyst was used twice in the esterification reaction.
Graphic Abstract
HPW was incorporated into the metal-organic framework Cu3(BTC)2 through a one-step hydrothermal synthesis method and used for catalyzing the esterification reaction of cyclohexene with formic acid. The crystal structure of composite HPW–Cu3(BTC)2 was modulated during the preparation process to convert the catalyst from the {111} crystal facets of the original regular octahedral structure to the {100} crystal facets of the cubic structure using p-toluic acid (pTA) as the modulator.

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Cyclohexanol is an important raw material that is used widely in the organic-chemical, paint, and textile industries. Global industrial production methods for cyclohexanol include cyclohexane oxidation [1], phenol hydrogenation [2], and cyclohexene hydration [3]. There are significant safety hazards in the phenol hydrogenation process, and few companies have used this method to prepare cyclohexanol. Cyclohexane oxidation is the most widely used preparation method of cyclohexanol in the World. The production route is relatively mature, but the process has higher temperature and pressure; the conversion rate of cyclohexane is only approximately 4%, and a large amount of waste alkali liquor causes serious environmental pollution.
The cyclohexene direct hydration process was developed by Asahi Kasei Corp. (Japan), and the main processes are as follows: Partial hydrogenation of benzene is performed to give a mixture of benzene, cyclohexene, and cyclohexane (n:n:n = 60:32:8). First, the mixture enters the de-watering section to take off the water, and then the benzene in the mixture is first separated by extraction distillation using dimethylacetamide (DMCA) as an extractant and recycled. The mixture of cyclohexene and cyclohexane is also separated through extractive distillation by the same extractant, DMCA. The isolated cyclohexene enters the hydration section through the cyclohexene refining column to produce cyclohexanol. The brief preparation process is shown in Fig. 1. At present, there are two major problems in the process: First, since the relative volatility of cyclohexene and cyclohexane is close to 1, the energy required to separate cyclohexene in the early stage is extremely large; second, the direct hydration method of cyclohexene is easily restricted by the thermodynamics of the reaction itself [4], and cyclohexene has low solubility in water. These shortcomings result in a single-pass conversion rate of cyclohexene of less than 10%. The method of cyclohexene indirect hydration to cyclohexanol [5] solves the problems in cyclohexene direct hydration, which is worthy of in-depth research and discussion. Indirect hydration uses a two-step synthetic route to subtly circumvent major shortcomings in direct hydration: esterification of formic acid with cyclohexene and hydrolysis of cyclohexyl formate to cyclohexanol. The relative volatility of cyclohexyl formate from the first-step reaction and cyclohexane is relatively large, and the two are easier to separate, which solves the problem of large separation energy in the direct hydration process. Since the equilibrium constant of the esterification reaction (K = 53.3, 25 °C) well solves the thermodynamic limitations of direct hydration, the conversion rate is improved qualitatively. The second-step hydrolysis is an easy-processing reaction.

Direct hydration production methods for cyclohexanol
The esterification reaction of cyclohexene with formic acid is a typical acid-catalyzed reaction. Conventional esterification catalysts involve inorganic acids such as H2SO4, HCl, HF, H3PO4, and ClSO2OH. However, due to their erosive nature, the excess acids must be neutralized after completion of the reaction, which causes disposal problems of the resulting neutralization products. Heteropolyacids [6] (HPAs), a solid species of higher Brønsted acidity, were introduced as a substitute catalyst featuring higher catalytic activity. Among various HPA structures, Keggin-type HPAs, especially Keggin phosphotungstic acid (H3PW12O40, HPW), have received increasingly more attention due to their simple preparation and high acidity [7]. The esterification reaction is currently carried out by HPW as a catalyst, for example, via esterification of acetic acid with alkyl alcohol [8], esterification of acetic acid with n-butanol [9], preparation of ethyl acetate [10], and esterification of acetic acid and glycerol [11], and so on. However, HPW also has the disadvantage of small surface area (< 10 m2/g) [12]. This drawback has been initially solved by its immobilization on magnetite nanoparticles [13, 14], tin oxide material [15], silica material [16], or a mesoporous molecular sieve [17, 18]. However, due to the high solubility of HPW in polar solvents, HPW physically adsorbed on the surface of the support can be easily leached out [19,20,21].
Metal–organic-framework materials (MOFs) refer to porous crystal materials with periodic network structure formed by self-coordination of organic ligands and metal ions, which have high porosity and surface area [22]. The structural unit contains metal-inorganic and -organic parts and has strong structural controllability and subsequent chemical modification characteristics. By changing the organic ligand or metal unit, fine adjustment of the material at the atomic scale can be achieved, as well as high regulation of the size, shape, and internal environment of the pore. MOF materials have great practical applications in, for example, gas storage and separation [23, 24] and catalysis [25]. MOF materials have been widely used today as a catalytically active component encapsulating carrier [26], which has the following advantages: (i) high tunable adsorption performance, (ii) ability to form active sites of different strength in the framework, and (iii) good accommodation properties for guest molecules and strong ion-exchange capacity. In recent years, the study of MOF loading of Keggin phosphotungstic acid has gained increasing attention due to the advantages of moderate dispersion of HPW, easy preparation and recovery, and less loss of active components. The stability and solubility of Cu3(BTC)2-supported HPW catalyst are greatly affected by solvents [12, 27], so its catalytic reaction under polar conditions commands special attention. What is more, due to the interaction between HPW and Cu2+ (under acidic conditions) [28] during the catalyst synthesis process, some of the active sites of HPW are masked by Cu2+ ions, which results in lower activity of the catalyst than pure HPW.
In recent years, the morphology of crystal materials and the control of crystal planes have received significant attention. The crystal properties can be further enhanced and optimized by changing or regulating the atomic structure of the crystal surface. In this vein, the synthesis of crystalline MOFs with specific sizes and/or morphologies is increasingly active, the most representative of which are MOF-5 [29], ZIF-8 [30], and Cu3(BTC)2 [29, 31]. However, these materials are only used in the storage or separation of gases. There are fewer applications in catalysis. Liu et al. [32] obtained good experimental results in the study of the influence of different crystal morphologies on the catalytic activity of MOFs. The conversion rate of fatty acid (C12–C22) in the reaction of catalytic biodiesel reached over 90% ({100} facets) and < 22% ({111} facets). This suggests that it is feasible to increase the exposure probability of active sites by regulating the crystal-plane structure of MOFs. Umemura et al. [33] used lauric acid as a modulator to control the crystal morphology of Cu3(BTC)2 and explored the transformation process of crystal and the mechanism of crystal growth. This provides a theoretical basis for the crystal transformation of HPW–Cu3(BTC)2. The competitive action of the monohydroxy additive results in a change in the nucleation process, and the rate of the nucleation process can be adjusted by adjusting the concentration of the modulator. However, lauric acid has a disadvantage of being used in a large amount as a modulating agent, and the catalyst yield is low.
In the work reported in this study, the composite catalyst material HPW–Cu3(BTC)2 was prepared by a thermal synthesis method, reported by Chowdhury [34]. To improve the catalytic activity of the composite in the esterification of cyclohexene and formic acid, pTA [32] was used as a modulator to control the growth rate of the crystal face from the {111} facets of the triangle to the {100} facets of the square, which is beneficial for the exposure of active ingredients of HPW. The influence of modulator dosage on the structure of the composite catalyst was investigated, and the catalytic performance of the obtained samples was evaluated in the esterification reaction of cyclohexene and formic acid.
2 Experimental Section
2.1 Materials and Chemicals
Phosphotungstic acid (HPW) [analytical reagent (AR) grade], trihydrate and copper nitrate (AR grade, ≥ 99.5%), 1,3,5-benzenetricarboxylic acid (AR grade, > 98%), and pTA (AR grade, > 98%) were purchased from Shanghai Macklin Biochemical Co., Ltd (China). Benzene, cyclohexane, cyclohexene, and formic acid (AR grade, 98%) were purchased from Aladdin Chemical Reagent Co., Ltd. (China). Ethanol, acetone, chloroform, and chlorobenzene were purchased from Sinopharm Chemical Reagent Co., Ltd. (China).
2.2 Cu 3 (BTC) 2 Synthesis
Cu(NO3)2·3H2O (2.4 g) was dissolved in 40 mL of absolute ethanol and H3BTC (2.0 g) was dissolved in 40 mL of absolute ethanol and stirred separately for 30 min. The two solutions were then mixed and stirred 30 min. The mixtures were transferred into a Teflon™-lined autoclave and subjected to crystallization at 393 K for 8 h under static conditions. After the desired time was reached, the synthesis mixture was cooled naturally to room temperature. The solid product was filtered and washed with ethanol and dried at 373 K for 12 h in vacuum. The resulting material is referred to as Cu3(BTC)2.
2.3 HPW-Cu 3 (BTC) 2 Synthesis
Cu(NO3)2·3H2O (0.24 g), HPW (0.2 g), and pTA (0, 0.4, 1.0, 2.0, and 4.0 g) were dissolved in 40 mL of absolute ethanol, stirred for 30 min, and then added into H3BTC (0.2 g) (m(pTA)/m(BTC) = 0, 2, 5,10, and 20). This mixture was stirred for an additional 30 min and then transferred into a Teflon™-lined autoclave and subjected to crystallization at 393 K for 8 h under static conditions. The resulting solid was washed with a mixture of absolute ethanol and deionized water and dried at 373 K for 12 h in vacuum. The resulting blue solids are referred to as HPW–Cu3(BTC)2, HPW–Cu3(BTC)2–P2, HPW–Cu3(BTC)2–P5, HPW–Cu3(BTC)2–P10, and HPW–Cu3(BTC)2–P20.
2.4 Characterization
Fourier-transform infrared (FTIR) spectra were measured using a Nicolet-380 FTIR spectrometer (Thermo Nicolet Corporation, USA) at room temperature. Analyses were performed using a KBr pellet technique in the spectral range 400–4000 cm−1. X-ray diffraction (XRD) was carried out using a Rigaku D/Max-2500 X-ray diffractometer and Cu Kα radiation at 40 kV and 30 mA. XRD patterns were recorded between 2θ = 0.7°–5.0° and 5.0°–40°. The specific surface area and pore structure of the samples were measured by N2 adsorption using a NOVA-2200e instrument. The surface area was calculated by the Brunauer–Emmett–Teller (BET) method. The catalyst morphology was characterized by scanning electron microscopy (SEM) using a JEOL-6610LV field-emission scanning electron microscope. Temperature-programmed desorption of ammonia (NH3-TPD) measurements were carried out with 100-mg samples on a multitask TPD system (Chembet 3000, Quantachrome Instruments, Boynton Beach, FL, USA). After pre-treatment in He (473 K, 100 mL/min, 60 min), the samples were cooled to 398 K, and the carrier gas switched to a NH3–He gas mixture and the NH3 adsorption of the sample started. After the ammonia gas was saturated, the mixture gas was switched to He carrier gas, the sample purged for 0.5 h, and then placed in the He gas stream. The temperature was programmed to 773 K for analysis (temperature ramp of 10 K/min). The catalyst thermostability was measured by thermogravimetric analysis (TGA) using a NETZSCH TG209 analyzer. The temperature was increased from ambient temperature to 1273 K under air/N2 flow at 10 K/min. The detection of W content in the catalyst was carried out by inductively-coupled-plasma (ICP) analysis using an ICP atomic emission spectrometer (Thermo Jarrell Ash IRIS Advantage 1000, USA).
2.5 Catalytic-Activity Test
Benzene, cyclohexane, and cyclohexene were mixed as raw material according to the actual ratio of the industrial production process. Esterification with formic acid was then carried out in a 50-mL flask fitted with a magnetic stirrer. In a typical experiment, 8.2 g of cyclohexene (0.1 mol), 14.6 g of benzene, 2.1 g of cyclohexane (n:n:n = 32:60:8), and 13.3 g of formic acid (0.3 mol) were introduced into the flask with 3.1 g of catalyst. The system was heated to 343 K for 8 h with stirring. After the reaction, the catalyst was separated by centrifugation and the solid washed with acetone and chloroform to eliminate impurities. The separated reaction solution was dissolved in acetone and stirred sufficiently to form a phase. The organic phases were then identified and quantified by gas chromatography using an Agilent 7890 instrument with an HP-5 capillary column and flame ionization detector (FID) indicator, using chlorobenzene as an internal standard.
3 Results and Discussion
3.1 SEM Morphology
Figure 2 shows scanning electron micrographs of the catalyst sample. It can be seen in Figs. 2a, b that the Cu3(BTC)2 and HPW–Cu3(BTC)2 samples are typical regular octahedral structures. It is apparent that a significant change in the crystal form of the catalyst occurs as the initial amount of pTA is increased. When m(pTA)/m(BTC) = 2, the crystal form of the catalyst still maintains the shape of a regular octahedron with a grain size of approximately 35 μm, as shown in Fig. 2c. This indicates that pTA participates in the coordination process during crystal nucleation, but the growth rate of the {111} crystal facets is much smaller than that of the {100} crystal facets, resulting in the catalyst crystals all being {111} crystal facets. As m(pTA)/m(BTC) increases, the crystal form of the catalyst gradually transforms from the octahedron of the {111} crystal facets to the cube of the {100} crystal facets. When m(pTA)/m(BTC) = 20, as shown in Fig. 2f, the catalyst is completely covered by the {100} crystal facets, the crystal grains are approximately 1 μm in size, and the crystal form of the catalyst is uniform.

SEM micrographs of samples: a Cu3(BTC)2; b HPW–Cu3(BTC)2; c HPW–Cu3(BTC)2–P2; d HPW–Cu3(BTC)2–P5; e HPW–Cu3(BTC)2–P10; f HPW-Cu3(BTC)2-P20
3.2 XRD Analysis
The diffraction peaks appearing at 2θ = 6.72°, 11.54°, 13.4°, 17.4°, 19.0°, and 25.9° are typical diffraction peaks of Cu3(BTC)2 obtained by XRD (Fig. 3). In Fig. 3a, the small diffraction peak at 5.6° corresponds to (111) reflection. This variation has been also observed in a previous contribution [35, 36] and can be associated with the degree of hydration. After the addition of HPW, the diffraction peak at 5.6° is significantly enhanced, which may be due to the crystal water in HPW has an enhanced effect on the hydration degree of Cu3(BTC)2. When pTA is not added, due to the addition of HPW, which results in a weak de-protonation ability of the H3BTC, the diffraction peak of 6.72° is not obvious. With increasing m(pTA)/m(BTC), the diffraction peak intensity is gradually increased for 2θ = 6.72°. It is pointed out in the literature [32] that the diffraction peak appearing at 6.72° is that of the {200} crystal facets. The ratio of the (200)/(111) diffraction peaks gradually increases, which indicates that the crystal facets of the catalyst are gradually changing from {111} to {100} crystal facets. This indicates that the addition of the new ligand pTA can indeed achieve a crystal form transformation and that XRD and SEM characterizations can be cross-validated.

XRD patterns of samples: a Cu3(BTC)2; b HPW–Cu3(BTC)2; c HPW–Cu3(BTC)2–P2; d HPW–Cu3(BTC)2–P5; e HPW–Cu3(BTC)2–P10; f HPW–Cu3(BTC)2–P20
3.3 Nitrogen Adsorption–Desorption Isotherms
Figure 4 shows nitrogen physical adsorption–desorption curves of the catalyst samples. Figure 4a shows a typical microporous adsorption–desorption isotherm. When m(pTA)/m(BTC) = 2 or 5, the catalyst still maintains the H1-type hysteresis loop. Because the amount of pTA added is low, the de-protonation ability is weak, and some through-and-through holes still appear in the formed pore structure. As m(pTA)/m(BTC) increases, the hysteresis loop of the catalyst sample becomes increasingly smaller, which indicates that the pore structure of the catalyst is more uniform when the crystal facets of the catalyst are gradually converted into {100} crystal facets. As m(pTA)/m(BTC) is increased from 2 to 5, the specific surface area of the catalyst is increased from 533 to 712 m2/g, and the pore volume is gradually increased, as shown in Table 1. This indicates that the crystal face change of the catalyst has a certain influence on the pore structure.

Nitrogen physical adsorption–desorption curves of catalyst samples: a Cu3(BTC)2; b HPW–Cu3(BTC)2; c HPW–Cu3(BTC)2–P2; d HPW–Cu3(BTC)2–P5; e HPW–Cu3(BTC)2–P10; f HPW–Cu3(BTC)2–P20
3.4 FTIR Characterization
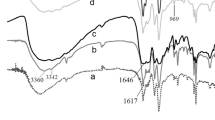
The catalyst sample structure was examined by FTIR spectroscopy Fig. 5a. Bands at 1080, 985, 887, and 804 cm−1, which are fingerprints of the Keggin structure of HPW, are usually assigned to vas (P–O), vas (W=O), and vas (W–Ob–W) in corner-shared octahedra, and vas (W–Oc–W) in edge-shared octahedra [37]. Cu3(BTC)2 shows IR bands at 1641 cm−1, which are assigned to vas V–(C=O) (stretching vibration), while those at 1589 and 1373 cm−1 are assigned to vas V–(C=O–OH) (asymmetric and symmetric stretching vibrations) and 1450 and 792 cm−1 to vas –CH on benzene (stretching and flexural vibrations). The IR band at 492 cm−1 was assigned to Cu–O (flexural vibration) separately [38]. By comparing the IR spectrum of the catalyst material loaded with HPW and the pure carrier, it can be found that the supported catalyst has the characteristic absorption peak of phosphotungstic acid and the carrier, which indicates that the HPW has been successfully loaded onto the carrier Cu3(BTC)2.

a FTIR spectra of catalyst samples and b partially amplified FTIR spectra of the catalyst sample
The IR characterization map was locally enlarged at 700–1100 cm−1 to obtain Fig. 5b. vas W=O, W–Ob–W, and W–Oc–W are all shifted. vas W=O is shifted to low wave number (985–979 cm−1), a phenomenon that indicates that there is hydrogen bonding [39] between the central atom of the HPW and the carbonyl atom of the organic ligand trimesic acid. vas W–Ob–W and W–Oc–W are shifted to high wave numbers (887–900 and 804–829 cm−1, respectively), which indicates that the bridge oxygen atom forms a chemical bond with Cu2+. This phenomenon indicates that the HPW bridge oxygen atom plays a coordination role with the metal ion during the formation of the catalyst. Primarily, the electron cloud density of the bridging oxygen atom in the heteropolyanion is larger than that of the terminal oxygen atom, which can attract Cu2+ and produce a coordination effect. The terminal oxygen atom is mainly responsible for the B acidic site of HPW, which also proves that during the synthesis of the catalyst the acidic sites of catalyst are not destroyed, and the main catalytic effect is still the B acidic site of the HPW. The shift of these groups indicates that there is a strong interaction between HPW and the support. The introduction of HPW is not only a simple encapsulation effect, but, more likely, HPW participates in the nucleation process of the catalyst [40, 41]. The synthesis process diagram of the catalyst and the influence of pTA dosage on the crystal surface of the catalyst are shown in Fig. 6. First, Cu2+ is attracted to the bridging oxygen atom of the heteropolyacid. With the addition of pTA and H3BTC, pTA will first be covered around Cu2+ and HPW, due to methylbenzoic acid having only one carboxyl group and that it cannot form a cage structure, which is gradually exchanged by H3BTC. The crystal growth mode of the catalyst changes during the exchange process. When m(pTA)/m(BTC) = 20, the growth rate of the {100} crystal facets is less than that of the {111} crystal facets. Finally, a catalyst mainly composed of {100} crystal facets is formed.

Proposed modulation mechanism of pTA for catalyst synthesis
3.5 Thermogravimetric Analysis
Figure 7 presents a graph showing the thermogravimetric and differential thermal curves of the catalyst samples. It can be seen from the TGA curve that the catalyst sample has three stages of weight loss, approximately 346–364 K for the removal of physically adsorbed water, approximately 443 K for the removal of chemisorbed water from the catalyst sample, and approximately 603–633 K for the removal of HPW and the collapse of the catalyst skeleton, because HPW is the nucleation center of the catalyst. Li et al. [42] reported that the addition of hydrophobic functional groups would strengthen the water stability of MOFs. It can be seen from the figure that in the range 346–364 K, as the amount of pTA increases, the physical adsorption water of the catalyst gradually decreases. It can be seen from the figure that in the range 346–364 K, as the amount of pTA increases, the physical adsorption water of the catalyst gradually decreases. The weight loss percentage of physically adsorbed water for HPW–Cu3(BTC)2 is 28.9%, while that for HPW–Cu3(BTC)2–P20 is 19.7%. The reason is that the hydrophobicity of the methyl group leads to a decrease in the water-absorption performance of the catalyst, indicating that pTA has a participating effect. The chemisorbed water at approximately 443 K is mainly the coordination of metal ions with water molecules, which causes the metal active site (L acid) of the catalyst to be covered. With an increasing amount of pTA, the crystal form of the catalyst changes, and the chemical adsorption water content of the catalyst decreases, which indicates that the {100} crystal facets can better expose the active sites of the catalyst. The desorption peak at approximately 603–633 K is attributed to the collapse of the catalyst structure. When the temperature is 603 K, the skeleton of catalyst HPW–Cu3(BTC)2 completely collapsed, while in the catalyst samples containing PTA, the skeleton collapse temperature increased by 30 K is 633 K. As the amount of pTA is increased, the temperature at which the catalyst collapses increases, indicating that the addition of pTA can increase the thermal stability of the catalyst. The weight loss percentage of HPW–Cu3(BTC)2–P20 is 37.7% at the temperature of 633 K. With the decrease of pTA, the weight loss percentage of HPW–Cu3(BTC)2–P10, HPW–Cu3(BTC)2–P5, and HPW–Cu3(BTC)2–P2 gradually decreased (35.0%, 31.4%, and 31.0%, respectively). HPW-Cu3(BTC)2 had a weight loss percentage of 34.9%, owing to the much higher content of HPW in its skeleton.

Thermogravimetric analysis of the catalyst sample: a HPW–Cu3(BTC)2; b HPW–Cu3(BTC)2–P2; c HPW–Cu3(BTC)2–P5; d HPW–Cu3(BTC)2–P10; e HPW–Cu3(BTC)2–P20
3.6 NH 3 –TPD Analysis
NH3–TPD is an effective method for characterizing solid acid catalysts, especially for characterizing acid strength, and it has been used to determine the acidity of phosphotungstic-acid-based materials. The acid sites can be classified as a function of the desorption temperature of ammonia as weak (423–523 K), medium (523–623 K), strong (623–773 K), and very strong (> 773 K). Figure 8 shows two NH3 desorption peaks in the catalyst, namely the weak-acid-site peak of Cu2+ at 464–523 K and the strong-acid-site peak of HPW at 609–650 K. By increasing the ratio of m(pTA)/m(BTC), it is found that the adsorption peak of the weak acid site moves toward the low-temperature direction, which indicates the decrease of the chemisorbed water on the surface of Cu2+. This can be confirmed by the thermogravimetric analysis. A large adsorption peak appeared at 633 K due to the desorption of NH3 from HPW. From the thermogravimetric results, it was found that the decomposition of the carrier occurred at this time. The mass spectrometry of the outlet of NH3–TPD of the catalyst sample with m(pTA)/m(BTC) = 20 also indicates that the desorption peak at this time also has a small amount of CO2 (Fig. 8). This indicates that the decomposition of the carrier did occur at 633 K. As the amount of pTA increases, the width of the adsorption peak at higher temperatures becomes increasingly larger, indicating that the active sites of the catalyst are increasingly more exposed as pTA is added.

Temperature-programmed desorption of ammonia mass spectrum of NH3–TPD gas outlet of HPW–Cu3(BTC)2–P20 (top left): a HPW–Cu3(BTC)2–P2; b HPW–Cu3(BTC)2–P5; c HPW–Cu3(BTC)2–P10; d HPW–Cu3(BTC)2–P20
3.7 ICP Characterization of Samples
Table 2 shows the ICP characterization of the catalysts. The W and P contents in the catalysts were determined. Owing to HPW being the main catalytically active component, the content of HPW in the catalyst is critical. The ICP characterization shows that the amount of HPW on HPW–Cu3(BTC)2 is much higher than the sample when adding pTA, because of the relatively weak coordination ability of pTA. Among the samples when adding pTA, with increasing pTA content, the amount of HPW from HPW–Cu3(BTC)2–P2 to HPW–Cu3(BTC)2–P10 slightly increased and then decreased for the sample HPW–Cu3(BTC)2–P20, which achieved the transformation of crystal facets completely. A large amount of pTA present in the skeleton structure and the different crystal structure and coordination sites may be the reason for the decreasing amount of HPW.
3.8 Catalytic Performance and Reusability
The catalytic performances of the obtained samples were evaluated in the esterification of cyclohexene mixture with formic acid, and the results are summarized in Table 3. The four kinds of catalysts reported in the literature, namely peanut-shell-derived carbon solid acid (PSCSA), an HZSM-5 molecular sieve, cation exchange resin Amberlyst-15, and Nafion NR50, are also compared with these obtained samples in the same esterification reaction. It is obvious from Table 3 that HPW series catalysts can achieve a similar conversion rate at a lower temperature (343 K). The turn-over frequency (TOF) value of HPW–Cu3(BTC)2–P20 is the highest, which indicates that its performance is better than that of other catalysts in the table. The {100}-crystal-facet catalysts exhibit better activity than those of the {111} crystal facets with the same initial amount of HPW added. The conversion of cyclohexene at {100} crystal facets is 51% higher than that at {111} crystal facets, indicating that the {100} crystal facets can better expose the catalytically active sites of HPW; as is well known, the esterification of cyclohexene with formic acid is an electrophilic addition reaction. First, the double bond in cyclohexene is attacked by H+ ions in HPW to form a carbocation, and then the carbocation reacts with the nucleophile formic acid to form cyclohexyl formate. The Brønsted acid site from HPW plays the major role in the esterification reaction, and, after crystal facet transformation, {100} crystal facets with larger square faces will be more likely to expose more HPW active sites than {111} crystal facets with smaller triangular faces, so the catalyst with {100} crystal facets showed the better catalytic performance. The reason for the decrease in selectivity is that benzene and cyclohexene undergo a Friedel–Crafts reaction under the catalysis of L acid at a molar ratio close to 16:1. However, the active site of the {100}-crystal-facet catalyst Cu2+ is no longer covered by chemisorbed water, thus providing more active sites for the alkylation reaction.
The recovered catalysts were washed using mixed solutions of chloroform and acetone with a volume ratio of 1:1 and were re-used for two runs. Figure 9 shows SEM micrographs of the recovered catalyst. It can be seen from the figure that the catalyst still maintains a {100}-crystal-facet structure, but the recovered catalyst is agglomerated. Table 4 shows the N2 physical adsorption data and catalytic reaction evaluation of the recovered catalyst. It can be seen from the data in Table 4 that the catalyst exhibits a decrease in specific surface area after recovery, resulting in a decrease in catalytic activity. Combined with SEM results, this confirms that the main cause of the decrease in activity after catalyst recovery was the agglomeration of the catalyst. To further improve the reusability of the catalyst, the next stage of work will focus on solving the problem of catalyst agglomeration using appropriate synthesis methods.

SEM of recovered catalyst HPW–Cu3(BTC)2–P20: a primary recovered catalyst; b secondary recovered catalyst
4 Conclusions
In the work reported in this study, HPW was incorporated into the metal–organic framework Cu3(BTC)2 through a one-step hydrothermal synthesis method and used for catalyzing the esterification reaction of cyclohexene with formic acid. The crystal structure of composite HPW–Cu3(BTC)2 was modulated during the preparation process to convert the catalyst from the {111} crystal facets of the original regular octahedral structure to the {100} crystal facets of the cubic structure using p-toluic acid (pTA) as the modulator. When m(pTA):m(BTC) = 20, the crystal facets of the catalyst are completely converted into {100} crystal facets. Through crystal face transformation, the obtained composite HPW–Cu3(BTC)2 can expose more active sites of HPW and enhance the affinity for the reaction substrate. Compared with the {111} facets (35.0%), the single-pass conversion of cyclohexene for esterification reaction catalyzed by {100} facets was improved significantly (86.8%). However, because of the agglomeration phenomenon, the performance of the recovery catalyst has been reduced to a certain extent.
References
Wang Y, Zhang J, Wang X et al (2010) Boron-and fluorine-containing mesoporous carbon nitride polymers: metal-free catalysts for cyclohexane oxidation. Angew Chem Int Ed 49(19):3356–3359
Li Y, Xu X, Zhang P et al (2013) Highly selective Pd@ mpg-C3N4 catalyst for phenol hydrogenation in aqueous phase. Rsc Advances 3(27):10973–10982
Yuan PQ, Liu Y, Bai F et al (2011) Hydration of cyclohexene in sub-critical water over WOx–ZrO2 catalysts. Catal Commun 12(8):753–756
Chen BC, Yu BY, Lin YL et al (2014) Reactive-distillation process for direct hydration of cyclohexene to produce cyclohexanol. Ind Eng Chem Res 53(17):7079–7086
Ahamed Imam R, Freund H, Guit RPM et al (2013) Evaluation of different process concepts for the indirect hydration of cyclohexene to cyclohexanol. Org Process Res Dev 17(3):343–358
Shanmugam S, Viswanathan B, Varadarajan TK (2004) Esterification by solid acid catalysts-a comparison. J Mol Catal A Chem 223(1–2):143–147
Heravi MM, Derikvand F, Bamoharram FF (2007) Highly efficient, four-component one-pot synthesis of tetrasubstituted imidazoles using Keggin-type heteropolyacids as green and reusable catalysts. J Mol Catal A Chem 263(1–2):112–114
Zang Y, Shi J, Zhao X et al (2013) Highly stable chromium (III) terephthalate metal organic framework (MIL-101) encapsulated 12-tungstophosphoric heteropolyacid as a water-tolerant solid catalyst for hydrolysis and esterification. React Kinet Mech Catal 109(1):77–89
Zhang W, Leng Y, Zhu D et al (2009) Phosphotungstic acid salt of triphenyl(3-sulfopropyl) phosphonium: an efficient and reusable solid catalyst for esterification. Catal Commun 11(3):151–154
Dai C, Zhang A, Li J et al (2014) Synthesis of yolk-shell HPW@ Hollow silicalite-1 for esterification reaction. Chem Commun 50(37):4846–4848
Zhu S, Gao X, Dong F et al (2013) Design of a highly active silver-exchanged phosphotungstic acid catalyst for glycerol esterification with acetic acid. J Catal 306:155–163
Wee LH, Janssens N, Bajpe SR et al (2011) Heteropolyacid encapsulated in Cu3(BTC)2 nanocrystals: an effective esterification catalyst. Catal Today 171(1):275–280
Hosseini MS, Masteri-Farahani M (2019) Surface functionalization of magnetite nanoparticles with sulfonic acid and heteropoly acid: efficient magnetically recoverable solid acid catalysts. Chem Asian J 14(7):1076–1083
Ngu TA, Li Z (2014) Phosphotungstic acid-functionalized magnetic nanoparticles as an efficient and recyclable catalyst for the one-pot production of biodiesel from grease via esterification and transesterification. Green Chem 16(3):1202–1210
Sert E, Atalay FS (2012) Esterification of acrylic acid with different alcohols catalyzed by zirconia supported tungstophosphoric acid. Ind Eng Chem Res 51(19):6666–6671
Wu N, Li B, Ma W et al (2014) Synthesis of lacunary polyoxometalate encapsulated into hexagonal mesoporous silica and their catalytic performance in esterification. Microporous Mesoporous Mater 186:155–162
Wu M, Zhao QQ, Li J et al (2016) Tungstophosphoric acid-based mesoporous materials anchored to MCM-41: characterization and catalytic performance in esterification of levulinic acid with ethanol. J Porous Mater 23(5):1329–1338
Lu B, Wu Z, Ma L et al (2018) Phosphotungstic acid immobilized on sulphonic-acid-functionalized SBA-15 as a stable catalyst for the esterification of cyclohexene with formic acid. J Taiwan Inst Chem Eng 88:1–7
Zhang L, Jin Q, Shan L et al (2010) H3PW12O40 immobilized on silylated palygorskite and catalytic activity in esterification reactions. Appl Clay Sci 47(3–4):229–234
Mizuno N, Misono M (1998) Heterogeneous catalysis. Chem Rev 98(1):199–218
Jansen RJJ, Van Veldhuizen HM, Schwegler MA et al (1994) Recent (1987–1993) developments in heteropolyacid catalysts in acid catalyzed reactions and oxidation catalysis. Recl Trav Chim Pays-Bas 113(3):115–135
Eddaoudi M, Kim J, Rosi N et al (2002) Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 295(5554):469–472
Li Y, Yang RT (2007) Gas adsorption and storage in metal-organic framework MOF-177. Langmuir 23(26):12937–12944
Sun N, Wen X, Wang F et al (2010) Effect of pore structure on Ni catalyst for CO2 reforming of CH4. Energy Environ Sci 3(3):366–369
Song J, Zhang Z, Hu S et al (2009) MOF-5/n-Bu4 NBr: an efficient catalyst system for the synthesis of cyclic carbonates from epoxides and CO2 under mild conditions. Green Chem 11(7):1031–1036
Juan-Alcañiz J, Gascon J, Kapteijn F (2012) Metal-organic frameworks as scaffolds for the encapsulation of active species: state of the art and future perspectives. J Mater Chem 22(20):10102–10118
Janssens N, Wee LH, Bajpe S et al (2012) Recovery and reuse of heteropolyacid catalyst in liquid reaction medium through reversible encapsulation in Cu3(BTC)2 metal-organic framework. Chem Sci 3(6):1847–1850
Bajpe SR, Breynaert E, Mustafa D et al (2011) Effect of Keggin polyoxometalate on Cu (ii) speciation and its role in the assembly of Cu3(BTC)2 metal-organic framework. J Mater Chem 21(26):9768–9771
Biemmi E, Christian S, Stock N et al (2009) High-throughput screening of synthesis parameters in the formation of the metal-organic frameworks MOF-5 and HKUST-1. Microporous Mesoporous Mater 117(1–2):111–117
Lan X, Huang N, Wang J et al (2018) A general and facile strategy for precisely controlling the crystal size of monodispersed metal-organic frameworks via separating the nucleation and growth. Chem Commun 54(6):584–587
Mao Y, Su B, Cao W et al (2014) Specific oriented metal–organic framework membranes and their facet-tuned separation performance. ACS Appl Mater Interfaces 6(18):15676–15685
Liu Y, Liu S, He D et al (2015) Crystal facets make a profound difference in polyoxometalate-containing metal-organic frameworks as catalysts for biodiesel production. J Am Chem Soc 137(39):12697–12703
Umemura A, Diring S, Furukawa S et al (2011) Morphology design of porous coordination polymer crystals by coordination modulation. J Am Chem Soc 133(39):15506–15513
Chowdhury P, Bikkina C, Meister D et al (2009) Comparison of adsorption isotherms on Cu-BTC metal organic frameworks synthesized from different routes. Microporous Mesoporous Mater 117(1–2):406–413
Loera-Serna S, Oliver-Tolentino MA, de Lourdes L-Núñez M et al (2012) Electrochemical behavior of [Cu3(BTC)2] metal–organic framework: the effect of the method of synthesis. J Alloy Compd 540:113–120
Schlesinger M, Schulze S, Hietschold M et al (2010) Evaluation of synthetic methods for microporous metal–organic frameworks exemplified by the competitive formation of [Cu2(btc)3(H2O)3] and [Cu2(btc)(OH)(H2O)]. Microporous Mesoporous Mater 132(1–2):121–127
Anbia M, Faryadras M (2015) In situ Na·Cu3(BTC)2 and Li·Cu3(BTC)2 nanoporous MOFs synthesis for enhancing H2 storage at ambient temperature. J Nanostruct Chem 5(4):357–364
Xu Y, Chen D, Jiao X (2005) Fabrication of CuO pricky microspheres with tunable size by a simple solution route. J Phys Chem B 109(28):13561–13566
Pérez-Mayoral E, Čejka J (2011) [Cu3(BTC)2]: a metal-organic framework catalyst for the Friedländer reaction. ChemCatChem 3(1):157–159
Leng Y, Wang J, Zhu D et al (2009) Heteropolyanion-based ionic liquids: reaction-induced self-separation catalysts for esterification. Angew Chem Int Ed 48(1):168–171
Leng Y, Wang J, Zhu D et al (2009) Sulfonated organic heteropolyacid salts: Recyclable green solid catalysts for esterifications. J Mol Catal A Chem 313(1–2):1–6
Xu X, Lu Y, Yang Y et al (2015) Tuning the growth of metal-organic framework nanocrystals by using polyoxometalates as coordination modulators. Sci China Mater 58(5):370–377
Xue W, Zhao H, Yao J et al (2016) Esterification of cyclohexene with formic acid over a peanut shell-derived carbon solid acid catalyst. Chin J Catal 37(5):769–777
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Grant No. 21776237). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflicts of interest to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jiang, H., Lu, B., Ma, L. et al. Effect of Crystal Form Control on Improving Performance of Cu3(BTC)2 Immobilized Phosphotungstic Acid in Esterification of Cyclohexene with Formic Acid. Catal Lett 150, 1786–1797 (2020). https://doi.org/10.1007/s10562-019-03090-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-019-03090-1