
Abstract
Silica supported cinchona alkaloids were prepared by thio-ene coupled reaction so as to develop novel highly efficient heterogeneous organocatalysts for asymmetric Michael reaction. As-prepared supported cinchona alkaloids were used as heterogeneous catalysts to catalyze the asymmetric Michael reaction between 1,3-dicarbonyl compounds and N-benzylmaleimide, and their catalytic performance was evaluated. It was found that, when toluene is employed as the solvent, silica supported cinchona alkaloid catalysts can catalyze the aforementioned Michael reaction with medium enantiomeric excess (ee) values (up to 87 %) and significant diastereo ratio (dr) values (up to 96:4). In the meantime, they can be recovered and reused for at least five cycles while their stereo-selectivity remains almost unchanged. This means that the title catalysts could be highly efficient organocatalysts for the investigated Michael reaction.
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Asymmetric Michael reaction is one of the most effective approaches for constructing of stereo-selective C–C bond [1, 2]. Kinds of optically active compounds, which are important intermediates of medical compounds and natural product, can be synthesized by Michael addition Reaction easily (like 1,5-dicarbonyl compounds). This can well explain why scholars pay more and more attention to asymmetric Michael reaction [3–5].
Among various efficient asymmetric catalysts for Michael reaction [6–10] and many other reactions [11–13], Cinchona alkaloids and their derivatives are of special significance, because of their high efficiency and hypotoxicity. Unfortunately, the utility of Cinchona alkaloids and their derivatives is often limited by the difficulty of separating them from the reaction system. In order to overcome this drawback, researchers have tried to graft cinchona alkaloid-type organocatalysts with suitable supports thereby acquiring improved recyclability, simplified reaction set-up plus easy experimental procedure, and reduced toxicity [14–16]. Frequently-used supports for such a purpose are polymers, such as polystyrene [17–20], polyethyleneglycol [21, 22], copolymer [23], and even nature polymers [24, 25]. Nevertheless, few reports are currently available about grafting cinchona alkaloid-type organocatalysts with silica, although silica as an inorganic support exhibits high surface area, good thermal and mechanical stabilities, and good chemical inertness [26, 27].
Bearing those perspectives in mind and noticing that cinchona alkaloids are considered as active and highly stereo-selective catalysts for asymmetric Michael reaction, in the present research we choose quinine and cinchonine as original small catalysts to synthesize silica-supported cinchona alkaloid heterogeneous catalysts via thermal induced thiol–ene coupling [28]. The present approach is facile and competitive, and it could be utilized to synthesize organocatalysts possessing high catalytic activity while the catalytic centre of cinchona alkaloids is maintained. This paper reports the synthesis of silica supported cinchona alkaloids and the evaluation of their catalytic performance for asymmetric Michael reaction between 1,3-dicarbonyl compounds and N-benzylmaleimide. Moreover, the recyclability of the catalyst was also evaluated carefully.
2 Experiment
2.1 General
Commercial grade reagents and solvents were used as-received except that specific purification procedure was recommended. Thin layer chromatography (TLC) was conducted with GF254 silica gel plates. Silica and (3-Mercaptopropyl)trimethoxysilane were bought from Changzhou Chemistry Company. Infrared spectra were recorded with an Avatar360 Fourier transform infrared spectrometer (FTIR; Nicolet Company, USA). Nuclear magnetic resonance (NMR) spectra were obtained from Bruker Avance 400 M system, and the chemical shifts of 1H NMR spectra were reported in relation to tetramethyl silane (δ = 0). Analytical high performance liquid chromatography (HPLC) was performed with an Agilent 1100 system with a diode array ultraviolet detector and Daicel Chiralpak AD-H chiral columns.
2.2 Preparation of the Catalysts
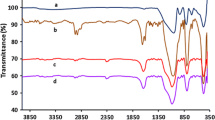
Silica-thiol was prepared according to the literature [29] and was characterized by FT-IR and elemental analysis (S 0.51 mmol%/g). SEM showed that silica–thiol appeared as random power-like, while this kind of silica was imporous. Routes to synthesis of silica supported Cinchona Alkaloids catalysts are outlined in Scheme 1. Silica–thiol (0.50 g, 0.25 mmol), quinine (0.24 g, 0.75 mmol), 2,2-azobisisobutyronitrile (denoted as AIBN; 0.08 g, 0.50 mmol), and anhydrous toluene (30 mL) were sequentially added into three-necked flask (50 mL). Resultant reaction system was saturated with N2 and heated to 70 °C with a water bath, followed by magnetic stirring at 70 °C for 24 h to allow generation of yellow powder. The yellow powder was collected by leaching and washing sequentially with toluene and chloroform, followed by drying in a vacuum oven until its weight remained unchanged to afford target product, silica-supported quinine catalyst (denoted as Cat.I). Silica-supported cinchonine catalyst (denoted as Cat.II) was prepared in the same manners while chloroform rather than anhydrous toluene was used as the solvent and the reaction temperature was kept around 61 °C (Reflux). As-synthesized catalysts were characterized by FT-IR. It could be seen from Fig. 1 (b, c) that the broad band at around 3100 cm−1 was assigned to hydroxyl and amino group, the absorption of 1600 cm−1 was caused by the aromatic rings of cinchona alkaloid. The catalyst load of as-prepared heterogeneous catalysts was also confirmed by elemental analysis (Ratio of S to N; Cat.I: 0.27 mmol/g; Cat.II: 0.23 mmol/g).

Routes to synthesis of silica supported Cinchona Alkaloids catalysts (Me refers to methyl)

FT-IR spectra of silica-SH (a), silica-supported quinine catalyst (b) and silica-supported cinchonine catalyst (c)
2.3 General Procedure for Michael Reaction
1,3-dicarbonyl compound (0.2 mmol), N-benzylmaleimide (0.24 mmol), and a proper amount of as-synthesized catalyst were added into solvent (1 mL). Resultant mixture was heated at pre-set temperature under magnetic stirring, and the reaction system was detected by TLC [ethyl acetate/petroleum ether = 1:2 (volume ratio)]. Upon completion of the reaction, the product was filtered and extracted with ethyl acetate, and then the organic layer was dried with anhydrous Na2SO4, filtered, concentrated and purified by TLC on a silica gel (ethyl acetate/petroleum ether) to afford desired target products.
2.3.1 Ethyl 1-(1-benzyl-2,5-dioxopyrrolidin-3-yl)-2-oxocyclopentanecarboxylate, 3a
Colourless foam. [Daicel Chiralpak AD-H, hexane/i-PrOH = 85/15, 0.75 mL/min, λ 214 nm, major diastereomer: τmajor = 17.2 min, τminor = 20.1 min; minor diastereomer: τmajor = 21.8 min, τminor = 24.2 min]. 1H NMR(CDCl3): δ = 1.22 (t, J = 7.2, 3H), 1.93–2.03 (m, 2H), 2.08–2.20 (m, 1H), 2.35–2.50 (m, 3H), 2.60 (dd, J = 6.0, 18.0, 1H), 2.80 (dd, J = 9.2, 18.0, 1H), 3.46 (dd, J = 6.0, 9.2, 1H), 4.17 (q, J = 7.2, 2H), 4.57 (AB, J = 14.2, 2H), 7.18–7.35 (m, 5H). 13C NMR (CDCl3): δ = 13.9 (CH3), 19.1 (CH2), 31.6 (CH2), 32.6 (CH2), 37.9 (CH2), 42.1 (CH), 42.2(CH2), 60.6 (C), 62.0 (CH2), 127.7 (CH), 128.3 (CH, 2C), 128.5 (CH, 2C), 135.4 (C), 169.5 (C), 175.0 (C), 177.0 (C), 213.5 (C). HRMS: m/z calcd for C19H21NO5: 343.14197; found: 343.14173.
2.3.2 3-(3-acetyl-2-oxotetrahydrofuran-3-yl)-1-benzylpyrrolidine-2,5-dione, 3b
White solid. [Daicel Chiralpak AD-H, hexane/i-PrOH = 80/20, 0.75 mL/min, λ 214 nm, major diastereomer: τminor = 27.9 min, τmajor = 33.2 min; minor diastereomer: τmajor = 20.0 min, τminor = 23.8 min]. 1H NMR (CDCl3): δ = 2.27 (s, 3H), 2.40–2.49 (m, 1H), 2.53 (dd, J = 6.4, 18.4, 1H), 2.68–2.75 (m, 1H), 2.81 (dd, J = 9.2, 18.4, 1H), 3.35 (dd, J = 6.4, 9.2, 1H), 4.27–4.34 (m, 2H), 4.64 (AB, J = 14.4, 2H), 7.23–7.33 (m, 3H), 7.32–7.36 (m, 2H). 13C NMR (CDCl3): δ = 25.9 (CH3), 28.7 (CH2), 31.7 (CH2), 42.1 (CH), 42.6 (CH2), 61.6 (C), 65.9 (CH2), 127.9 (CH), 128.5 (CH, 2C), 128.6 (CH, 2C), 135.3 (C), 173.6 (C), 174.4 (C), 176.1 (C), 200.7 (C). HRMS: m/z calcd for C17H17NO5: 315.11067; found: 315.11085.
2.3.3 Ethyl 2-(1-benzyl-2,5-dioxopyrrolidin-3-yl)-2-methyl-3-oxobutanoate, 3c
White foam. [Daicel Chiralpak AS-H column, hexane/i-PrOH = 80/20, 0.75 mL/min, λ 214 nm, major diastereomer: τminor = 21.3 min, τmajor = 26.2 min; minor diastereomer: τminor = 32.7 min, τmajor = 38.3 min]. 1H NMR (CDCl3): δ = 1.22 (t, J = 7.2, 3H), 1.50 (s, 3H), 2.24 (s, 3H), 2.44 (dd, J = 6.0, 18.4, 1H), 2.85 (dd, J = 9.2, 18.4, 1H), 3.37 (dd, J = 6.0, 9.2, 1H), 4.18 (q, J = 7.2, 2H), 4.64 (AB, J = 14.0, 2H), 7.23-7.40 (m, 5H). 13C NMR (CDCl3): δ = 13.9 (CH3), 18.9 (CH3), 26.8 (CH3), 32.4 (CH2), 42.4 (CH2), 44.9(CH), 61.2 (C), 62.2 (CH2), 127.8 (CH), 128.5 (CH, 2C), 128.7 (CH, 2C), 135.6 (C), 170.7 (C), 175.2 (C), 177.0 (C), 204.2(C). HRMS: m/z calcd for C18H21NO5: 331.14197; found: 331.14163.
2.3.4 3-(1-acetyl-2-oxocyclopentyl)-1-benzylpyrrolidine-2,5-dione, 3d
White foam. [Daicel Chiralpak AD-H, hexane/i-PrOH = 75/25, 0.75 mL/min, λ 214 nm, major diastereomer: τminor = 15.2 min, τmajor = 22.7 min; minor diastereomer: τminor = 11.4 min, τmajor = 13.8 min]. 1H NMR (CDCl3): δ = 1.80–2.00 (m, 3H), 2.19 (s, 3H), 2.35 (dd, J = 6.4, 18.4, 1H), 2.40–2.57 (m, 3H), 2.74 (dd, J = 9.2, 18.4, 1H), 3.55 (dd, J = 6.4, 9.2, 1H), 4.63 (AB, J = 14.0, 2H), 7.24-7.39 (m, 5H). 13C NMR (CDCl3): δ = 19.4 (CH2), 26.2 (CH3), 28.8 (CH2), 31.8 (CH2), 38.4 (CH2), 42.5 (CH2), 43.6 (CH), 68.7 (C), 128.0 (CH), 128.6 (CH, 2C), 128.7 (CH, 2C), 135.4 (C), 174.7 (C), 176.5 (C), 202.0 (C), 213.4 (C). HRMS: m/z calcd for C18H19NO4: 313.13141; found: 313.13121.
2.3.5 3-(2-acetyl-1-oxo-1,2,3,4-tetrahydronaphthalene-2-yl)-1-benzylpyrrolidine-2,5-dione, 3e
White solid. [Daicel Chiralpak AD-H, hexane/i-PrOH = 75/25, 0.75 mL/min, λ 214 nm, major diastereomer: τminor = 24.0 min, τmajor = 22.8 min; minor diastereomer: τminor = 25.8 min, τmajor = 20.3 min]. 1H NMR (CDCl3): δ = 2.19 (s,3H), 2.37–2.54 (m, 3H), 2.63 (dd, J = 6.0, 18.4, 1H), 2.98–3.03 (m, 2H), 3.30 (dd, J = 6.0, 9.2, 1H), 4.73 (AB, J = 14.4, 2H), 7.22–7.37 (m, 5H), 7.39–7.45 (m, 2H), 7.52 (t, J = 7.6, 9.2, 1H), 8.07 (d, J = 7.6, 9.2, 1H). 13C NMR (CDCl3, T = 50 °C): δ = 25.6 (CH2), 29.1 (CH3), 31.1 (CH2), 31.9 (CH2), 42.6 (CH2), 44.2 (CH), 65.5 (C), 127.4 (CH), 127.7 (CH), 128.1 (CH), 128.5 (CH, 2C), 128.6 (CH, 2C), 129.0 (CH), 132.1 (CH), 134.4 (C), 135.9 (C), 142.9 (C), 175.0 (C), 177.1 (C), 196.1 (C), 205.6 (C). HRMS: m/z calcd for C23H21NO4: 375.14706; found: 375.14643.
2.3.6 (S)-(+)-Methyl 2-carbomethoxy-4-nitro-3-phenyl-butyrate, 4a
White solid. [Daicel chiralcel AD-H, hexane/i-PrOH = 95/5, 1.0 ml/min, λ 254 nm, τmajor = 26.2 min, τminor = 40.8 min]. 1H NMR (400 MHz, CDCl3): δ = 7.34–7.22 (m, 5H), 4.95–4.85 (m, 2H), 4.25 (dt, J = 5.2 Hz, 8.8 Hz, 1H), 3.86 (d, J = 9.2 Hz, 1H), 3.77 (s, 3H), 3.57 (s, 3H). 13C NMR (100 MHz, CDCl3): δ = 167.8, 167.2, 136.1, 128.9, 128.3, 127.8, 77.3, 54.7, 52.9, 52.7, 42.9. HRMS: m/z calcd for (C13H15NO6 + H+) 282.0964, found 282.0969.
2.3.7 (+)-Methyl 2-carbomethoxy-4-nitro-3-(4-chloro-phenyl)-butyrate, 4b
White solid. [Daicel chiralcel AD-H, hexane/i-PrOH = 70:30, 1.0 ml/min, λ 220 nm, τmajor = 10.5 min, τminor = 15.6 min]. 1H NMR (400 MHz, CDCl3): δ 7.31 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 4.91 (dd, J = 4.8 Hz, 12.8 Hz, 1H), 4.85 (dd, J = 8.4 Hz, 13.6 Hz, 1H), 4.23 (dt, J = 4.8 Hz, 9.2 Hz, 1H), 8.83 (d, J = 9.2 Hz, 1H), 3.77 (s, 3H), 3.60 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 167.6, 167.0, 134.6, 134.4, 129.3, 129.2, 77.1, 54.4, 53.0, 52.9, 42.3. HRMS: m/z calcd for (C13H14ClNO6 + H+) 316.0583, found 316.0582.
2.3.8 (+)-Methyl 2-carbomethoxy-4-nitro-3-(4-methoxy-phenyl)-butyrate, 4c
Colorless foam. [Chiralpak AD-H, hexane/i-PrOH = 60:40, 1.0 ml/min, λ 220 nm, τminor = 7.6 min, τmajor = 11.9 min]. 1H NMR (400 MHz, CDCl3): δ 7.14 (d, J = 9.2 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 4.89 (dd, J = 4.8 Hz, 12.8 Hz, 1H), 4.83 (dd, J = 8.8 Hz, 13.6 Hz, 1H), 4.19 (dt, J = 4.8 Hz, 13.2 Hz, 1H), 3.82 (d, J = 9.2 Hz, 1H), 3.78 (s, 3H), 3.76 (s, 3H), 3.58(s, 3H). 13C NMR (100 MHz, CDCl3): δ 167.9, 167.3, 159.4, 129.0, 127.8, 114.3, 77.6, 55.2, 54.8, 52.9, 52.8, 42.2. HRMS: m/z calcd for (C14H17NO7 + H+) 312.1070, found 312.1076.
2.4 Evaluation of Recyclability of Catalyst
At the end of the Michael reaction, the used catalyst was recollected by filtering in vacuum, followed by washing with dichloromethane and drying in an oven for 24 h to afford recycled catalyst. As-obtained recycled catalyst was reused directly without further purification.
3 Results and Discussions
The catalytic performances of as-synthesized catalysts in different solvents are listed in Table 1. It can be seen that Cat.I possesses good stereo-selectivity and catalytic activity in low polar aprotic solvents such as toluene, dichloromethane, and chloroform (Entries 1–4), but it exhibits lower stereo-selectivity in polar aprotic solvents (Entries 5 and 6). Unsurprisingly, silica supported cinchona alkaloids do not adapt to protic solvent (Entry 7), due to the formation of hydrogen bonds via either the catalysts or the substrates. The effects of catalyst load on the stereo-selectivity and yield are also summarized in Table 1. It can be seen that decreasing catalyst load to 5 mol% leads to lower yield but has no effect on the stereo-selectivity (Entries 8 and 9). The suitability of Cat.II for different solvents was also explored, and it was found that Cat.II exhibits similar suitability for various solvents as Cat.I does. Summarizing the results listed in Table 1, we can conclude that toluene is the optimal solvent for the Michael reaction under investigation, and the optimal catalyst dosage is 10 mol%.

We further examined the conjugate addition of a variety of trisubstituted carbon Michael donors to N-benzylmaleimide in the presence of Cat.I (Table 2). It can be seen that Cat.I can catalyze the aforementioned Michael reaction successfully. Namely, when cyclic β-ketoesters (Entries 3 and 4) or acyclic β-ketoesters (Entries 5 and 6) are used as Michael donors, reactions proceed with moderate stereo-selectivity. When β-diketones, much more challenging substrates, are used as Michael donors, high diastereo-selectivity is obtained, but fair enantio-selectivity is acquired. Moreover, we carried out relevant reactions at ambient temperature and 0 °C and found that the catalyst possesses higher stereo-selectivity but lower catalytic activity at 0 °C (Table 2). The reaction between aimethyl malonate and N-benzylmaleimide was also been explored, but the Michael production was not obtained. Moreover, by comparing with quinine (Entries 11 and 12), we could draw a conclusion that the supported catalyst possessed similar stereoselectivity but lower catalytic activity.

The catalytic performances of Cat.II for different kinds of Michael reactions were also explored (Table 3). It is worth emphasizing that Cat.II allows access to the opposite enantiomer of the 1,4-adduct with a medium stereo-selectivity. Besides, Cat.II provides similar ee values as Cat.I does (Table 2), but the former exhibits greatly reduced dr values.

To broaden the scope of the methodology, we also adopted silica-supported cinchona alkaloid catalysts to catalyze the Michael reaction between β-alkenylstyrene and dimethyl malonate [30, 31]. As shown in Table 4, target Michael reaction products can successfully be obtained with a medium yield but a quite poor stereo-selectivity (the highest ee value is only about 30 %). But, by comparing with non-supported catalysts (Entries 11 and 12), we could draw a conclusion that the low stereoselectivity of Cat.I and Cat.II inherited from cinchona alkaloids.

Furthermore, the Michael reaction between ethyl 2-oxocyclopentanecarboxylate and N-benzylmaleimide was adopted as a model reaction to evaluate the recyclability of as-synthesized silica-supported cinchona alkaloid Cat.I (Table 5). It can be seen that as the recycle times rises, the reaction proceeds with slightly reduced stereo-selectivity and catalytic activity as well. Reactions which used filtered toluene as reaction solvent (Entries 6 and 7) implied that, although stirring might cause detachment of the alkaloid from the supporter during the aforementioned asymmetric Michael reactions, the detached catalyst was few and could not show catalytic effect. After all, silica-supported cinchona alkaloids as heterogeneous catalysts possess good catalytic activity and reusability.

4 Conclusion
Silica supported cinchona alkaloid catalysts have been successfully prepared by thio–ene coupled reaction. As-prepared silica supported cinchona alkaloids as heterogeneous catalysts exhibit desired performances for the asymmetric Michael reaction between 1,3-dicarbonyl compounds and N-benzylmaleimide, and both their catalytic activity and stereo-selectivity vary with varying reaction conditions such as the type of solvent, catalyst dosage, and reaction temperature. Particularly, they can well catalyze the title Michael reaction with medium ee values (up to 87 %) and significant dr values (up to 96:4) in the presence of toluene as the solvent. Besides, as-prepared silica supported cinchona alkaloids retain almost unchanged stereo-selectivity even after five cycle of recovery and reuse, showing promising potential as highly efficient organocatalysts for the aforementioned Michael reaction. Studies on the immobilization of 9-amino-9-deoxy-epi-cinchonine are underway in order to further improve the performance of the title catalysts.
References
Hashimoto T, Maruoka K (2007) Chem Rev 107:5656
Hélène P (2007) Tetrahedron 63:9267
Vicario JL, Badía D, Carrillo L (2007) Synthesis 2007:2065
Tsogoeva SB (2007) Eur J Org Chem 2007:1701
Mukherjee S, Yang JW, Hoffmann S, List B (2007) Chem Rev 107:5471
Jiang Z, Ye W, Yang Y, Tan CH (2008) Adv Synth Catal 350:2345
Lattanzi A, De Fusco C, Russo A, Poater A, Cavallo L (2012) Chem Commun 48:1650
Bartoli G, Bosco M, Carlone A, Cavalli A, Locatelli M, Mazzanti A, Ricci P, Sambri L, Melchiorre P (2006) Angew Chem Int Ed 45:4966
Luo J, Xu LW, Hay RAS, Lu Y (2008) Org Lett 11:437
Wang YF, Chen RX, Wang K, Zhang BB, Li ZB, Xu DQ (2012) Green Chem 14:893
Yeboah EMO, Yeboah SO, Singh GS (2011) Tetrahedron 67:1725
Czarnecki P, Plutecka A, Gawronski J, Kacprzak K (2011) Green Chem 13:1280
Nakamura S, Hayashi M, Hiramatsu Y, Shibata N, Funahashi Y, Toru T (2009) J Am Chem Soc 131:18240
Itsuno S, Parvez MM, Haraguchi N (2011) Polymer Chemistry 2:1942
Puglisi A, Benaglia M, Chiroli V (2013) Green Chem 15:1790
Trindade AF, Gois PMP, Afonso CAM (2009) Chem Rev 109:418
Qin Y, Yang G, Yang L, Li J, Cui Y (2011) Catal Lett 141:481
Arakawa Y, Haraguchi N, Itsuno S (2008) Angew Chem Int Ed 47:8232
Alvarez R, Hourdin MA, Cavé C, d’Angelo J, Chaminade P (1999) Tetrahedron Lett 40:7091
Thierry B, Plaquevent JC, Cahard D (2001) Tetrahedron Asymmetry 12:983
Danelli T, Annunziata R, Benaglia M, Cinquini M, Cozzi F, Tocco G (2003) Tetrahedron Asymmetry 14:461
Thierry B, Plaquevent JC, Cahard D (2003) Tetrahedron Asymmetry 14:1671
Mandoli A, Pini D, Agostini A, Salvadori P (2000) Tetrahedron Asymmetry 11:4039
Yang L, Zhou D, Qu C, Cui Y (2012) Catal Lett 142:1405
Qin Y, Zhao W, Yang L, Zhang X, Cui Y (2012) Chirality 24:640
Hong J, Lee I, Zaera F (2011) Top Catal 54:1340
Monge-Marcet A, Cattoen X, Alonso DA, Najera C, Man MWC, Pleixats R (2012) Green Chem 14:1601
Lowe AB, Harvison MA (2010) Aust J Chem 63:1251
Bortolini O, Caciolli L, Cavazzini A, Costa V, Greco R, Massi A, Pasti L (2012) Green Chem 14:992
Shi X, He W, Li H, Zhang X, Zhang S (2011) Tetrahedron Lett 52:3204
Li X, Zhang B, Xi ZG, Luo S, Cheng JP (2010) Adv Synth Catal 352:416
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhao, W., Zhang, Y., Qu, C. et al. Catalytic Performance of Silica Supported Cinchona Alkaloids as Heterogeneous Catalysts for Asymmetric Michael Reaction. Catal Lett 144, 1681–1688 (2014). https://doi.org/10.1007/s10562-014-1322-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-014-1322-5