
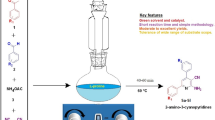
Abstract
Although l-proline derived aminothioureas are being used as catalyst in asymmetric synthesis, their applications in multicomponent reactions are not yet reported in the literature. We report herein a l-proline derived secondary aminothiourea as a multifunctional catalyst in multicomponent reaction for synthesis of coumarin-based unsymmetrical trisubstituted methanes whose prototype has been found to be acetylcholinesterase inhibitor. The method requires low catalyst loading (5 mol%), very short reaction time (15–30 min) to give excellent yields. For the first time, it is observed that our bifunctional catalyst significantly increases the rate of conversion in comparison to that of two cooperative organocatalysts having similar catalytic sites.
Graphical Abstract
l-Proline derived secondary aminothiourea organocatalyst in multicomponent reaction: synthesis of coumarin derived trisubstituted methanes

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Since the discovery of l-proline as a bifunctional organocatalyst for organic synthesis [1], many catalysts with similar functional features have come to the fore. Despite its huge catalytic applications, numerous efforts are being made to derivatize l-proline to address its solubility issues by keeping its stereochemical features, the bifunctional character and overall catalytic efficiency intact [2,3,4,5,6,7,8,9,10,11]. In recent years, aminothioureas have found immense applications as bifunctional catalysts for carbon-carbon bond forming reactions after the success of chincona alkaloid derived organothioureas [12, 13]. Although some l-proline derived thiourea organocatalysts are reported in the literature for stereoselective synthesis [14,15,16,17], there are hardly any report on use of l-proline derived aminothioureas to catalyze multicomponent reactions (MCRs). Since l-proline is used extensively for MCRs [18], we proposed to study simple, easily accessible and cost-effective l-proline derived aminothioureas to improve upon its catalytic efficiency in MCRs. Given the recent emphasis on using green solvents [19], we aimed to carry out the reaction preferably in water so that the purification can be done by simple filtration and recrystallization.
Coumarin is a privileged structural motif that exist in numerous pharmacophore derived from natural and synthetic sources [20]. In recent years, coumarins have been found to slow down the progression of alzheimer disease (AD) by inhibiting acetylcholinesterase (AChE). The AChE terminates the impulse transmission through rapid hydrolysis of the neurotransmitter acetylcholine (ACh), the progressive decline of which leads to pathogenesis of AD [21,22,23]. While various coumarin derivatives are reported for acetylcholinesterase inhibition, we have very recently observed that coumarin-based unsymmetrical trisubstituted methane (uTRSM), 3-(6-amino-1,3-dimethyluracilyl)benzyl-4-hydroxycoumarins (2a, Table 2) acts as a potent AChE inhibitor with IC50 value at 48.49 ± 5.6 nM which is much better than the reference drug donepezil (IC50 = 74.13 ± 8.3 nM) [24]. In contrast, the corresponding biscoumarin, 3,3′-methylene-bis(4-hydroxy coumarin), MHC showed relatively inferior inhibition (80.16 ± 7.1 nM). Encouraged by the findings, we planned to synthesize a series of coumarin based uTRSMs by MCR which could be screened for inhibition of AChE.
There are two reports for synthesis of 3-(6-amino-1,3-dimethyluracilyl)benzyl-4-hydroxycoumarins derived from multicomponent reaction of 4-hydroxy coumarin, aryl aldehydes and 6-aminouracil employing l-proline [25] and a tertiary aminothiourea [26]. In both the cases, the reaction took high catalyst loading (20 mol%) and long reaction time (3–12 h) at reflux temperature. Since most of the tertiary aminothiourea catalyzed reactions involve activation of unactivated ketones [13], we hardly saw any reason to employ the tertiary amine group for activation of highly acidic 4-hydroxy coumarin (pKa 5.3) in the said reaction. We were curious to know if simpler organothiourea would be enough to accomplish the reaction without needing the amine altogether. Our study led to development of a new secondary aminothiourea catalysed method for synthesis of coumarin-based uTRSMs (Scheme 1).

Synthesis of coumarin based trisubstituted methanes
2 Experimental Section
2.1 Materials and Instruments
The chemicals and reagents were available commercially and that were used without further purification. (S)-2-[[3,5-Bis(trifluoromethyl)phenyl]thioureido]-N-benzyl-N,3,3-trimethyl-butanamide was purchased from Sigma Aldrich. The products were characterized by IR, 1H NMR, 13C NMR, Mass spectroscopy and elemental analysis. The IR spectra were recorded on a Perkin Elmer spectrophotometer. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were obtained on a Bruker AC-400 using CDCl3 as solvent and TMS as internal standard, unless otherwise stated. The mass spectra were obtained from Waters ZQ 4000 mass spectrometer by the ESI method, and the elemental analyses of the complexes were performed on a Perkin–Elmer-2400 CHN/S analyzer. HPLC analysis was carried out on an Waters M515 equipped with Chiracel OD-H and Chiralcel AD-H columns using n-hexane and 2-propanol as mobile phase at room temperature.
2.2 Synthesis of 2-(3-Phenylthioureido)ethyl prolinamide, 1
To a stirred solution of Boc- l-proline (3 g, 13.95 mmol) in anhydrous CH2Cl2 at − 5 °C, triethyl amine (1.94 mL, 13.95 mmol) and ethyl chloroformate (1.46 mL, 15.34 mmol) were added dropwise. After 20 min of stirring, the reaction mixture was washed successively with a cold solution of citric acid (5% aqueous, 20 mL), a solution of sodium bicarbonate (10%, 20 mL) and water (20 mL). The organic layer was dried over Na2SO4, filtered, and diluted with CH2Cl2 to a volume of 200 mL. This solution was added dropwise (30–60 min) to a vigorously stirred solution of ethylenediamine (4.66 mL, 69.77 mmol, 5 equiv.) in CH2Cl2 (200 mL) previously cooled to − 78 °C. When the addition was completed, the mixture was stirred for 10 min at that temperature and then warmed to room temperature and stirred overnight. The reaction mixture was concentrated in vacuo to a volume of about 50 mL, and extracted with 1 M aqueous HCl solution (2 × 20 mL). The aqueous layer was basified with concentrated ammonia and it was thoroughly extracted with CHCl3 (10 × 25 mL). The organic phases were combined, dried over Na2SO4, filtered, and the solvent was evaporated in vacuo to give the product 1a as colourless oil (3.15 g, 87%). Part of the crude product (1a) (2.056 g, 8 mmol) was dissolved in anhydrous CH2Cl2 (20 mL) and phenyl isothiocyanate (0.96 mL, 8 mmol) was added dropwise at 0 °C with continuous stirring. The reaction was allowed to stir for 1 h at room temperature for complete conversion (part of the crude product was purified by column chromatography to get the 2-(3-phenylthioureido)ethyl-N-Boc-prolinamide as white solid). The remaining solution was cooled to 0 °C and TFA (2 mL) was added with stirring. After the consumption of the starting material, as indicated by TLC analysis, the reaction mixture was diluted with H2O (10 mL) and the resulting solution was neutralized with aqueous NaHCO3 solution. The reaction mixture was extracted with CH2Cl2 (2 × 20 mL) and the combined organic layer was washed with brine, dried over Na2SO4, filtered and the solvent was removed in vacuo. The solid crude product was recrystallized from methanol to get the pure product 1 as white solid in 77% overall yield (2.079 g, 7.12 mmol). M.p. 110–112 °C; IR (KBr): ν 3435, 2923, 1650, 1525, 1104 cm−1; 1H NMR (400 MHz, CDCl3, δ ppm): 1.65–2.09 (m, 6H, 2-CH2, 2NH), 2.90–3.00 (m, 2H, –CH2), 3.47–3.77 (m, 5H, 2-CH2, –CH), 6.81 (m, 1H, ArH), 7.22–7.43 (m, 4H, ArH), 7.87 (s, 1H, –NH), 7.99 (s, 1H, –NH); 13C NMR (100 MHz, CDCl3, δ ppm): 25.67, 31.99, 38.58, 45.18, 46.90, 60.20, 122.71, 125.03, 126.72, 128.32, 129.63 136.70, 173.25, 181.16; ESI-MS (m/z): 292.12 (M+).
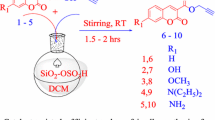
2.3 General Procedure for the Synthesis of uTRSMs
To an equimolar solution of 6-amino-1,3-dimethyluracil (0.155 g, 1 mmol), aldehyde (1 mmol) and 4-hydroxycoumarin (0.162 g, 1 mmol) in 1 mL of water, 5 mol% of thiourea catalyst was added and the solution was stirred under reflux condition. In about 5 min, the product started forming (as evident from TLC). The stirring was continued until completion of the reaction (monitored by TLC). The resulting mixture was cooled to room temperature and the solid that precipitated out of the solution was filtered off, washed with water and then with ethanol twice to remove any unreacted starting materials and trace of catalyst present. The solid crude product was then dissolved in ethanol and kept overnight to get the pure recrystallized product.
3 Results and Discussion
Initially, we optimized the reaction conditions for a model reaction of an equimolar mixture of 6-amino-1,3-dimethyluracil, m-nitrobenzaldehyde and 4-hydroxycoumarin (1 mmol each). When the model reaction was carried out in 1 mL of water in the presence of 10 mol% of N,N′-diphenylthiourea at reflux temperature (Table 1, entry 1), it was completed in 2 h to give 80% isolated yield. Catalysis of the model reaction by N-Benzyl-N′-phenyl thiourea also took similar reaction time with no appreciable difference in yield. The observations revealed that organothiourea alone can catalyze the reaction in the absence of an amine source and the role of the tertiary amine group is very nominal in the reaction reported by Parvin et al. [26]. This might be due to the fact that the tertiary amine groups in organothioureas basically acts a base to catalyze the reaction via enolization of unactivated ketone (Fig. 1). In this case, the 4-hydroxy coumarin is naturally an enol and hardly requires any base catalyst for activation to undergo addition reaction at reflux temperature. The fact that a secondary amine can work both as base and nucleophile, we proposed to add a pyrrolidine source along with organothiourea as catalysts to accelerate the rate of the reaction between 4-hydroxy coumarin and the aldehyde (via iminium salt formation) further.

2°/3°—aminothiourea catalysed C–C bond forming reaction
We planned to study whether an equimolar mixture of secondary amine and thiourea can positively affect the model reaction. To that effect, we treated the reaction mixture with N,N′-diphenylthiourea (10 mol%) and pyrrolidine (10 mol%) in water (Table 1, entry 3). The reaction time was reduced (1.5 h) slightly to generate the desired product in 87% yield. The fact that nucleophilicity of l-proline in water is slightly higher than pyrrolidine [27], the reaction mixture was refluxed in water with l-proline (10 mol%) as well. The reaction took similar time (1.5 h) to obviate 90% isolated yield of the desired product, but it was comparable to l-proline (Table 1, entry 6) with no visible contribution from N,N′-diphenylthiourea. We reasoned that the thiourea group in N,N′-diphenylthiourea and the carboxylate group in l-proline might have engaged in H-bonding interaction leading to partial functional deactivation of the thiourea group [28]. Similar observation was made when the model reaction was carried out with phenyl prolinamide as catalyst, but significant reduction of reaction time (1 h) was noted. The same catalyst completed the reaction within 40 min in the presence of 10 mol% N,N′-diphenylthiourea as co-catalyst. These observations confirmed that the presence of both functionalities may be critical to achieve faster completion of the reaction. But when the reaction was carried out with N-benzyl-N′-phenyl prolinamide with N,N′-diphenylthiourea as co-catalyst (entry 8), the reaction took almost the same time as that of N,N′-diphenylthiourea as the sole catalyst (entry 1). It shows that the presence of the secondary amine in pyrrolidine accelerated the reaction much faster than that of the tertiary amine (entry 9) in the presence of thiourea unit. In order to see if a bifunctional catalyst having the same catalytic sites could be a better catalyst than cooperative catalysis by separate pyrrolidine moiety and thiourea, we decided to synthesize a secondary aminothiourea 1 from l-proline (Scheme 2). The catalyst (1) was synthesized by converting Boc-protected l-proline to the amide derivative (A) followed by treatment with phenyl isothiocyanide in methylene chloride and then TFA in three simple steps [29].

Synthesis of the catalyst
With the secondary aminothiourea in hand, we carried out the model reaction with 10 mol% of the catalyst 1 in 1 mL of water by heating at reflux temperature. Gratifyingly, we observed complete conversion of the starting aldehyde within 20 min to obviate the desired product with an excellent isolated yield of 93%. It was interesting to observe that the reaction with the catalyst 1 took much lesser time than those where equimolar mixture of pyrrolidine and thiourea were used. This proves for the first time that catalytic efficiency of bifunctional catalyst is better than cooperative catalysis by two monofunctional catalysts.
In order to see the effect of solvents on efficacy of the reaction, we first screened the solvents like ethanol, methanol, water, ethanol-water (1:1), CH3CN, THF, CHCl3, and DCM at various reaction temperatures (Table 1S in supplementary materials). The best result was obtained in water where the reaction was completed within 20 min of heating at reflux temperature to obviate the desired product in 93% isolated yield.
Then we went on to optimize the catalyst loading (Table 2S in the supplementary materials) for the reaction. Under catalyst-free condition, only a trace amount of product was obtained upon stirring for 12 h. The same reaction when carried out employing 1 mol% of the catalyst resulted in 70% yield of the desired product within 1 h of stirring under reflux. But, a significant increase in reaction rate was observed upon increasing the catalyst loading to 5 mol% to obtain the desired product with 93% yield within 20 min. However, further enhancement of catalyst loading to 10–15 mol% did not reduce the reaction time appreciably and gave almost similar yields.
3.1 Substrate Scope
Having achieved the optimum reaction conditions, we expanded the scope of our protocol by varying the aldehyde in the model reaction. The results are summarized in Table 2. The reaction worked extremely well with almost all the aldehydes. Aromatic aldehydes having electron-withdrawing substituents (entries 2–10) as well as the ones containing electron-donating substituents (entries 11–13) gave very good to excellent yields. Thus the nature and position of the substituents did not have much of an effect on the reaction rate. The reaction also worked well with a heteroaromatic aldehyde (entry 17), ethyl glyoxalate (entry 18) and an aliphatic aldehyde (entry 19) producing the desired products in good to excellent yields. When the reaction was expanded to the substituted 4-hydroxy coumarins, we noted excellent conversion under the optimised conditions (Entries 20–21).
3.2 Study of Asymmetric Induction
Although this l-proline derived bifunctional amidothiourea catalyst is asymmetric in nature, it did not give any stereoselectivity probably due to the high reaction temperature. In our bid to achieve enantiomeric induction by conducting the reaction at room temperature, we observed the reaction for 12 h to get 70% yield and no enantioselectivity (Table 3, entry 1). The reaction at 10 °C did not give the desired product at all within 48 h. Interestingly, the reaction failed to show any enantioselective induction when the reaction was catalysed by N-phenyl l-prolinamide in the presence of a chiral thiourea catalyst, (S)-2-[[3,5-bis(trifluoromethyl)phenyl]thioureido]-N-benzyl-N,3,3-trimethylbutanamide at room temperature (Table 3, entry 3–4).
3.3 Mechanistic Perspective
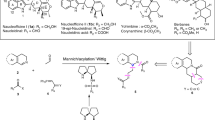
While trying to unfold the mechanism of the reaction, we carried out two control experiments by reacting m-nitrobenzaldehyde with 4-hydroxycoumarin and 6-amino-1,3-dimethyluracil separately under the optimized reaction conditions. The formation of symmetrical trisubstituted methanes (3) from the reaction of 4-hydroxycoumarin took only 30 min for complete conversion, while the reaction of 6-amino-1,3-dimethyluracil with the same aldehyde under similar reaction conditions took almost double the reaction time (1 h) for complete conversion to the corresponding symmetrical TRSM, 4 (Scheme 3). On the other hand, the reaction with other coumarin derivatives such as 4-methylcoumarin, and 4-methoxycoumarin did not generate unsymmetrical TRSMs 5 and 6, but the symmetrical TRSM, 4 only.

Control experiments
Above observations suggest that (i) the hydroxy group at 4-position of the coumarin is vital for the reaction, (ii) the pyrrolidine moeity has acted as a base to activate 4-hydroxy coumarin to form the Knoevenagel product, and (iii) the activation of the aldehyde by the thiourea for addition of the pyrrolidine nucleophile is true in all the cases. The base catalyzed activation of 6-amino-1,3-dimethyluracil does not arise at all, rather it is driven by the amino group in it. The thiourea scaffold might have also catalyzed the formation of the iminium salt through H-bond activation of aldehyde for nucleophilic attack by pyrrolidine moeity of the catalyst 1. Due to greater electrophilicity of H-bond activated aldehyde, the formation of iminium ion with proline derived pyrrolidine is much faster which ultimately affects overall reaction time. It may be noted that the transformation of the carbonyl compound into an iminium species increases the electrophilicity of the carbonyl compound for the subsequent attack of nucleophiles [30]. The higher electrophilicity of the resultant iminium ion might have facilitated easier C–C bond forming reaction with C-3 carbon of 4-hydroxy coumarin to yield the Knoevenagel product. Since 6-amino-1,3-dimethyluracil is a softer nucleophile than 4-hydroxy coumarin, the former will undergo 1,4-addition reaction preferentially with the Knoevenagel product to give the desired product. With all probabilities, the Michael addition of 6-aminouracil to the Knoevenagel product may be very fast and requires no catalyst. As a result, the reaction did not show any stereoselective induction in the presence of the chiral catalysts (Table 3).The plausible mechanim is schematically presented in the Fig. 2.

Plausible reaction mechanism
4 Conclusion
For the first time, we have reported the catalytic application of l-proline derived aminothiourea 1 in a multicomponent reaction. The bifunctional catalyst 1 demonstrated multiple catalytic role in the multicomponent synthesis of coumarin-based unsymmetrical trisubstituted methanes whose prototype has been found to be acetylcholinesterase inhibitor. The secondary aminothiourea core of the l-proline derived catalyst has shown excellent catalytic activity in the synthesis of coumarin based trisubstituted methanes. The better activity of the catalyst 1 is explained to be the result of dual catalytic role of the secondary amine group as base and nucleophile to accelerate the reaction rate. The method requires low catalyst loading (5 mol%) and very short reaction time (15–30 min) to give excellent yields. The use of water as a reaction medium facilitated isolation of the product by simple filtration and therefore chromatographic separation employing hazardous organic solvents could be avoided. Nevertheless, we report for the first time that catalytic efficiency of bifunctional catalyst is better than cooperative catalysis by two monofunctional catalysts.
References
Mukherjee S, Yang JW, Hoffmann S, List B (2007) Chem Rev 107:5471
Saito S, Nakadai M, Yamamoto H (2001) Synlett 1245
Mase N, Tanaka F, Barbas CF III (2003) Org Lett 5:4369
Mase N, Tanaka F, Barbas CF III (2004) Angew Chem Int Ed 43:2420
Tang Z, Jiang F, Yu L-T, Cui X, Gong L-Z, Mi A-Q, Jiang Y-Z, Wu Y-D (2003) J Am Chem Soc 125:5262
Tang Z, Jiang F, Cui X, Gong L-Z, Mi A-Q, Jiang Y-Z, Wu Y-D (2004) Proc Natl Acad Sci USA 101:5755
Torii H, Nakadai M, Ishihara K, Saito S, Yamamoto H (2004) Angew Chem Int Ed 43:1983
Berkessel A, Koch B, Lex J (2004) Adv Synth Catal 346:1141
Silva F, Sawicki M, Gouverneur V (2006) Org Lett 8:5417
Obregón-Zúñiga A, Milán M, Juaristi E (2017) Org Lett 19:1108
Hernández JG, Juaristi E (2012) Chem Commun 48:5396
Serdyuk OV, Heckel CM, Tsogoeva SB (2013) Org Biomol Chem 11:7051
Siau W-Y, Wang J (2011) Catal Sci Technol 1:1298
Fu J-Y, Huang Q-C, Wang Q-W, Wang L-X, Xu X-Y (2010) Tetrahedron Lett 51:4870
Demir AS, Basceken S (2013) Tetrahedron Asymm 24:515
Kokotos C (2012) J Org Chem 77:1131
Fotaras S, Kokotos CG, Kokotos G (2012) Org Biomol Chem 10:5613
Khandelwal S, Tailor YK, Kumar M (2016) Current Organocatal 3:176
Clarke CJ, Tu W-C, Levers O, Bröhl A, Hallett JP (2018) Chem Rev 118:747
Peng XM, Damu GLV, Zhou C-H (2013) Curr Pharm Des 19:3884
Craig LA, Hong NS, McDonald RJ (2011) Neurosci Biobehav Rev 35:1397
Francis PT, Palmer AM, Snape M, Wilcock GK (1999) J Neurol Neurosurg Psychiatry 66:137
Peters J, Trovaslet M, Trapp M, Nachon F, Hill F, Royer E, Tehei M (2012) Phys Chem Chem Phys 14:6764
Baruah P, Basumatary G, Yesylevskyy SO, Aguan K, Bez G, Mitra S (2018) J Bimol Struct Dyn. https://doi.org/10.1080/07391102.2018.1465853
Bharti R, Parvin T (2015) Synth Commun 45:1442
Bharti R, Parvin T (2015) RSC Adv 5:66833
Bentley TW (2011) Org Biomol Chem 9:6685
Wang W-H, Abe T, Wang X-B, Kodama K, Hirose T, Zhang G-Y (2010) Tetrahedron 21:2925
Pedrosa R, Andrés JM, Gamarra A, Manzano R, Pérez-López C (2013) Tetrahedron 69:10811
Appel R, Chelli S, Tokuyasu T, Troshin K, Mayr H (2013) J Am Chem Soc 135:6579
Acknowledgements
The analytical services provided by Sophisticated Analytical Instrumentation Facility (SAIF), North Eastern Hill University, Shillong, India are acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Basumatary, G., Mohanta, R. & Bez, G. l-Proline Derived Secondary Aminothiourea Organocatalyst for Synthesis of Coumarin Derived Trisubstituted Methanes: Rate Enhancement by Bifunctional Catalyst over Cooperative Catalysis. Catal Lett 149, 2776–2786 (2019). https://doi.org/10.1007/s10562-019-02809-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-019-02809-4