
Abstract
An efficient, rapid, one-pot synthesis of tetrahydrobenzo[b]pyran is achieved via a three-component reaction of aldehydes, 1,3-diketone, malononitrile in 20% ethanol using anhydrous potassium phosphate as a catalyst at room temperature. The key advantages are the short reaction time, high yields, simple work-up, inexpensive catalyst and purification of products by non-chromatographic methods, i.e. by simple recrystallization from ethanol.
Graphical Abstract
An efficient, rapid, one-pot synthesis of tetrahydrobenzo[b]pyran is achieved via a three-component reaction of aldehydes, dimedone, malononitrile in 20% ethanol using anhydrous potassium phosphate as a catalyst at room temperature. The key advantages are the short reaction time, high yields, simple work-up, inexpensive catalyst and purification of products by non-chromatographic methods, i.e. by simple recrystallization from ethanol.

Similar content being viewed by others
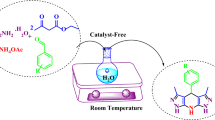
Avoid common mistakes on your manuscript.
1 Introduction
Tetrahydrobenzo[b]pyrans have recently attracted attention as an important class of heterocyclic scaffolds in the field of drugs and pharmaceuticals. These compounds are widely used as anti-coagulant, diuretic, spasmolytic, anticancer and anti-anaphylactin agents [1–5]. Also they can be used as cognitive enhancers for the treatment of neurodegenerative disease including alzheimer’s disease, amyoprophic lateral sclerosis, Huntington’s disease, Parkinson’s disease, AIDS associated dementia and Down’s syndrome as well as for the treatment of schizophrenia and myoclonus [6]. Other than their biological importance, some 2-amino-4H-pyrans have been widely used as photoactive materials [7]. In addition, the tetrahydrobenzo[b]pyran nucleus is an important structural motif of a series of natural products [8, 9] and can be converted into pyridine systems which relate to pharmacologically important calcium antagonists of the dihydropyridine(DHP) [10, 11] type. The importance of these compounds has led the scientific community to synthesize them using the bicomponent condensation [12, 13] of dimedone with α-cyanocinnamonitriles or multicomponent condensation [14–19] of dimedone with aromatic aldehydes and malononitrile.
A detailed literature survey towards the multicomponent synthesis of tetrahydrobenzo[b]pyran revealed that most of the protocols employed for this reaction operate under high thermal activation [14–19], microwave activation [15, 20, 21] and ultrasonic irradiation [22, 23]. Recently, Fotouhi et al. [24] reported electrochemical synthesis of tetrahydrobenzo[b]pyran. There are a few protocols operable at room temperature using N-methylimidazole [25], (S)-proline [26] and (d,l)-proline [27]. Each of the above method has its own merit with at least one of the limitations of low yields, commercially unavailable catalysts, long reaction times, harsh reaction conditions and tedious work-up procedures. Hence, improved methods for multicomponent synthesis of tetrahydrobenzo[b]pyran using inexpensive and less toxic reagents coupled with simple reaction conditions and easier work-up procedures are required.
In our laboratory, we have studied alkylations of Meldrum’s acid [28], nitroaldol condensation [29] and Michael addition of thiols to α,β-unsaturated ketones [30] using anhydrous K3PO4 as a base. Also, K3PO4 has been utilized for oxidative coupling of thiols to disulfides [31], dithiocarbamates [32], aza-Markovnikov addition of N-heterocycles to vinyl esters [33], while hydrated K3PO4 has frequently been used in transition metal catalyzed cross coupling reactions [34]. Thus we explored its efficacy in multicomponent synthesis of tetrahydrobenzo[b]pyrans (Scheme 1).

Three-component synthesis of tetrahydrobenzo[b]pyran
In continuation with our earlier experience with potassium phosphate in Michael additions [27], we envisaged that K3PO4 could be a suitable catalyst for the present transformation, as potassium is oxophilic, the central K+ will make a strong co-ordinate bond with ‘O’ of 1,3-diketone to form its enolate ion (6). The counteranion PO4 3− is sufficiently basic for the formation of cyanoolefin (5) and subsequent Michael addition of enolate of 1,3-diketone (6) on cyanoolefin (5), followed by cyclocondensation to form corresponding tetrahydrobenzo[b]pyran (4). To make the mechanism clear, we have carried out reaction of dimedone and aldehyde (1:1) in ethanol using K3PO4 at room temperature for an hour. However, we get reactants as it is instead of Knoevenagel adduct which illustrated reaction sequence. A plausible mechanism for the multicomponent synthesis of tetrahydrobenzo[b]pyran using K3PO4 in ethanol is proposed (Scheme 2).

A plausible Mechanism for formation of tetrahydrobenzo[b]pyran
Initially, the reaction of anisaldehyde, dimedone, malononitrile and potassium phosphate was carried out in ethanol medium at room temperature. The corresponding 2-amino-3-cyano-7,7-dimethyl-5-oxo-4-(4′-methoxy phenyl)-5,6,7,8-tetrahydro-4H-benzo[b]pyran was obtained rapidly in excellent yield. Encouraged by this result, we then employed this reaction as a template to optimize the reaction conditions (Table 1).
A brief screening of solvents showed that water, chloroform, methanol, acetonitrile and ethanol were less effective than mixed solvent system H2O + C2H5OH (80:20, v/v). We also found that the reaction carried out with other potassium sources such as KH2PO4, K2HPO4, K2CO3 also gave inferior results. Upon examining the influence of the amount of anhydrous K3PO4 on the reaction, it was found that 15 mol% of anhydrous K3PO4 was sufficient to promote the reaction. In the presence of less than this amount, the yield dropped dramatically, even if longer reaction times were used (Table 1, entry 9). When the amount of anhydrous K3PO4 was increased over 15 mol% equivalent, neither the yield nor the reaction time was improved (Table 1, entry 10).
Using the optimized reaction conditions, a range of substituted tetrahydrobenzo[b]pyrans 4a–m were synthesized (Table 2). This method was found to be equally effective for aromatic aldehydes bearing either electron-donating (Table 2, entries 4c–e, 4k) or electron-withdrawing substituents (entries 4g–j, l, m), cyclic aldehyde (entry 4b) as well as for heterocyclic aldehyde (entry 4f). Moreover, the variants of 1,3-diketones could be successfully used for the synthesis of tetrahydrobenzo[b]pyrans. The advantage of this method is its easy work-up, which includes pouring of the reaction mixture into ice water to precipitate a solid, which is filtered off to give sufficiently pure compounds. The products were isolated in better yields and in less reaction time than previously reported methods.
In summary, we disclose here an efficient method for multicomponent synthesis of tetrahydrobenzo[b]pyran at ambient temperature using anhydrous K3PO4 as an inexpensive catalyst. This procedure offers several advantages including mild condition, high yields, inexpensive catalyst, wide scope of substrates and operational simplicity, simple work-up, and purification of products by non-chromatographic methods, i.e., by simple recrystallization from ethanol.
2 Experimental
2.1 General
IR spectra were recorded on a Perkin–Elmer FT-IR 783 spectrophotometer. NMR spectra were recorded on a BrukerAC-300 spectrometer in CDCl3 using tetramethylsilane as internal standard. Melting points are uncorrected.
3 Typical Procedure
A mixture of aldehyde (1 mmol), malononitrile (1 mmol), 1,3-diketone (1 mmol) and K3PO4 (21 mg, 15 mol%) in 20% ethanol (5 mL) was stirred at r.t. for the time indicated in Table 2. The reaction mixture was poured into ice water and just filtered to yield corresponding tetrahydrobenzo[b]pyran. The residue was purified by recrystalization in ethanol to provide the desired product 4 (Table 2).
3.1 Spectral Data of Unknown Compounds
Entry 4b mp 209–210 °C; IR (KBr): 3414, 3327, 3249, 2923, 2193, 1675, 1654, 1594, 1380, 1251, 693 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.11 (s, 3H,CH3), 1.16 (s, 3H, CH3), 1.24–1.71 (m, 11H), 2.9 (s, 2H, –CH2), 2.37 (s, 2H, –CH2), 3.09 (d, J = 2.7 Hz, 1H, –CH), 4.53 (s, 2H, NH2); 13C NMR (75 MHz, CDCl3): δ 26.19, 26.31, 26.56, 27.41, 27.84, 29.27, 30.52, 32.05 34.74, 40.67, 43.79, 50.84, 58.91, 114.18, 120.23, 159.87, 163.12, 196.46; EIMS: m/z 300 (M+).
Entry 4d mp 198–200 °C; IR(KBr): 3369, 3181, 2984, 2185, 1656, 1509, 1469, 1408, 1363, 1249, 778, 696 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.05 (s, 3H, CH3), 1.11 (s, 3H, CH3), 1.19 (s, 3H, CH3), 1.21 (s, 3H, CH3), 2.22 (s, 2H, –CH2), 2.45 (s, 2H, –CH2), 2.83 (m, 1H, CH3–CH–CH3), 4.37 (s, 1H), 4.52 (s, 2H, NH2), 7.12(s, 4H, Ar–H); 13C NMR (75 MHz, CDCl3): δ 23.86, 27.75, 28.79, 29.64, 32.16, 33.63, 35.02, 40.66, 50.67, 63.59, 114.15, 118.81, 126.60, 127.29, 140.50, 147.42, 157.50, 161.46, 195.93; δ EIMS:m/z 336 (M+).
Entry 4e mp 240–242 °C; IR(KBr): 3409, 3315, 3045, 2963, 2927, 2191, 1659, 1692, 1500, 1461, 1404, 1367, 814, 769 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.06 (s, 3H, CH3), 1.11 (s, 3H, CH3), 2.21(s, 2H, –CH2), 2.22 (s, 3H, –CH3), 2.47 (s, 2H, –CH2), 2.52 (s, 3H, –CH3), 4.49 (s, 2H, NH2), 4.63 (s, 1H, CH), 6.73 (s,1H), 6.87 (d, J = 8 Hz, 1H), 7.0 (d, J = 8 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ 19.07, 21.13, 27.58, 28.95, 30.95, 32.29, 40.63, 50.60, 114.67, 127.75, 128.04, 130.41, 132.65, 135.56, 141.66, 157.13, 161.56,161.56, 195.95; EIMS: m/z 322 (M+).
Entry 4m mp 234–236 °C; IR(KBr): 3423, 3334, 3190, 2918, 2228, 2199, 1679, 1653, 1602, 1498, 1456, 1416, 1332, 762 cm−1; 1H NMR (300 MHz, CDCl3): δ = 2.05 (m, 2H, –CH2–CH2–CH2), 2.37(t, 2H, –CH2), 2.61 (t, 2H, CO–CH2), 4.47 (s, 1H), 4.66 (s, 2H, NH2), 7.37(d, J = 8 Hz, 2H, Ar–H), 7.59(d, J = 8 Hz, 2H, Ar–H); 13C NMR (75 MHz, CDCl3): δ 20.09, 27.04, 35.73, 36.61, 100, 110, 114.31, 118.41, 119.81, 128.82, 132.64, 148.61, 158, 163.89, 195.84; δ EIMS: m/z 290 (M+).
References
Foye WO (1991) Prinicipi di Chemico Farmaceutica Piccin. Padova, Italy, p 416
Andreani LL, Lapi E (1960) Bull Chim Farm 99:583
Zhang YL, Chen BZ, Zheng KQ, Xu ML, Lei XH, Yao Xue Xue Bao (1982) 17:17 Chem Abstr 96(1982)135383e
Bonsignore L, Loy G, Secci D, Calignano A (1993) Eur J Med Chem 28:517
Witte EC, Neubert P, Roesch A (1986) Ger Offen DE, 3427985 Chem Abstr 104(1986) 224915f
Konkoy CS, Fick DB, Cai SX, Lan NC, Keana JFW, PCT Int Appl WO 0075123, (2000); Chem Abstr 134(2001)29313a
Arnesto D, Horspool WM, Martin N, Ramos A, Seaone C (1989) J Org Chem 54:3069
Hatakeyama S, Ochi N, Numata H, Takano S (1988) J Chem Soc Chem Commun 21:1202
Gonzalez R, Martin N, Seoane C, Soto J (1985) J Chem Soc, Perkin Trans.1 1202
Martin N, Martin G, Seoane AC, Maco JL, Albert A, Cano FH (1993) Ann Chem 7:801
Maco JL, Martin N, Grau AM, Seoane C, Albert A, Cano FH (1994) Tetrahedron 50(11):3509
Hassanien AA, Zahrran MA, El-Gaby MSA, Ghorab MM (1999) J Indian Chem Soc 76:350
Svarez M, Salfran E, Verdecia Y, Ochoa E, Alba L, Martin N, Martinez R, Quinteiro M, Seoane A, Novoa H, Blaton N, Peeters OM, Ranter CD (2002) Tetrahedron 58:953
Bandgar SB, Bandgar BP, Korbad BL, Totre JV, Patil S (2007) Aust J Chem 60(4):305
Saini A, Kumar S, Sandhu JS (2006) Synlett 5:1928
Bhosale RS, Magar CV, Solanke KS, Mane SB, Choudhary SS, Pawar RP (2007) Synth Commun 37:4353
Seifi M, Sheibani H (2008) Catal Lett 126:275
Wang LM, Shao JH, Tian H, Wang YH, Liu B (2006) J Fluorine Chem 127:97
Balalie S, Bararjanian M, Sheikh-Ahmadi M, Hekmat S, Salehi P (2007) Synth Commun 37:1097
Tu S-J, Gao Y, Guo C, Shi D, Lu Z (2002) Synth Commun 32:2137
El-Rahaman NMA, El-Kateb AA, Mady MF (2007) Synth Commun 37(22):3961
Tu SJ, Jiang H, Zhuang QY, Miao CB, Shi DQ, Wang XS, Gao Y (2003) Chin J Org Chem 23:488
Li JT, Xu WZ, Yang LC, Li TS (2004) Synth Commun 34:4565
Fotouhi L, Heravi MM, Fatehi A, Bakhtiari K (2007) Tetrahedron Lett 48:5379
Lian X-Z, Huang Y, Li Y-Q, Zheng W-J (2008) Monatshefte fur Chemie 139:129
Balalaie S, Bararjanian M, Amani AM, Movassagh B (2006) Synlett 2:263
Guo S-B, Wang S-X, Li J-T (2007) Synth Commun 37:2111
Desai UV, Pore DM, Mane RB, Solabannavar SB, Wadgaonkar PP (2004) Synth Commun 34:25
Desai UV, Pore DM, Mane RB, Solabannavar SB, Wadgaonkar PP (2004) Synth Commun 34:19
Pore DM, Soudagar MS, Desai UV, Thopate TS, Wadagaonkar PP (2006) Tetrahedron Lett 47:9325
Joshi AV, Bhusare S, Baidossi M, Qafisheh N, Sasson Y (2005) Tetrahedron Lett 46:3583
Li RT, Ding PY, Han M, Cai MS (1998) Synth Commun 28:295
Lin S-W, Sun Q, Li R-T, Cheng T-M, Ge Z-M (2007) Synthesis 13:1933
Urawa Y, Ogura K (2003) Tetrahedron Lett 44:271
Acknowledgments
Authors DMP and KAU thank UGC, New Delhi for financial assistance [F.2-3/2007(Policy/SR)] and for the Research Fellowship, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pore, D.M., Undale, K.A., Dongare, B.B. et al. Potassium Phosphate Catalyzed a Rapid Three-Component Synthesis of Tetrahydrobenzo[b]pyran at Ambient Temperature. Catal Lett 132, 104–108 (2009). https://doi.org/10.1007/s10562-009-0074-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-009-0074-0