
Abstract
In the present study, a new series of ester analogues of substituted coumarin-3-carboxylic acids were synthesized which were typically accessed via a facile esterification reaction between propargyl alcohol and appropriately substituted coumarin-3-carboxylic acids (1–5). This new environmentally benign solid acid catalyst catalyzed, synthetic eco-friendly approach resulted in a noteworthy progress in synthetic efficiency (89–94 % yield), high purity, operational simplicity, mild reaction conditions, cleaner reaction profiles, recyclability of the catalyst and minimizing the production of chemical wastes without using highly toxic reagents for the synthesis. The molecular structure of compound 6 was authenticated by single crystal X-ray crystallographic analysis. The structure and morphology of the catalyst has been established on the basis of FT-IR, scanning electron microscopy–energy dispersion X-ray spectrometry and transmission electron microscopy. The promising bioactive score against enzymatic inhibition prompted us to carry out acetylcholinesterase inhibition screening of the synthesized compounds (6–10). A computer-aided molecular docking study was carried out to validate the specific binding mode of the newly synthesized compounds into the active site of receptor to bear out the specific binding modes of the compounds.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Over the years, coumarins have been established as the well-known naturally occurring oxygen containing heterocyclic compounds isolated from various plant sources as well as have been synthesized chemically [1]. They are structural subunits in many complex natural products and exhibit wide spectrum of pharmacological activities such as, antitumor [2], anti-HIV (NNRTI) [3], antioxidant [4], tumor necrosis factor-a (TNF-a) inhibition [5], antimicrobial [6], anti-inflammatory and antipyretic [7], antibacterial [8], antifungal [9], serine protease inhibition [10] and anticancer activities [11]. The studies have also shown that naturally occurring as well as the chemically synthesized coumarin analogs exhibit potent acetylcholinesterase (AChE) inhibitory activity [12]. Furthermore, functionalization of the coumarin nucleus has led to the development of novel analogs that are capable of inhibiting Aβ aggregation [13]. The recognition of key structural features within coumarin template has helped in designing and synthesizing new analogs with improved AChE inhibitory activity and additional pharmacological activities including beta secretase (BACE) inhibition associated with decreased Aβ deposition [14, 15] and monoamine oxidase (MAO) inhibition. Preliminary studies by employing Torpedo californica AChE revealed that coumarin and its derivatives are capable of interacting and inhibiting AChE by binding to PAS in a reversible manner [16, 17]. It led the scientists to make modifications in coumarin moiety to synthesize potent AChE inhibitors as potential candidates for managing Alzheimer’s disease (AD) [18].
The synthesis of coumarin derivatives has previously been achieved by the use of various methods reported in the literature including the Pechmann [19], Perkin [20], Knoevenagel [21], Claisen [22], Reformatsky [23] and Wittig reactions [24]. In quest to achieve higher efficiency, several catalysts and reaction conditions were tried which include the use of microwave [25], nano-crystalline ZnO [26], heteropoly acids [27], tetrabutylammonium bromide [28], InCl3 [29], molecular I2 [30], PYBOX-DIPH-Zn(OTf)2 complex [31], Cu(OTf)2 [32], SiCl4/EtOH [33], bases such as K2CO3 [34], NaH [35], palladium-catalyzed base [36]. Earlier investigations required belated reaction times, use of costly and hazardous reagents and tedious workup procedures. In view of immense biological applications of coumarin derivatives, the development of simple and convenient protocol is of considerable interest.
In recent years, the use of silica supported catalysts [37–40] has received considerable attention due to growing environmental concerns. Such reagents not only simplify purification processes but also help in preventing the discharge of toxic reaction residues into the environment. In this respect silica supported sulfuric acid (SiO2–OSO3H) is an attractive candidate. It is a well known catalyst in organic synthesis and has been documented in several organic transformations [41–48]. Silica supported sulfuric acid (SiO2–OSO3H) catalyst was prepared by employing standard procedures depicted in the literature (Scheme 1) [49]. It possesses environmentally benign properties such as non-toxicity, biocompatibility, recyclability, physiological inertness, inexpensiveness, thermal stability. This new synthetic strategy resulted in a remarkable improvement in synthetic efficiency, minimizing the production of chemical wastes without using highly toxic reagent for the synthesis. In addition to efficacy, a major requirement of novel supported reagents concerns their reusability, a factor that has significant environmental and economic impact since the most costly components in a chemical reaction are often not the starting materials but the catalyst [50, 51]. The silica supported sulfuric acid is believed to be good proton source, which can give rise to lewis acid center on carbonyl carbon, followed by simultaneous attack of nucleophile to give corresponding coumarin derivatives.

Synthetic pathway for the synthesis of catalyst
Thus, based on the above findings and in continuation of our interest in the development of efficient, economical and new methodologies, we therefore, report a novel methodology for the synthesis of novel biologically active coumarin-3-carboxylic acid derivatives using silica supported sulfuric acid as the heterogeneous, environmentally benign solid acid catalyst. The efficacy of the catalyst was also examined. After a simple filtration, the catalyst was reused five times without a significant loss in yield. The structure and morphology of the catalyst was established on the basis of FT-IR, scanning electron microscopy–energy dispersion X-ray spectrometry (SEM–EDX) and transmission electron microscopy (TEM). The compounds were also screened for AChE inhibitory activity against standard drug tacrine. Moreover physicochemical calculations have been carried out in order to determine the relationship between the electronic properties and enzymatic inhibition activity of the synthesized compounds (6–10). The promising bioactive score of the synthesized compounds against enzymatic inhibition prompted us to carry out AChE inhibition screening of the synthesized compounds (6–10). A computer-aided molecular docking study was carried out to validate the specific binding mode of the newly synthesized compounds into the active site of receptor to bear out the specific binding modes of the compounds.
2 Experimental
2.1 Materials and General Methods
Chemicals were purchased from Merck and Sigma-Aldrich as ‘synthesis grade’ and used without further purification. Human recombinant AChE (EC: 3.1.1.7) lyophilized powder was purchased from Sigma-Aldrich. The IR spectra were recorded with Shimadzu IR-408 Perkin–Elmer 1800 (FTIR) and its values are given in cm−1. 1H NMR and 13C NMR spectra were run in DMSO-d 6 on a Bruker Avance-II 400 MHz instrument with TMS as internal standard, J values are measured in Hertz. Chemical shifts are reported in ppm (δ) relative to the TMS. Mass spectra were recorded on a JEOL D-300 mass spectrometer. Melting points were determined on a Kofler apparatus and are uncorrected. Elemental analysis (%) C, H, N was conducted using Carlo Erba analyzer model 1108. Thin layer chromatography (TLC) glass plates (20 × 5 cm) were coated with silica gel G (Merck) and exposed to iodine vapors to check the homogeneity as well as progress of reaction.
2.2 Preparation of (SiO2–OSO3H) Catalyst
A 500 mL suction flask fitted with a constant pressure dropping funnel containing chlorosulphonic acid (23.3 g, 0.2 mol) and a gas inlet tube for conducting HCl gas over an absorbing solution (water). It was charged with silica gel (60.0 g) and chlorosulphonic acid was added drop wise over a period of 30 min at room temperature, evolution of profuse amounts of HCl occurred instantaneously. After, the addition was completed the mixture was shaken well for 30 min, a white solid of silica-sulfuric acid (78.5 g) was obtained [49]. The various catalysts i.e. HClO4–SiO2 [52], NaHSO4–SiO2 [53], NH4OAc–SiO2 [54], P2O5–SiO2 [55] and NH2SO3H–SiO2 [56] used for the comparative study with respect to silica-sulfuric acid have been synthesized according to the previously published standard procedures.
2.3 Titration Analysis of (SiO2–OSO3H) Catalyst
The amount of H+ in the silica-sulfuric acid catalyst was determined by acid–base titration according to the following reaction (Scheme 2). The librated H3O+ was titrated by standard NaOH and the amount of H+ in silica-sulfuric acid catalyst was calculated (0.05 g of silica sulfuric acid equal to 0.13 mmol).

Estimation of H+ ion concentration in catalyst by acid base titration
2.4 General Method for the Synthesis of Prop-2-ynyl Derivatives of Substituted Coumarin-3-Carboxylic Acids
To a mixture of substituted coumarin-3-carboxylic acids (1–5) and propargyl alcohol (1 mmol) each, in dichloromethane (20 mL) was added silica supported sulfuric acid (2.5 mol %). The reaction mixture was allowed to stirr at room temperature for 1.5–2 h. During stirring, the clear solution of reaction mixture began to turn thick and solid product precipitated. After completion of the reaction, as evident from TLC, the solid formed was filtered, washed with hot methanol to recover the catalyst. The filtrate containing soluble product was evaporated under reduced pressure to obtain crude product. The crude product obtained was washed with appropriate solvents, filtered, dried and crystallized from appropriate solvents. The catalyst was reused as such without a significant loss in yield.
2.4.1 Prop-2-yn-1-yl-2-oxo-2Hchromene-3-carboxylate, 6
Compound 6 recrystallized from CHCl3–MeOH as colorless crystals [57]; Yield: 93 %, mp 163 °C; Anal. Calc. for C13H8O4; C, 68.42; H, 3.53; found: C, 68.44; H, 3.54. IR \(\nu_{\hbox{max} }^{KBr}\) cm−1: 2,118 (C≡C), 1,735 (C=O, ester), 1,713 (C=O, α-pyrone), 1,615, 1,563 (C=C). 1H NMR (400 MHz, DMSO-d 6, δ, ppm): 8.81 (s, 1H, vinylic-H), 7.86–7.82 (m, 1H, H-8), 7.75–7.72 (m, 1H, H-5), 7.44–7.40 (m, 2H, H-6, H-7), 4.93–4.94 (d, 2H, OCH2), 3.31 (s, 1H, acetylenic proton). 13C NMR (100 MHz, DMSO-d 6, δ, ppm): 163.5 (C=O, ester), 161.8 (C=O, α-pyrone), 154.7 (8a), 134.8 (CH), 130.5 (CH), 124.9 (CH), 117.8 (C-4), 116.7 (C-4a), 115.3 (C-3), 114.5 (CH), 78.2, 78.1 (acetylinic carbons), 52.8 (OCH2). MS (ESI) m/z: 228 [M + H] +.
2.4.2 Prop-2-yn-1-yl-7-hydroxy-2-oxo-2H-chromene-3-carboxylate, 7
Compound 7 recrystallized from CHCl3–MeOH as yellowish solid; Yield: 90 %, mp 245–246 °C; Anal. Calc. for C13H8O5; C, 63.94; H, 3.30; found: C, 63.95; H, 3.34. IR \(\nu_{\hbox{max} }^{KBr}\) cm−1: 3,285 (OH), 2,123 (C≡C), 1,730 (C=O, ester), 1,710 (C=O, α-pyrone), 1,605, 1,561 (C=C). 1H NMR (400 MHz, DMSO-d 6, δ, ppm): 8.79 (s, 1H, vinylic-H), 8.15 (s, 1H, OH), 7.89 (s, 1H, H-8), 7.52–7.48 (m, 1H, H-6), 7.45 (d, 1H, H-5), 4.63–4.64 (d, 2H, OCH2), 3.12 (s, 1H, acetylenic proton). 13C NMR (100 MHz, DMSO-d 6, δ, ppm): 162.3 (C=O, ester), 160.9 (C=O, α-pyrone), 157.2 (C–OH), 154.4 (8a), 140.8 (CH), 132.5 (CH), 128.6 (CH), 118.4 (C-4), 115.2 (C-4a), 114.7 (C-3), 76.2, 75.5 (acetylinic carbons), 51.4 (OCH2). MS (ESI) m/z: 244[M + H] +.
2.4.3 Prop-2-yn-1-yl-7-methoxy-2-oxo-2H-chromene-3-carboxylate, 8
Compound 8 recrystallized from CHCl3–MeOH as colorless crystalline solid; Yield: 94 %, mp 176 °C; Anal. Calc. for C14H10O5; C, 65.12; H, 3.90; found: C, 65.15; H, 3.92. IR \(\nu_{\hbox{max} }^{KBr}\) cm−1: 2,110 (C≡C), 1,725 (C=O, ester), 1,712 (C=O, α-pyrone), 1,615, 1,564 (C=C), 1,225 (C–O), 1H NMR (400 MHz, DMSO-d 6, δ, ppm): 8.52 (s, 1H, vinylic-H), 7.78 (s, 1H, H-8), 7.55–7.51 (m, 2H, H-5, H-6), 4.71(d, 2H, OCH2), 3.85 (s, 3H, CH3), 3.32 (s, 1H, acetylenic proton). 13C NMR (100 MHz, DMSO-d 6, δ, ppm): 165.1 (C=O, ester), 160.2 (C=O, α-pyrone), 153.2 (C-7), 150.1 (8a), 130.4 (CH), 125.6 (CH), 122.3 (CH), 117.8 (C-4), 116.1 (C-4a), 113.4 (C-3), 78.4, 76.8 (acetylinic carbons), 56.1 (CH3), 53.3(OCH2). MS (ESI) m/z: 258 [M + H] +.
2.4.4 Prop-2-yn-1-yl-7-(diethylamino)-2-oxo-2H-chromene-3-carboxylate, 9
Compound 9 recrystallized from CHCl3–MeOH as yellowish solid; Yield: 91 %, mp 190 °C; Anal. Calc. for C17H17NO4; C, 68.21; H, 5.72; N, 4.68; found: C, 68.22; H, 5.71; N, 4.67. IR \(\nu_{\hbox{max} }^{KBr}\) cm−1: 2,120 (C≡C), 1,720 (C=O, ester), 1,710 (C=O, α-pyrone), 1,606, 1,558 (C=C), 1,277 (C–N). 1H NMR (400 MHz, DMSO-d 6, δ, ppm): 8.12 (s, 1H, vinylic-H), 7.43 (s, 1H, H-8), 7.35–7.31 (m, 2H, H-5, H-6), 4.59 (d, 2H, OCH2), 3.45 (q, 4H, N–CH2), 3.20 (s, 1H, acetylenic proton), 1.21 (t, 6H, –CH3). 13C NMR (100 MHz, DMSO-d 6, δ, ppm): 164.1 (C=O, ester), 162.4 (C=O, α-pyrone), 156.5 (C-8a), 149.3 (C-7), 127.4 (CH), 124.5 (CH), 123.0 (CH), 118.8 (C-4), 114.8 (C-4a), 113.4 (C-3), 78.7, 76.4 (acetylinic carbons), 52.5 (OCH2), 45.4 (2 × CH2), 12.3 (2 × CH3). MS (ESI) m/z: 299 [M + H] +.
2.4.5 Prop-2-yn-1-yl-7-amino-2-oxo-2H-chromene-3-carboxylate, 10
Compound 10 recrystallized from CHCl3–MeOH as white solid; Yield: 89 %, mp 210 °C; Anal. Calc. for C13H9NO4; C, 64.20; H, 3.73; N, 5.76; found: C, 64.22; H, 3.71; N, 5.77. IR \(\nu_{\hbox{max} }^{KBr}\) cm−1: 3,305 (NH2), 2,130 (C≡C), 1,735 (C=O, ester), 1,715 (C=O, α-pyrone), 1,598, 1,553 (C=C), 1,267 (C–N). 1H NMR (400 MHz, DMSO-d 6, δ, ppm): 8.09 (s, 1H, vinylic-H), 7.41 (s, 1H, H-8), 7.36–7.33 (m, 2H, H-5, H-6), 5.07 (s, 2H, NH2, D2O exchangeable), 4.55 (d, 2H, OCH2), 3.17 (s, 1H, acetylenic proton). 13C NMR (100 MHz, DMSO-d 6, δ, ppm): 163.6 (C=O, ester), 161.7 (C=O, α-pyrone), 155.2 (C-8a), 147.2 (C-7), 129.3 (CH), 122.1 (CH), 121.4 (CH), 116.2 (C-4), 114.3 (C-4a), 110.2 (C-3), 77.4, 76.2 (acetylinic carbons), 50.4 (OCH2). MS (ESI) m/z: 243 [M + H] +.
2.5 Single crystal X-ray Crystallographic Studies of Compound (6)
Single crystal X-ray data of compound 6 was collected at 100 K on a Bruker SMART APEX CCD diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). The linear absorption coefficients, scattering factors for the atoms and the anomalous dispersion corrections were taken from the International Tables for X-ray crystallography [58]. The data integration and reduction were carried out with SAINT software [59]. Empirical absorption correction was applied to the collected reflections with SADABS and the space group was determined using XPREP [60]. The structure was solved by the direct methods using SHELXTL-97 and refined on F2 by full-matrix least-squares using the SHELXL-97 [61] program package. All non-hydrogen atoms were refined anisotropically. Pertinent crystallographic data for compound 6 is summarized in Table 1.
2.6 In Vitro Acetylcholinesterase Inhibition Activity
The ability of coumarin-3-carboxylic acid (COM-3) and synthesized compounds (6–10) to inhibit AChE activity was assessed by Ellman’s method [62]. AChE stock solution was prepared by dissolving human recombinant AChE (EC: 3.1.1.7) lyophilized powder (Sigma-Aldrich) in 0.1 M phosphate buffer (pH 8.0) containing Triton X-100 (0.1 %). Five increasing concentrations of test compounds were assayed to obtain % inhibition of the enzymatic activity in the range of 20–80. The assay solution consisted of a 0.1 M phosphate buffer pH 8.0, with the addition of 340 µM 5,5′-dithio-bis(2-nitrobenzoic acid), 0.02 unit/mL of human recombinant AChE from human serum and 550 µM of substrate (acetylthiocholine iodide, ATCh). Increasing concentration of tested inhibitor were added to the assay solution and pre-incubated for 5 min at 37 °C with the enzyme followed by the addition of substrate. Initial rate assays were performed at 37 °C with a Jasco V-530 double beam Spectrophotometer. Absorbance value at 412 nm was recorded for 5 min and enzyme activity was calculated from the slope of the obtained linear trend. Assays were carried out with a blank containing all components except AChE to account for the non-enzymatic reaction. The reaction rates were compared and the percent inhibition due to the presence of tested inhibitors was calculated. Each concentration was analyzed in triplicate, and IC50 values were determined graphically from log concentration–inhibition curves (GraphPad Prism 4.03 software, GraphPad Software Inc.). Tacrine was used as a standard inhibitor. AChE inhibition activity of synthesized compounds is presented in Table 2.

2.7 Computational Methods
Molinspiration online database was used to calculate fourteen descriptors (www.molinspiration.com), which are logP, polar surface area, molecular weight, number of hydrogen bond donor, number of hydrogen bond acceptor, number of violation to Lipinski’s rule, number of rotatable bonds, volume, drug likeness includes G protein coupled receptor (GPCR) ligand, ion channel ligand, kinase inhibitor, nuclear receptor ligand, protease inhibitor and enzyme inhibitor, for all synthesized ligands. The method for calculation of MilogP was developed by Molinspiration (miLogP 2.2—2005) programme, based on group contributions. Group contributions have been obtained by fitting calculated logP with experimental logP for a training set of more than twelve thousand of drug-like molecules. Molecular polar surface area (TPSA) was calculated relied based on the published methodology [63], which is virtually a sum of fragment-based contributions. The maps of molecular lipophilicity potential (MLP) and polar surface area (TPSA) were viewed in Molinspiration Galaxy 3D Structure Generator.
2.8 Molecular Docking
The retrieved protein (PDB: 4ey4) used for this purpose was improved by using import and preparation option of MVD software [64] and missing bond order, hybridization state, angle and flexibility for achieving reliable potential binding site in the receptor. All the compounds were designed and structures were analyzed using Chem Draw Ultra3D software and then these structures were energetically minimized using MM2 force field with RMS gradient set to 0.0001 and coordinates of compounds were checked using PRODRG programme [65]. Hex6.1 [66], Discovery studio 4.0 Client [67] and iGEMDOCK [68] softwares were used to sort out top ten molecular docking poses of ligand (compound)–receptor interactions, perform visualization of docked ligands and illustration of basic features of the docked interface and compute energy calculation of docked ligands, respectively.
3 Results and Discussions
3.1 Characterization of the Silica-Sulfuric Acid Catalyst
The FT-IR spectrum of the catalyst is shown in Fig. 1. The IR spectrum was recorded using the KBr disk technique. For silica (SiO2), the major peaks are broad anti symmetric Si–O–Si stretching from 1,200 to 1,000 cm−1 and symmetric Si–O–Si stretching near 800 cm−1. For sulfuric acid functional group, the FT-IR absorption range of the O=S=O asymmetric and symmetric stretching modes lies in 1,120–1,230 and 1,010–1,080 cm−1 respectively, and that of the S–O stretching mode lies in 600–700 cm−1. FT-IR spectrum displays the overlap asymmetric and symmetric stretching bands of SO2 with Si–O–Si stretching bands in the silica sulfuric acid. The spectrum also shows a broad OH stretching absorption around 3,440 cm−1.

FT-IR spectrum of catalyst SiO2–OSO3H
To study the surface morphology of the catalyst, SEM analysis of the catalyst was employed. An examination of SEM images (Fig. 2) showed that catalyst particles were of uneven shape and size, well distributed and no conglomeration of catalyst particles was found on the surface of the silica gel. The increased reactivity of sulfuric acid supported on silica material could be possibly due to the catalyst–support interactions and the resultant changes in the surface properties of the reactive sites. TEM image (Fig. 3) further showed uniform distribution of catalyst particles as black dots on the surface of silica, confirming the formation of the expected catalytic system. EDX analysis (Fig. 4) of the catalyst showed the presence of Si, S and O elements suggesting the formation of SiO2–OSO3H catalytic system.

SEM image of catalyst SiO2–OSO3H

TEM image of catalyst SiO2–OSO3H

Energy dispersive X-ray (EDX) analysis of catalyst SiO2–OSO3H
3.2 Chemistry
The synthetic pathway of a series of new ester derivatives of coumarin-3-carboxylic acid (6–10) is shown in Scheme 3. Herein series of compounds were typically accessed via a facile esterification reaction of substituted coumarin-3-carboxylic acids (1–5) and propargyl alcohol. All the compounds were obtained in excellent yields (89–94 %) with high purity. The molecular structure of compound (6) was further supported by single crystal X-ray crystallographic analysis.

Synthetic pathway for the synthesis of ester derivatives of substituted coumarin-3-carboxylic acids (6–10)
The structural elucidation of the synthesized compounds (6–10) was established on the basis of elemental analysis, IR, 1H NMR, 13C NMR and Mass spectral studies. The analytical results for C, H, N, were within ±0.3 % of the theoretical values. All the synthesized compounds displayed characteristic peaks for α-pyrone carbonyl of coumarin nucleus, ester carbonyl and acetylenic group, resonating at around 1,710–1,715, 1,720–1,735 and 2,110–2,130 cm−1 respectively. Moreover absorption bands resonating at 3,285 and 3,305 cm−1 in compounds 7 and 10, are ascribed to OH and NH2 groups respectively.
In 1H NMR, each compound displayed a sharp singlet around δ 8.09–8.81, attributed to vinylic proton (H-4) of coumarin nucleus, this prominent downfield shift displayed by H-4 protons is attributed to their hydrogen bonding with adjacent ester carbonyl oxygen. A sharp singlet and a doublet peak, integrating for one and two protons each, resonating at δ 3.12–3.31 and 4.55–4.94 is ascribed to acetylinic (≡C–H) and methyleneoxy (–OCH2) protons respectively. Similarly peaks for aromatic protons are already discussed in experimental section. In 13C NMR, characteristic absorption bands resonating at around δ 160.2–160.4, 162.3–165.1, 50.4–53.3 and 75.5–78.7 were ascribed to carbonyl moiety of coumarin nucleus, ester carbonyl, methyleneoxy carbon (–OCH2) and acetylinic carbons (–C≡CH), respectively. A sharp absorption signal at δC 157.2 in case of compound 7 is ascribed to hydroxylated (C-7) carbon. Besides, a series of signals emerging at around δ 121.4–140.8 is ascribed to aromatic carbons. Mass spectral analysis was also in agreement with the proposed structure of the compounds.
In order to develop eco-friendly approach, we explored efficacy of silica supported sulfuric acid (SiO2–OSO3H) by carrying out the reaction of propargyl alcohol with a variety of substituted coumarin-3-carboxylic acids in equimolar ratio (1:1), at room temperature. The reaction proceeded smoothly and resulted in the formation of corresponding products (6–10) in excellent yields (89–94 %) within (1.5–2 h) as monitored by TLC (Table 3). The plausible reaction mechanism for the synthesis of target compounds (6–10) is shown in Scheme 4.

Plausible mechanistic catalytic loop for the synthesis of compounds (6–10)
In order to optimize the reaction conditions a model reaction was conducted using coumarin-3-carboxylic acid (1, 1 mmol) and propargyl alcohol (1 mmol) under various reaction conditions (including loading of catalyst, effect of solvents in terms of yields and time). In order to establish the best reaction conditions, we performed an optimization study using model substrate in the presence of varying amounts of catalyst (SiO2–OSO3H) (Table 4). The model reaction was primarily, tested in absence of catalyst, it was found that reaction took prolonged time for completion with meager yield. It can be inferred from (Table 4, entry 6) that 2.5 mol% of the catalyst is sufficient to get optimum yield in shorter reaction time. Using less than 2.5 mol% catalyst (0.5, 1, 1.5, 2 mol%) moderate yield of the product (65–85 %) was obtained with higher reaction time, while with excess mol% of catalyst (>2.5 mol%) there was no further increment in the yield of the product probably due to the saturation of the catalyst surface. In order to study the solvent effect, the model reaction was carried out in different solvent systems. The reaction was initially, tested under solvent-free condition by grinding method, however only traces of product were obtained (Table 5, entry 1). When the reaction was performed in CH3OH and CH3CH2OH, moderate yield of the products were obtained after prolonged time period (Table 5, entry 5, 6). This can be attributed to the nucleophilic nature of these solvents, due to presence of lone pair of electrons on oxygen atom. Thus nucleophilic competition between these solvents (MeOH, EtOH) and propargyl alcohol for eletrophilic carbonyl carbon of acid will eventually resulted in low yield of desired product. Whereas in acetic acid, reaction again took longer time period (Table 5, entry 4) but there was an improvement in yield, probably CH3COOH facilitates the formation of carbocation by activating the carbonyl group of coumarin acid, thus enhances the electrophilicity of carbonyl carbon, rendering it more feasible for nucleophilic attack. However there was noteworthy improvement in the yield when tetrahydrofuran (THF) was used as a solvent (Table 5, entry 3). When the reaction was carried out in CH2Cl2 (Table 5, entry 2), the yield of the product improved significantly in shorter time period. A comparative study of variety of silica supported catalysts was conducted to confirm the superiority of silica-sulfuric acid as a heterogeneous acid catalyst. The model reaction was initially conducted in absence of catalyst, the reaction took prolonged time and yield was less than 50 % (Table 6, entry 1). When the model reaction was investigated with heterogeneous catalysts such as NaHSO4–SiO2, NH4OAc–SiO2, P2O5–SiO2 and NH2SO3H–SiO2, the reaction took extended time period, and yields were not satisfactory (Table 6, entry 4–7). It was found that, the yield of the product increased significantly when HClO4–SiO2 was added to the model reaction, probably due to its facile proton release which catalyzes the reaction (Table 6, entry 3). However there was noteworthy improvement in the yield and reduction in reaction time when HO3SO–SiO2 was employed as a catalyst (Table 6, entry 2).
The reusability of the silica-chloride catalyst was also examined on model reaction. The catalyst was reused five times and the results demonstrate that catalyst can be reused as such without a significant loss in yield (Table 7). After the first fresh run with 93 % yield, the catalyst was removed by filtration. The recovered catalyst was dried under vacuum at 120 °C for 10 h and tested up to five more reaction cycles. Recycling and reuse of the catalyst showed minimal decreases in yields. The product 6 was obtained in 90, 88, 85, 81, 81 % yields after successive cycles. (Table 7, entries 2–6), thus proving the catalyst’s reusability. To ascertain the variation in morphological features of the recovered catalyst, we carry out its SEM–EDX analysis. It was observed that the composition of the catalytic system was almost consistent with the fresh catalyst (Fig. S1, see supplementary information) and also there was no significant change in the morphology of the catalyst (Fig. 5) as compared to the fresh catalyst.

SEM image of recovered catalyst after five runs at different magnifications
3.3 X-ray Crystallographic Study
Compound 6, once isolated, was found to be air-stable and soluble in all common organic solvents but insoluble in water. X-ray crystallographic analysis reveals that compound 6 crystallizes in the monoclinic structure with space group P21/c. The asymmetric unit of compound 6 is shown in Fig. 6, while other crystallographic parameters are listed in Table S1, S2, S3, S4 and S5 (see supplementary information).

Asymmetric unit showing (a) thermal ellipsoid (50 %) plot and (b) ball and stick model of compound (6)
3.4 AChE Inhibition Results
The parent coumarin-3-carboxylic acid (COM-3) and all the synthesized compounds (6–10) substituted with different groups were analyzed for AChE inhibition activity. It can be inferred from the data shown in Table 2, that compound 10, exhibited the strongest inhibition to AChE with an IC50 value of 0.21 µM, followed by compound 7 (IC50 = 0.24 µM), 8 (IC50 = 0.33 µM), 6 (IC50 = 0.35 µM) and 9 (IC50 = 0.54 µM). The results indicate that all the tested compounds displayed significant AChE inhibitory activity except compound 9. On close examination of the data reported in Table 2, it can be observed that the substituent at C-7 position of the coumarin nucleus inflict prominent effect on the AChE inhibition. When the hydrogen atom at C-7 position in compound 6 (IC50 = 0.35 µM) is replaced by OH group (compound 7), there was significant increase in the AChE inhibitory activity by ±0.11 value. The replacement of hydrogen atom in compound 6 at C-7 by methoxy (OCH3) group (compound 8) leads to a slight increase in the activity by ± 0.02 value. However when the same hydrogen atom was replaced by diethyl amine N(CH3CH2)2 group (compound 9), there was prominent decrease in AChE activity by ±0.19 value. This significant dip in activity of compound 9 is probably due to the bulky substituent N(CH3CH2)2 at C-7 position of coumarin nucleus which restricts it to fit better inside the cavity of AChE. It was observed that the substitution of hydrogen atom in compound 6 by amine (NH2) group (compound 10), exceptionally enhanced the AChE activity by ±0.14 value with IC50 value of 0.21 µM almost comparable to standard drug tacrine (IC50 = 0.19 µM).
In order to study the effect of appended propargyl group in coumarin esters (6–10) on the AChE activity, we have also analyzed parent coumarin-3-carboxylic acid (1) for AChE inhibition. It can be inferred from the data reported in the Table 2, that there has been prominent increase in AChE inhibition of compound 6 by appending propargyl group to the parent coumarin-3-carboxylic acid (1) by ±0.24 value. This suggests that incorporation of propargyl group inflict a noticeable effect on AChE inhibition. The enzyme inhibition activity of parent coumarin acid (1) (IC50 = 0.59 µM) was found to be less in comparison to all the synthesized compounds (6–10). It was found that the nature of substituent and appended propargyl group in the synthesized coumarin esters (6–10) are believed to inflict effect on AChE activity. The results obtained were well supported by the docking studies in which parent coumarin acid (1) displayed poor interaction with the target protein (PDB: 4ey4).
3.5 Molecular Properties Prediction and Drug-Likeness Results
The prediction of absorption, distribution, metabolism and excretion (ADME) properties of the parent coumarin acid (1) and synthesized compounds (6–10) were calculated to depict molecular properties essential for a drug pharmacokinetics in the human body. The physicochemical parameter such as Polar surface area (TPSA), MilogP, number of hydrogen bond donor and acceptor atoms, number of rotatable bonds, molecular volume and violations of Lipinski’s rule of five were premeditated using Molinspiration online property calculation toolkit. Basically, Molinspiration is a cheminformatic software tool which computes the molecular properties of any chemical structure as well as bioactivity score for the most important drug targets such as GPCR ligands, ion channel ligand, kinase inhibitors, nuclear receptor ligand, protease inhibitor and enzyme inhibitor. Membrane permeability and bioavailability are associated with some basic molecular descriptors such as partition coefficient (MiLogP), molecular weight (MW), hydrogen bond acceptors (HBA) and hydrogen bond donors (HBD) count in a molecule. Moreover, number of rotatable bonds is also important to predict the conformational changes and flexibility for binding with receptor or channels. The TPSA is a signifier for the anticipation of passive molecular transport through membranes. The combined parameters, TPSA and molecular volume are inversely proportional to percentage absorption (% ABS) and predict the nature of transport properties of drugs in the intestines and blood–brain barrier crossing. All the compounds (6–10) showed zero violations of Lipinski’s “Rule of Five” which indicated their presence for bioavailability. The molecular lipophilicity potential (MLP) map and polar surface areas (TPSAs) of compounds are shown in Fig. 7 and Table 8. On the close inspection of Table 8, all the synthesized compounds were found in good agreement with Lipinski’s rules for new chemical entity to have good oral bioavailability with no violations. The MilogP valves of all the compounds was found within the range of 0.986–2.764 (<5), without exception, suggesting that these will be soluble in aqueous solution and hence will be able to gain access to membrane surfaces. Moreover the values of TPSA are with the limit i.e. 82 Å, indicating a good permeability of the drug in the cellular plasma membrane. The upper limit for TPSA for a molecule to penetrate the brain is around 90 Å.

3D sketch of the compounds (6–10) with molecular lipophilicity (left side) and polar surface pockets (right side) showing the most lipophilic area (pink color), intermediate lipophilic area (green color), most hydrophobic area (blue color), nonpolar area (gray white color) and polar area (red color) (Color figure online)
The bioactivity score was also calculated for GPCR ligand, ion channel ligand, kinase inhibitor, nuclear receptor ligand, protease inhibitor and enzyme inhibitor. For an average drug the probability is, if the bioactivity score is more than 0.00 then it is active, if −0.50 to 0.00 then moderately active and if less than −0.50 then inactive. On comparing the relative bioactivity scores of tacrine with synthesized compounds (6–10) for various classes, all the compounds displayed moderate to better results towards enzyme inhibition as evident from the Table 9, where compound 10 exhibited promising bioactivity score in comparison to other synthesized compounds. The standard drug tacrine showed highest bioactivity score 0.43 towards enzyme inhibition followed by compounds 10 > 7 > 8 = 6 > 9. We have also compared the bioactivity score of the synthesized compounds (6–10) with the parent coumarin acid (1). The results obtained suggest that the parent coumarin acid (1) displayed lowest bioactivity score (−1.21) towards enzyme inhibition. It was conclude that the appending of propargyl group to the coumarin acids enhance their enzyme inhibition potency. The lowest bioactivity score and AChE inhibition of parent coumarin acid (1) was in good agreement with the docking study where parent coumarin acid (1) displayed unsatisfactory results. The bioactivity score data obtained for synthesized compounds was almost in collaboration with the AChE enzyme inhibition results where compound 10 showed promising AChE inhibition further validated by docking studies. The possible pharmacological effects and clarification of their potential as prodrugs will be an aim to another research.
3.6 Acetylcholinesterase Inhibition Docking Studies
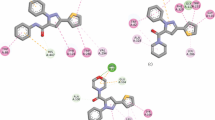
Molinspiration calculations and prediction of molecular properties, values are within the limits, following Lipinski’s rule, fulfilling the requirements of solubility, low polar surface area and total hydrogen bond count are important predictors of good oral bioavailability. On the basis of lipophilicity, the synthesized compounds (6–10) are considered to be as oral drug/lead. The synthesized compounds were subjected to docking studies using Hex6.1, Discovery studio 4.0 Client and iGEMDOCK softwares to predict the binding mode of compounds towards target enzyme (PDB: 4ey4). The docking studies and their scoring functions gave crucial information regarding the orientation of the compounds and the strength of the non-covalent interaction (binding affinity) between molecules (ligand and receptor) in the binding pockets. On the basis of docking simulations, the strong binding affinity of compound 10 with AChE can be explained on the basis of hydrogen bonding as well as orientation and electronic features of the substituents towards the active site of the target enzyme Fig. 8f. The hydrogen bonds formed with amino acids of the protein showed good agreement with the predicted binding affinities obtained by molecular docking studies as verified by AChE inhibition activity data where compound 10 was found to be most potent AChE inhibitor (IC50 = 0.21 µM). The improved activity of the compound 10 in comparison to compounds 6, 7, 8 and 9 can be explained on the basis of its skeleton, that the presence of amine group at C-7 position of coumarin nucleus of compound 10, enhances activity due to the formation of additional hydrogen bonds with amino acid residues PHE 531 and ALA 377 of the protein and easily perform as guest relation with receptor protein (host) Fig. 8f. In addition to this ester carbonyl in compound 10, displayed hydrogen bonding interaction with the amino acid residue TRP 385. The docking studies revealed that the coumarin nucleus plays a major role in stabilizing the ligand–receptor complex by pi-cation interactions with amino acid residue of the target protein as shown in Fig. 8. Moreover, anticholinesterase activity of the target compounds revealed that inhibition properties depend largely on the steric hindrance, orientation and electronic features of the substituents towards the active site and the combined effect of these features can determine the activity of the compounds. This steric effect can be seen operative in case of compound 9 with bulky N(CH3)2 substituent, rendering it less accessible to the active site of the target protein. Additionally, the π–π stacking interactions between the aromatic rings of coumarin nucleus with amino acid residues of the target enzyme further stabilizes the orientation of the molecule into the cavity of receptor as shown in Fig. 9. These extra interactions minimize the energy profile of the docked ligands and stabilize the position of ligands in the cavity of receptor (Fig. 10). Compound 6, 7, 8 and 9 displayed moderate profile of AChE inhibition. The AChE inhibition potency of compounds based on experimental biological assay was seen in the order: Compound 10 > compound 7 > compound 8 > compound 6 > compound 9. The empirical scoring function of iGemDOCK is the estimated sum total of vander waals and H-bonding energies. From the combined table and chart shown in Fig. 10, it is obvious that compound 10 demonstrated better affinity to receptor and showed maximum docking score as it was buried well inside the cavity of target protein.

Available binding sites for parent coumarin acid (1) and compounds (6–10; UNK) and target enzyme (PDB: 4ey4) displaying various interactions with amino acids residues at active site of target protein (a = 3D active Site and b = 2D ligand–receptor interaction diagrams)

π–π interaction of compound 10 with amino acid residues PHE535 and HIS381 of target enzyme

Estimated binding affinities of synthesized compounds (6–10) based on docked poses within active site of target enzyme (PDB: 4ey4)
4 Conclusions
The present work reports the facile, convenient and eco-friendly, silica-sulfuric acid assisted synthesis of bioactive ester derivatives of substituted coumarins. The present protocol offers attractive features such as excellent yields of the products in short times, mild reaction conditions, simple work-up procedure, environmentally benign, economic viability and reusability of the catalyst. We believe that this synthetic approach provides a better scope for the synthesis of ester derivatives of coumarin-3-carboxylic acids and will be a more practical alternative to the other existing methods. Moreover, information obtained from the results, suggest that these synthesized ester derivatives of coumarin-3-carboxylic acids can be used as templates for future research, for designing new entities through modification and derivatization with an improved AChE inhibition affinities for therapeutic purposes.
5 Supplementary information
Crystallographic data for structural analysis has been deposited with the Cambridge Crystallographic Data Centre, (CCDC) bearing no. 996160.
References
Ajani OO, Nwinyi OC (2010) J Heterocycl Chem 47:179
Weber US, Steffen B, Siegers CP (1998) Res Commun Mol Pathol Pharmacol 99:193
Patil AD, Freyer AJ, Drake SE, Haltiwanger RC, Bean MF, Taylor PB, Caranfa MJ, Breen AL, Bartus HR, Johnson RK, Hertzberg RP, Westley JW (1993) J Med Chem 36:4131
Yun BS, Lee IK, Ryoo IJ, Yoo ID (2001) J Nat Prod 64:1238
Cheng JF, Ishikawa A, Ono Y, Arrhenius T, Nadzan A (2003) Bioorg Med Chem Lett 13:3647
Zaha AA, Hazem A (2002) Microbiologica 25:213
Backhouse CN, Delporte CL, Negrete RE, Erazo S, Zuniga A, Pinto A, Cassels BK (2001) J Ethnopharmacol 78:27
Tada Y, Shikishima Y, Takaishi Y, Shibata H, Higuti T, Honda G, Ito M, Takeda Y, Kodzhimatov OK, Ashurmetov O, Ohmoto Y (2002) Phytochemistry 59:649
Stein AC, Alvarez S, Avancini C, Zacchino S, Poser GV (2006) J Ethnopharmacol 107:95
Whittaker M, Floyd CD, Brown P, Gearing AJH (1999) Chem Rev 99:2735
Maly DJ, Leonetti F, Backes BJ, Dauber DS, Harris JL, Craik CS, Ellman JA (2002) J Org Chem 67:910
Changwong N, Sabphon C, Ingkaninan K, Sawasdee P (2012) Phytother Res 26:392
Piazzi L, Cavalli A, Colizzi F, Belluti F, Bartolini M, Mancini F, Recanatini M, Andrisano V, Rampa A (2008) Bioorg Med Chem Lett 18:423
Garino C, Pietrancosta N, Laras Y, Moret V, Rolland A, Quéléver G, Kraus JL (2006) Bioorg Med Chem Lett 16:1995
Ortega DDS, Murphy BP, Velasquez FJG, Wilson KA, Xie F, Wang Q, Moss MA (2011) Bioorg Med Chem 19:2596
Radić Z, Reiner E, Simeon V (1984) Biochem Pharmacol 33:671
Radić Z, Reiner E, Taylor P (1991) Mol Pharmacol 39:98
Rudolf VS, Kovarik Z, Radić Z, Reiner E (1999) Chem Biol Interact 119–120:119
Pechmann VH, Duisberg C (1884) Chem Ber 17:929
Perkin WH, Henry WS (1875) J Chem Soc 28:10
Brufola G, Fringuelli F, Piermatti O, Pizzo F (1996) Heterocycles 43:1257
Cairns N, Harwood LM, Astles DP (1994) J Chem Soc Perkin Trans 1:3101
Shriner RL (1942) The Reformatsky reaction. Wiley, London, p 1:15
Yavari I, Shoar RH, Zonouzi A (1998) Tetrahedron Lett 39:2391
Al-Zaydi KM (2003) Molecules 8:541
Ghosh PP, Das AR (2012) Tetrahedron Lett 53:3140
Khoobi M, Ramazani A, Foroumadi AR, Hamadi H, Hojjati Z, Shafiee A (2011) J Iran Chem Soc 8:1036
Khurana JM, Kumar S (2009) Tetrahedron Lett 50:4125
Rao P, Konda S, Iqbal J, Oruganti S (2012) Tetrahedron Lett 53:5314
Khan AT, Das DK, Islam K, Das P (2012) Tetrahedron Lett 53:6418
Ray SK, Singh PK, Molleti N, Singh VK (2012) J Org Chem 77:8802
Bagdi AK, Majee A, Hajra A (2013) Tetrahedron Lett 54:3892
Salama TA, Ismail MA, Khalil AGM, Elmorsy SS (2012) ARKIVOC ix:242
Karami B, Khodabakhshi S, Eskandari K (2012) Tetrahedron Lett 53:1445
Jung JC, Lee JH, Oh S, Lee JG, Park OS (2004) Bioorg Med Chem Lett 14:5527
Zhang XS, Li ZW, Shi ZJ (2014) Org Chem Front 1:44
Karimian R, Piri F, Safari AA, Davarpanah SJ (2013) J Nanostruct Chem 3:52
Datta B, Pasha MA (2013) ISRN Org Chem 2013:1
Chavan F, Madje B, Bharad J, Ubale M, Ware M, Shingare M, Shinde N (2008) Bull Catal Soc India 7:41
Gawande MB, Hosseinpour R, Luque R (2013) Curr Org Synth 11:526
Heravi MM, Ajami D, Ghassemzadeh M (1999) Synth Commun 29:1013
Oskooie HA, Heravi MM, Sadnia A, Jannati F, Behbahani FK (2008) Monatsh Chem 139:27
Wu H, Shen Y, Fan LY, Wan Y, Zhang P, Chen CF, Wang WX (2007) Tetrahedron 63:2404
Shaterian HR, Ghashang M, Feyzi M (2008) Appl Catal A Gen 345:128
Baltork IM, Mirkhani V, Moghadam M, Tangestaninejad S, Zolfigol MA, Alibeik MA, Khosropour AR, Kargar H, Hojati SF (2008) Catal Commun 9:894
Zolfigol MA, Veisi H, Mohanazadeh F, Sedrpoushan A (2011) J Heterocycl Chem 48:977
Veisi H (2010) Tetrahedron Lett 51:2109
Shirini F, Zolfigol MA, Salehi P (2006) Curr Org Chem 10:2171
Zolfigol MA (2001) Tetrahedron 57:9509
Gawande MB, Brancoa PS, Varma RS (2013) Chem Soc Rev 42:3371
Gawande MB, Rathi AK, Nogueira ID, Varma RS, Branco PS (2013) Green Chem 15:1895
Bandgar BP, Gawande SS, Muley DB (2010) Green Chem Lett Rev 3:49
Breton GW (1997) J Org Chem 62:8952
Gupta R, Gupta M, Paul S, Gupta R (2009) Bull Korean Chem Soc 30:2419
Hasaninejad A, Zare AK, Sharghi H, Niknam K, Shekouhy M (2007) ARKIVOC xiv:39
Aoyama T, Suzuki T, Nagaoka T, Takido T, Kodomari M (2013) Synth Commun 43:553
Pramitha P, Bahulayan D (2012) Bioorg Med Chem Lett 22:2598
International tables for X-ray crystallography, vol III. Kynoch Press, Birmingham, England (1952)
SAINT, Version 6.02, Bruker AXS, Madison, WI (1999)
XPREP, Version 5.1, Siemens Industrial Automation Inc., Madison, WI (1995)
Sheldrick GM (1997) SHELXL-97: program for crystal structure refinement. University of Göttingen, Göttingen
Ellman GL, Courtney KD, Andres VJ, Feather-Stone RM (1961) Biochem Pharmacol 7:88
Ertl P, Rohde B, Selzer P (2000) J Med Chem 43:3714
Thomsen R, Christensen MH (2006) J Med Chem 11:3315
Schuttelkopf AW, Aalten DMFV (2004) Acta Cryst D60:1355
Mustard D, Ritchie DW (2005) Struct Funct Bioinform 60:269
Discovery Studio v4.0 client Copyright @2005-12 Accelrys software Inc
Hsu KC, Chen YF, Lin SR, Yang JM (2011) BMC Bioinform 12:1
Acknowledgments
Authors thank the Chairman, Department of Chemistry, A.M.U, Aligarh, for providing necessary research facilities, University Sophisticated Instrument Facility (USIF), AMU, Aligarh for providing SEM–EDX facilities, SAIF Panjab University Chandigarh for TEM analysis and spectral studies, Division of Bioscience, Dongguk University, Gyeongju, South Korea is acknowledged for bioassay and X-ray analysis. UGC is also gratefully acknowledged for research fellowship to Faheem Ahmad and Ali Mohammed Malla.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Parveen, M., Ahmad, F., Malla, A.M. et al. Catalyst Promoted Synthesis, Computational and Enzyme Inhibition Studies of Coumarin Esters. Catal Lett 144, 2091–2106 (2014). https://doi.org/10.1007/s10562-014-1381-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-014-1381-7