
Abstract
Two different hollow structured Co3O4 micro-polyhedrons, derived from Zeolitic imidazolate frameworks (ZIF)-67 crystals, were fabricated by tuning the synthetic strategy (positive or reverse addition). The results demonstrate that the synthetic strategy significantly affect catalyst structure and properties. Compared with Co3O4-A nanocatalyst, the Co3O4-B nanocatalyst exhibit superior catalytic performance, showing 100% CO conversion at the conditions of T = 110 °C and SV = 30,000 h−1. The higher catalytic performance could be attributed to its higher surface area, lower temperature reducibility, and abundant surface Co3+ and adsorbed oxygen.
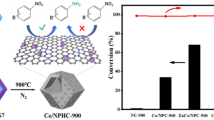
Graphic Abstract
Two different hollow structured Co3O4 micro-polyhedrons, derived from Zeolitic imidazolate frameworks (ZIF)-67 crystals, were fabricated by tuning the synthetic strategy (positive or reverse addition). The Co3O4-B nanocatalyst exhibit superior catalytic performance, which should be attributed to its higher surface area, lower temperature reducibility, and abundant surface Co3+ and adsorbed oxygen.

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Hollow structured nanomaterials have attracted a broad interest in recent years due to their unique properties, such as high specific surface area, well-defined active site, inner void space, and tunable mass transfer rate [1, 2]. Controllable calcination of Metal–organic frameworks (MOFs) is a effective strategy for the preparation of hollow structural materials [3,4,5,6,7,8]. And MOFs-derived hollow structural metal oxides, composing of special composites and developed pore channels, showed superior performance in catalysis and electrochemical applications. ZIF-67 derived hollow structured Co3O4 nanocomposites, have been reported with superior performance in electrochemical and gas sensing applications [9,10,11,12,13]. High porosity and inner voids endows it with high performance, which might be more meaningful in heterogeneous catalysis.
Co3O4 is considered as one of the most potential catalysts in CO oxidation due to its special morphology, structural and size effects [14,15,16,17]. Co3O4 nanobelts and nanocubes as model catalysts were well controllable synthesized to evaluate CO oxidation [14]. It was found that the Co3O4 nanobelts which have a predominance of exposed {011} planes are more active than Co3O4 nanocubes with exposed {001} planes. Compared with commercial Co3O4, mesoporous cobalt oxides showed improved activity in CO oxidation, which was ascribed to the lattice oxygen mobility of the catalysts promoted by the surface oxygen vacancy and large surface area. Besides, the particle size of Co3O4 nanocatalyst have shade an important effect on CO oxidation activity [16, 17]. Iablokov et al. [16] clarified the size effect of Co3O4 nanocatalyst in CO oxidation by well tuning the particle size from 3.5 to 12.2 nm. They found that the maximum reaction rates were observed for Co3O4 particles with a size in the range of 5–8 nm. These results clearly confirm that controlling the morphology of nanostructured cobalt oxides is beneficial for enhanced catalytic performance.
Herein, we prepared hollow structured Co3O4 nanocatalysts by annealing of ZIF-67 crystals in air atmosphere. Through tuning the synthetic strategy, two different Co3O4 nanocatalysts were fabricated. On the basis of catalytic performance for CO oxidation and various characterizations, a structure–activity relationship was well established.
2 Experiment
2.1 Synthesis
Porous Co3O4 hollow structural catalysts were prepared via a two-step route, involving the fabrication of Co-MOF (ZIF-67) nanocubes and the subsequent calcination treatment in air atmosphere. Synthetic route A Co(NO3)2·6H2O (0.87 g) and 2-methylimidazole (1.97 g) were dissolved in 50 mL methanol respectively. Then the solution was mixed by pouring 2-methylimidazole solution into Co3+ solution under thoroughly stirring. The resulting mixture was aged for 24 h at room temperature. Then the purple precipitate was collected by centrifugation, washed with methanol three times, and dried at 80 °C for overnight. Finally, the as-prepared ZIF-67 precursors were heated to 350 °C with a heating rate of 1 °C·min−1. The resulting product was denoted as Co3O4-350-A. Synthetic route B the synthetic process is similar to the above process. The only change is that the solution was mixed by pouring Co3+ solution into 2-methylimidazole solution under thoroughly stirring. And the final product was denoted as Co3O4-350-B.
2.2 Materials Characterization
The structural parameter, pore size distribution and N2 adsorption isotherms of the samples were tested at liquid nitrogen temperature using an Autocorb apparatus. The samples were pretreated at 200 °C for 3 h before testing. The N2 adsorption–desorption isotherm used the Barrett–Joyner–Halenda (BJH) method.
Thermogravimetric analysis (TGA) was conducted on SDT 2960 with a heating rate of 10 °C min−1 in air flow.
X-ray diffraction (XRD) patterns were obtained with a Bruker D8 diffractometer using Cu Kα radiation (λ = 1.540589 Å). The crystal size of samples was calculated from the width of diffraction pro- files, referring to the full width at half maximum (FWHM) of the crystalline phase using the Debye–Scherrer formula.
Scanning electron microscope (SEM) was performed on a JEOL JSM-6700F field emission SEM, which was operated at the accelerating voltage of 15 kV and the detector current of 10 mA. Transmission electron microscopy (TEM) was conducted on a JEOL JEM 2100 microscope operated at 200 kV.
The XPS patterns were measured on a PHI-5300/ESCA electronic energy spectrum at 300 W using Mg Kα X-rays as the excitation source. The data were processed by the XPS- PEAK software, and surface element contents were calculated through XPS peak areas. The inding energies (BE) of the elements were calibrated relative to the carbon impurity with a C 1 s at 284.8 eV. Laser Raman spectra were measured on a Renishaw Laser Raman spectrometer. Ar+ (514.5 nm) and He–Cd lasers (325 nm) were used as the excitation source.
Temperature-programmed reduction (H2-TPR) measurement was conducted on a TPDRO 1100 apparatus supplied by the Thermo-Finnigan company. Typically, 50 mg of the sample was heated from room temperature to 700 °C at a rate of 10 °C/min. A gas mixture of H2/N2 containing 5% H2 (volume percentage) was used as a reductant at a flow rate of 20 mL/min.
Temperature-programmed desorption of CO (CO-TPD) was performed on the same apparatus with TCD detector. First, 0.1 g of samples was pretreated under N2 flow at 120 °C for 1 h. After that, the temperature was cooled to 30 °C, and the flow gas was switched to 15% CO/He (30 mL/min). The CO adsorption continued at 30 °C for 1 h, and then were purged with He (30 mL/min) to remove the physisorbed CO. The CO-TPD experiment was carried out by elevating temperature up from 30 to 300 °C with a heating rate of 10 °C/min.
2.3 Measurement of Catalytic Activity
The catalytic activity was measured by a fixed-bed flow microreactor at atmospheric pressure. In a typical experiment process, a 50 mg sample was loaded into a quartz tube (id = 6 mm) and then placed into a microreactor. The gas mixture of CO/O2/N2 (1.6:21.0:77.4) with a flow rate of 25 mL/min was introduced into the reactor, and the corresponding space velocity is 30,000 h−1. Before test, the catalyst was pretreated in feed gas at 150 °C for 1 h. After cooling to room temperature, the CO oxidation was evaluated. The CO oxidation reaction was stabilized for 15 min, and the effluent gas from the reactor was analyzed by a gas chromatograph (GC-9860) equipped with a thermal conductivity detector and TDX-01 packed column.
3 Results and Discussion
Different hollow structured Co3O4 nanocomposites can be obtained through the calcination of ZIF-67 in air atmosphere via a tunable synthetic routes (Scheme 1). The powder X-ray diffraction (XRD) patterns of as-synthesized ZIF-67 sample is presented in Fig. 1a, which is matched well with the simulated and published XRD patterns, confirming the formation of pure ZIF-67 crystals [18]. Notable that the crystallinity of ZIF-67 prepared from synthetic route B is stronger than that from synthetic route A. This is in accordance with the SEM results. After calcination in air atmosphere, the spinel-type Co3O4 species were obtained, further confirmed by XRD characterizations Fig. 1b. The diffraction peaks located at around 31.3°, 36.8°, 38.6°, 44.8°, 55.7°, 59.4°, 65.2° and 77.3° were recorded, assigning as the (220), (311), (222), (400), (422), (511), (440) and (533) crystalline planes of Co3O4 (JCPDS No. 01-073-1701). In addition, it can be observed that the intensity of reflection peaks is different over Co3O4-A to Co3O4-B samples, meaning that the primary crystallites in the products are different in particles size. According to the Scherrer equation, the mean crystallite size of the Co3O4-A and Co3O4-B samples are calculated to be 11.4 and 6.8 nm using the FWHMs of the (311) line of Co3O4(Table 1). Additionally, through the comparison of the diffraction of ZIF-67 crystals and their derives, we can easily find that the ZIF-67(B) has good crystallinity, whereas its derives (Co3O4-B) show weak diffraction peaks, indicating smaller nanoparticles confirmed by TEM analyses. This result should be attributed to different synthetic route. In route A, 2-MI was poured into Co ions solution. Some uncoordinated 2-MI molecules would locate inside the structure of ZIF-67, and few residual Co ions might remain in the surface of ZIF-67, resulting in weak crystallinity of ZIF-67 and larger particle sizes of Co3O4. In route B, the reverse process was performed, some 2-MI might remain in the surface of ZIF-67, resulting in better crystallinity of ZIF-67 and smaller particle sizes of Co3O4.

Tunable synthetic strategy for hollow structured Co3O4 nanocomposites

XRD patterns of ZIF-67 crystals a, and b different hollow structured Co3O4 nanocomposites: Co3O4-A and Co3O4-B. c TGA curves of the ZIF-67(A) and ZIF-67(B) in air flow
The thermographic (TG) behaviors of the different ZIF-67 samples were measured in air flow. Seen from Fig. 1c, there exists a weak weight loss below 300 °C, which corresponds to the volatilization of methanol in the pores of ZIF-67. Above 300 °C, the weight loss becomes obvious, drops sharply at approximately 420 °C and finally flattens at approximately 480 °C. The latter weight loss might be attributed to the oxidation and decomposition of 2-methylimidazole. Notably, the weight loss of ZIF-67(B) took place at a relatively lower temperature that of ZIF-67(A). This result agrees well with the above analyses. For ZIF-67(B), some residual 2-MI remain on the surface of ZIF-67 during synthetic process, resulting in relatively lower decomposition temperature.
The morphology of the ZIF-67 samples and their corresponding calcined products are presents in Fig. 2. Whether synthetic A or B, the final ZIF-67 crystals are both composed of similar monodispersed rhombic dodecahedrons. By calcining the ZIF-67 crystals at 350 °C for 3 h, the typical morphology of Co3O4-A and Co3O4-B samples are presented in Fig. 2c and d. Clearly, the calcined particles retain the similar size and shape, along with inner hollow structure and rough porous shell. Some hollow structures are broken due to thermal effects. The microstructures of the prepared Co3O4-A and Co3O4-B samples are further characterized by HRTEM analysis (Fig. 3). Obviously, hollow structural dodecahedra, composed of numerous primary crystallites, are observed over the Co3O4-A and Co3O4-B samples (Fig. 3a, b, e and d). The average particle size of Co3O4-A (8.8 ± 2.6 nm) is larger than that of Co3O4-B samples (6.5 ± 1.2 nm) (Inset of Fig. 3b and e). This is consistent with above XRD results. Besides, typical (311) and (111) crystallographic planes of Co3O4 are also determined in Fig. 3c and f. Comparing Fig. 3a and d, it can be easily find that the formed Co3O4-A is assembled with core–shell structure, whereas the Co3O4-A has obvious hollow voids. This means that the different structure is originated from the different synthetic routes. In route A, more Co ions remaining in the surface of ZIF-67 are easily dropped into voids space during the oxidation process, forming an attached core.

SEM images of hollow structured Co3O4 nanocomposites: Co3O4-A (a, b) and Co3O4-B (c, d)

HR(TEM) images of hollow structured Co3O4 nanocomposites: Co3O4-A (a, b, c) and Co3O4-B (d, e, f). The inset of B and E are their corresponding particle size distribution
N2 sorption was also employed to characterize the surface areas and pore structure of Co3O4-A and Co3O4-B samples. Seen from Fig. 4a, both of the two samples display a characteristic Type IV adsorption isotherm with a H3-type hysteresis loop, indicating the presence of the mesoporous structure. A wide pore-size distribution is observed over these samples. The average pore sizes are 33.8 and 22.0 nm for Co3O4-A and Co3O4-B samples respectively (Table 1). This result is well consistent with TEM observations. Besides, their BET surface areas and pore properties are summarized in Table 1. The Co3O4-B sample exhibit much higher surface areas (72 m2/g) than that of Co3O4-A sample (41.7 m2/g). On basis of XRD and TEM results, the higher surface area is mainly originated from smaller primary particles of Co3O4, which generates more active sites for CO oxidation.

Characterizations of hollow structured Co3O4 nanocomposites including Co3O4-A and Co3O4-B: a Nitrogen sorption isotherms and pore diameter distributions (Inset), b H2-TPR profiles, and XPS spectra c Co 2p, d O1 s
H2-TPR was performed to investigate the redox properties. Seen from Fig. 4b, the reduction profiles essentially present two obvious step-wise reduction peaks over Co3O4-A and Co3O4-B samples, which can be attributed to the reduction process of Co3+ to Co2+ and Co2+ to metallic Co0 [19, 20]. Obviously, compared with Co3O4-A catalyst, the Co3O4-B catalyst exhibit better reducibility at lower temperature, with one reduction peak at 310 °C and the other at 425 °C. It suggests that promoted oxygen mobility took place over Co3O4-B catalyst [21], resulting in better catalytic performance toward CO oxidation.
XPS was employed to obtain an insight into the surface composites and chemical state of different Co3O4-A and Co3O4-B samples, and the XPS spectra of Co 2p and O1 s are shown in Fig. 4c and d. The XPS datum are presented in Table 1. In Fig. 4c, two major with binding energies at ca. 780.0 and 795.0 eV corresponding to the Co2p3/2 and Co2p1/2, respectively [22, 23]. The Co 2P2/3 peak is deconvoluted into two components peaks Co3+ and Co2+, and their binding energies are centered at around 780.0 and 781.5 eV. We found that the relative surface content of Co3+ in the Co3O4-B sample is 47.3%, which is larger than that in Co3O4-A sample (33.8%). This result conforms to the H2-TPR results. Figure 4c shows the O1 s XPS spectra of the contrast Co3O4-A and Co3O4-B catalysts, which can be fitted into three components peaks at ca. 530.1 and 531.5 eV. The two components peaks are sequentially ascribed to surface lattice oxygen (Olatt) species and surface adsorption oxygen (Oads) respectively [24]. It can be seen that the relative surface content of Oads in Co3O4-B (51.3%) is larger than that in Co3O4-B (43.8%). Thus, the surface contents of Co3+ and Oads are obviously influenced by preparation method. According to previous studies [25, 26], more abundant of surface Co3+ and Oads species contributed to higher catalytic performance. This finding is well in agreement with our study.
Figure 5a shows the catalytic activities of CO oxidation as a function of reaction temperature for Co3O4-A and Co3O4-B catalysts. It can be observed that there exhibit a large difference between Co3O4-A and Co3O4-B catalysts. And a complete 100% CO conversion took place at 110 °C for Co3O4-B catalyst, which is lower than that for Co3O4-A catalyst (T100 = 170 °C). The calculated specific rates at 110 °C are presented in Table 1. The specific rates are 1.29 × 10−8 and 8.27 × 10−8 mol/(m2s) for Co3O4-A and Co3O4-B respectively. It can be seen that the Co3O4-B catalyst has 7 times higher reaction rate than that of the Co3O4-A catalyst. Besides the contribution of surface area to activity, the surface concentrations of Co3+ and oxygen vacancies also contribute to higher activity. On the basis of above characterizations, there is no significant difference in morphology. As we know, CO species are adsorbed on the surface-exposed Co3+ sites and react with weakly bound oxygen species such as superoxide ion (O2−) to form CO2 [27]. The CO-TPD results are shown in Fig. 5b. It indicates that the Co3O4-B sample has more CO chemisorption uptakes, corresponding to abundant surface Co3+ contents. The active oxygen species are formed on surface oxygen vacancies which are increased by pretreatment in a reducing or oxidizing atmosphere [27, 28]. The higher catalytic performance of Co3O4-B catalyst, which can be attributed to higher surface area, lower temperature reducibility, and abundant surface Co3+ and adsorbed oxygen. Additionally, the used catalysts after reaction were further characterized by XPS techniques (Fig. 5c and d). By comparing the catalysts before and after reaction, we observe that the surface contents of Co3+ and adsorption oxygen (Oads) exhibit an obvious rise for Co3O4-A catalyst, whereas there are only a slight decrease for Co3O4-A catalyst. It indicates that different treatments (e.g. reaction atmosphere) can effectively tune the surface chemical components at above 150 °C [27]. Especially for Co3O4-A catalyst, surface oxygen vacancies increase significantly after reaction, leading to more surface adsorbed oxygen. Partial adjacent Co2+ were also oxidized to Co3+, as showed in Table 2.

a Catalytic activities of Co3O4-A and Co3O4-B samples in CO oxidation. b CO-TPD of Co3O4-A and Co3O4-B. And XPS spectra c Co 2p, d O1 s of the Co3O4-A and Co3O4-B after reaction
4 Conclusions
In summary, two different hollow structured Co3O4 nanocatalyst were facilely prepared by tuning synthetic strategy. The results indicate that preparation method plays an important effect on CO oxidation activities. The superior Co3O4-B nanocatalyst exhibit higher performance and 100% CO conversion at 110 °C and 30,000 h−1. The higher catalytic performance should be attributed to higher surface area, lower temperature reducibility, and abundant surface Co3+ and adsorbed oxygen. The results provide a scientific basis for the design of Co3O4-based oxidation catalysts.
References
Wang X, Feng JI, Bai Y, Zhang Q, Yin Y (2016) Chem Rev 116(18):10983–11060
Zhu W, Chen Z, Pan Y, Dai R, Wu Y, Zhuang Z, Li Y (2018) Adv Mater. https://doi.org/10.1002/adma.201800426
Zhang L, Wu HB, Lou XW (2013) J Am Chem Soc 135(29):10664–10672
Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM (2013) Science 341(6149):1230444
Dang S, Zhu QL, Xu Q (2018) Nat Rev Mater 3(1):17075
Xia W, Mahmood A, Zou R, Xu Q (2015) Energy Environ Sci 8(7):1837–1866
Shieh FK, Wang SC, Yen CI, Wu CC, Dutta S, Chou LY, Tsung CK (2015) J Am Chem Soc 137(13):4276–4279
Chen JE, Fan MS, Chen YL, Deng YH, Kim JH, Alamri HR, Wu KCW (2017) Chem A Eur J 23(54):13284–13288
Lü Y, Zhan W, He Y, Wang Y, Kong X, Kuang Q, Zheng L (2014) ACS Appl Mater Interfaces 6(6):4186–4195
Li S, Peng S, Huang L, Cui X, Al-Enizi AM, Zheng G (2016) ACS Appl Mater Interfaces 8(32):20534–20539
Kaneti YV, Dutta S, Hossain MS, Shiddiky MJ, Tung KL, Shieh FK, Yamauchi Y (2017) Adv Mater 29(38):1700213
Zhou S, Ye Z, Hu S, Hao C, Wang X, Huang C, Wu F (2018) Nanoscale 10(33):15771–15781
Zhong G, Liu D, Zhang J (2018) J Mater Chem A 6(5):1887–1899
Hu L, Sun K, Peng Q, Xu B, Li Y (2010) Nano Res 3(5):363–368
Li Y, Shen W (2014) Chem Soc Rev 43(5):1543–1574
Iablokov V, Barbosa R, Pollefeyt G, Van Driessche I, Chenakin S, Kruse N (2015) ACS Catal 5(10):5714–5718
Li L, Yao Y, Tang Z, Ji W, Dai Y, Shen X (2016) J Nanosci Nanotechnol 16(7):7573–7578
Banerjee R, Phan A, Wang B, Knobler C, Furukawa H, O’Keeffe M et al (2008) Science 319:939
Ma CY, Mu Z, Li JJ, Jin YG, Cheng J, Lu GQ, Qiao SZ (2010) J Am Chem Soc 132(8):2608–2613
Wang J, Yang N, Tang H, Dong Z, Jin Q, Yang M, Wang D (2013) Angew Chem Int Ed 125(25):6545–6548
Luo JY, Meng M, Zha YQ, Guo LHJ (2008) Phys Chem C 112(23):8694–8701
Liu B, Liu Y, Hou H, Liu Y, Wang Q, Zhang J (2015) Catal Sci Technol 5(12):5139–5152
Bai G, Dai H, Deng J, Liu Y, Wang F, Zhao Z, Au CT (2013) Appl Catal A 450:42–49
Song W, Poyraz AS, Meng Y, Ren Z, Chen SY, Suib SL (2014) Chem Mater 26(15):4629–4639
Ren Q, Mo S, Peng R, Feng Z, Zhang M, Chen L, Ye D (2018) J Mater Chem A 6(2):498–509
Bai B, Li J (2014) ACS Catal 4(8):2753–2762
Yu YB, Takei T, Ohashi H, He H, Zhang XL, Haruta M (2009) J Catal 267:121–128
Widmann D, Leppelt R, Behm RJ (2007) J Catal 251:437–442
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21503184), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (18KJA610004), and the Natural Science Foundation of Jiangsu Province-General Program (BK20171273).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, C., Chu, W., Jiang, R. et al. ZIF-67 Derived Hollow Structured Co3O4 Nanocatalysts: Tunable Synthetic Strategy Induced Enhanced Catalytic Performance. Catal Lett 149, 3058–3065 (2019). https://doi.org/10.1007/s10562-019-02871-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-019-02871-y