
Abstract
Purpose
We assessed the effects of TAK-491 (a newly designed potent and selective ARB) alone and in combination with pioglitazone (PIO) on myocardial infarct size (IS).
Methods
Rats received TAK-491 (0.3, 1, 3, or 10 mgkg−1d−1), PIO (1.0 or 2.5 mgkg−1d−1), or PIO 2.5 mgkg−1d−1 with TAK-491 (1 or 3 mgkg−1d−1) for 4 days. On day 5 rats underwent 30-minute coronary artery occlusion and 4-hour reperfusion. Area at risk (AR) was assessed by blue dye and IS by TTC. Left ventricular (LV) dimensions and function was assessed by echocardiography 35 days after infarction.
Results
TAK (1.0–10 mgkg−1d−1), PIO (1.0 to 2.5 mgkg−1d−1), PIO2.5+TAK1.0, and PIO2.5+TAK3.0 significantly reduced IS. IS was the smallest in the TAK 10.0, followed by PIO+TAK 3.0. The protective effects of TAK and PIO were additive, as IS was smaller in the PIO2.5+TAK1.0 than in PIO 2.5 alone (p = 0.008) or TAK1.0 alone (p = 0.002); and in PIO2.5+TAK3.0 than in PIO alone (p < 0.001) or TAK3.0 alone (p < 0.001). TAK, PIO and their combination tended to attenuate LV remodeling and improved LV function 35 days after infarction; however, the differences among individual groups were not statistically significant. Both TAK-491 and PIO increased calcium-dependent nitric oxide synthase activity, whereas only PIO increased COX2 expression and activity. Both PIO and TAK-491 increased Akt, ERK 1/2 and eNOS phosphorylation and inhibited BAX activation.
Conclusions
TAK-491 and PIO independently limited myocardial IS in a dose-dependent fashion; and the effects were additive. The mechanism of protection and the role of TAK-491 in this clinical setting should be further investigated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Angiotensin II receptor blockers (ARBs) are commonly used to treat hypertension [1], diabetes mellitus [2] and heart failure [3], and are commonly prescribed to patients with left ventricular dysfunction after an acute myocardial infarction who are intolerant or unresponsive to angiotensin-converting enzyme inhibitors (ACE-I) to prevent adverse remodeling [4]. However, the role of ARBs in attenuating ischemia-reperfusion (IR) injury is less clear. Several studies have concluded that the ARB losartan was ineffective in limiting infarct size (IS) in the rabbit [5, 6] and rat [7–10]. These studies used isolated heart models [5, 6] or administered losartan intravenously before reperfusion [7] or at 10–30 min before ischemia [9]. To our knowledge, in only three studies the drug was administered orally prior to ischemia and two found that losartan was ineffective in limiting IS [8, 10]. One study reported that 6 week pretreatment with losartan (40 mgkg−1d−1, added to the drinking water) reduced IS in rats exposed to 17-minute ischemia and 2-hour reperfusion [11]. On the other hand, several studies have reported that parenteral administration of the ARB candesartan reduces IS in dogs [12], pigs [13] and rats [14]. However, one study suggested that candesartan does not affect IS and even increases myocardial apoptosis in rats subjected to acute IR [15]. Intravenous valsartan limits IS in rats and dogs [16]. Oral olmesartan (another ARB), started 1 week before infarction and continued for 6 weeks was more effective than the ACE-I ramipril in attenuating remodeling and inflammation in the rat [17]. However, this study did not separate the effects of pretreatment on IR injury from the favorable post-infarction therapeutic effects on remodeling. Regarding other ARBs, pretreatment with oral L-158809 for 10 weeks failed to reduce IS in normocholesterolemic and hypercholesterolemic rabbits, whereas enalapril tended to reduce IS [18].
Several studies suggested that the protective effects of ARBs against IR injury are dependent on activation of angiotensin II type 2 (AT2) receptors, while the AT1 receptors are blocked [13]. Thus, reduced selectivity for AT1 receptors may explain the differences between different ARBs. We found no study that was conducted to assess the effects of pretreatment with oral selective ARB on myocardial IS.
Oral pioglitazone (PIO) is a peroxisome proliferator-activated receptor-gamma agonist that is used to treat diabetes mellitus. Pio protects against IR injury and limits IS in rats [19, 20] and mice [21]. Numerous patients with diabetes mellitus are receiving combination therapy with hypoglycemic agents and ARBs. Therefore, we asked whether pretreatment with oral TAK-491, a new selective AT1 receptor blocker, limits IS in the rat and whether it has additive or synergistic effects with PIO.
1 Methods
1.1 Animal care
Experiments were conducted on male Sprague-Dawley rats. All animals received humane care in compliance with ‘The Guide for the Care and Use of Laboratory Animals’ published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2 Materials
TAK-491 and PIO were provided by Takeda Pharmaceutical Company Limited, Osaka, Japan. Enzyme-linked immunosorbent assay kit for cyclooxygenase (COX) activity and nitric oxide synthase (NOS) activity kit were purchased from Cayman Chemicals (Ann Arbor, MI). Anti-Akt, anti-Thr-308 P-Akt and anti-Ser-473 P-Akt antibodies were purchased from R&D Systems (Minneapolis, MN); anti-endothelial NOS (eNOS), anti-Ser-1177 P-eNOS, anti-inducible NOS (iNOS), anti-extracellular signal regulated kinases (ERK) 1/2 and anti-P-ERK 1/2 from Cell Signaling (Beverly, MA); anti-BAX and anti-6A7 Bax antibodies from Abcam Inc (Cambridge, MA); anti-COX-2 antibodies from Cayman Chemicals (Ann Arbor, MI); anti-P-p38 mitogen-activated protein kinase (MAPK) and monoclonal anti-β-actin antibodies from Sigma-Aldrich (St. Louis, MO).
2.1 Drugs and pretreatment
Rats received 4-day pretreatment with TAK-491 (0.3, 1, 3, or 10 mgkg−1d−1), PIO (1.0 or 2.5 mgkg−1d−1), or PIO 2.5 mgkg−1d−1 with TAK-491 (1 or 3 mgkg−1d−1). Medications were given once a day by oral gavage. Control rats received water by oral gavage. On the 5th day rats underwent 30-minute coronary artery occlusion followed by 4 h reperfusion (IS protocol).
For enzyme activity and protein levels, rats received 4-day pretreatment with water alone, TAK 3.0 mgkg−1d−1, PIO 2.5 mgkg−1d−1, or TAK 3.0 mgkg−1d−1 + PIO 2.5 mgkg−1d−1 as described above. Hearts were explanted without being subjected to IR or after 30-minute ischemia and 4-hour reperfusion, as in the IS protocol (n = 4 in each group).
2.2 Surgical protocol
The rat model of myocardial IR injury has been described in detail [19, 20]. On day 5 rats were anesthetized with ketamine and xylazine, intubated and ventilated (FIO2 = 30%). The left carotid artery was cannulated. The chest was opened and the left coronary artery was encircled with a suture and ligated for 30 min. Isoflurane (1–2.5% titrated to effect) was added after the beginning of ischemia to maintain anesthesia. Then, the snare was released and myocardial reperfusion was verified as a change in the color of the myocardium. The chest was closed and the rats were allowed to recover from anesthesia. Four hours after reperfusion the rats were re-anesthetized, the coronary artery was occluded again, 1.5 ml of Evan’s blue dye 3% was injected into the right ventricle and the rats were euthanized under deep anesthesia. The pre-specified exclusion criteria were a lack of signs of ischemia during ligaton of the coronary artery, lack of signs of reperfusion after the snare was released, prolonged ventricular arrhythmia with hypotension, and an area at risk (AR)≤ 10% of the LV weight.
Heart rate and mean blood pressure were noted at baseline (10 min after the completion of surgery), just before coronary artery occlusion; at 25 min of ischemia; and at 20 min of reperfusion.
2.3 Determination of AR and IS
Hearts were excised and the left ventricle was sliced transversely into six sections. The slices were incubated for 10 min at 37°C in 1% buffered (pH = 7.4) 2,3,5-triphenyl-tetrazolium-chloride (TTC), fixed in a 10% formaldehyde solution and photographed in order to identify the ischemic AR (not colored by the blue dye), the IS (unstained by TTC), and the non-ischemic zones (colored by blue dye). The area of AR and IS in each slice were determined by planimetry, converted into percentages of the whole for each slice, and multiplied by the weight of the slice and the results summed to obtain the weight of the myocardial AR and IS [19–21].
2.4 Echocardiography
Rats were treated as above and underwent 30 min coronary artery ligation followed by reperfusion. Thirty-five days later, rats were anesthetized in an induction chamber using 2.5% isoflurane, and underwent echocardiographic examination using Vevo 770 ultrasound machine equipped with a 25 MHz transducer-710B (Visualsonics-Toronto,Canada). Standard M-mode images were taken in the short axis and long axis position at the level of the papillary muscles for each animal. Ejection fraction was measured by a single-plane area length using 2D parasternal long axis images [22].
2.5 NOS activity
Myocardial samples from the LV of rats not subjected to IR and ARs of rats subjected to 30 min ischemia and 44 h reperfusion were homogenized and centrifuged at 100,000 Xg for 60 min. The supernatant, containing the soluble enzyme inducible NOS (iNOS), and the pellet, containing the membrane-bound endothelial NOS (eNOS) and neuronal NOS (nNOS) [calcium dependent NOS (cNOS)] were separated. The pellet was resuspended in homogenization buffer. NOS activity was determined by measuring the conversion of L-[14C]-arginine to L-[14C]-citrulline by using a commercial kit. For assessing cNOS activity CaCl2 was added to the samples. To assess calcium independent (ciNOS) activity, CaCl2 was omitted from the solution. NOS activity was defined as counts per minute (cpm) [19].
2.6 COX activity
Myocardial samples were rinsed with 0.05 M Tris buffer (PH 7.4) to remove any red blood cells and clots. Samples were homogenized in 5–10 ml of cold buffer (0.1 M Tris-HCL, pH 7.8 containing 1 mM ethylenediaminetetraacetic acid) per gram of tissue, centrifuged at 10,000Xg for 15 min at 4°C, and the supernatant was collected and stored on ice. The COX activity assay kit measures the peroxidase activity of COX, assayed colorimetrically by monitoring the appearance of oxidized N,N,N′,N′-tetramethyl-p-phenylenediamaine (TMPD) at 590 nm. Each myocardial sample was tested in triplicate (the first without an inhibitor; the second with DuP-697, a specific COX2 inhibitor; and the third with Sc560, a specific COX1 inhibitor. COX1 activity was calculated as the difference between total COX activity in the sample without an inhibitor and the sample with Sc560, and COX2 activity as the difference between total COX activity in the sample without an inhibitor and the sample with DuP-697 [19, 21].
2.7 Immunoblotting
The control group comprised rat hearts that were treated with water by means of oral gavage for 4 days and were not subjected to the IR protocol. Myocardial samples from the previously ischemic risk zone of the left ventricular wall were homogenized in in lysis buffer (in mMol): 25 Tris·HCl (pH 7.4), 0.5 EDTA, 0.5 EGTA, 1 phenylmethylsulfonyl fluoride, 1 dithiothreitol, 25 NaF, 1 Na3VO4, 1% Triton X-100, 2% SDS and 1% protease inhibitor cocktail. The lysate was centrifuged at 10,000Xg for 15 min at 4°C. The resulting supernatants were collected. Protein (50 µg) was fractionated by SDS-PAGE (4–20% polyacrylamide gels) and transferred to PVDF membranes (Millipore, Bedford, MA). The membranes were incubated overnight at 4°C with primary antibodies. Bound antibodies were detected using the chemiluminescent substrate (NEN Life Science Products, Boston, MA). The protein signals were quantified with an image-scanning densitometer, and the strength of each protein signal was normalized to the corresponding β-actin signal. Data are expressed as percent of the expression in the control group.
Apoptosis in the border zone was determioned by TUNEL staining, as previously described [23].
2.8 Immunoprecipitations
Samples were homogenized in 0.5-ml of lysis buffer (10 mM HEPES (pH 7.4), 20 mM KCl, 5 mM MgCl2, 0.2 mM ethylenediaminetetraacetic acid) at 4°C and centrifuged at 14,000xg in a precooled centrifuge for 15 min. The supernatant fraction was transferred to a fresh centrifuge tube. We added 40 nM of 6A7 Bax antibody, specific for conformationally changed Bax protein [24, 25] and performed immunoprecipitation overnight on a rotator. Immunoprecipitates were collected by incubating the samples with protein G-Sepharose for 2 h, flowered by pulse centrifugation (5 s in the microcentrifuge at 14,000 rpm). The supernatant was discharged, the beads were washed two times with wash buffer A (10 mM HEPES (pH 7.4), 150 mM NaCl, 2% CHAPS), three times with wash buffer B (10 mM HEPES (pH 7.4), 150 mM NaCl, 0.2% CHAPS), then an additional three times with wash buffer C (100 mM Tris-HCl (pH 8), 100 mM NaCl). Immunoprecipitates were released from the beads in SDS loading buffer and analyzed by means of Western blotting with the Bax 2D2 antibody.
2.9 Statistical analysis
Data are presented as mean ± standard error of the mean. The significance level (α) was 0.05. Body weight, left ventricular weight, the size of the AR and IS, enzyme activity, echocardiographic parameters and protein expression were compared using analysis of variance (ANOVA) with Sidak corrections for multiple comparisons. The differences in heart rate (HR) and mean blood pressure (MBP) were compared using two way repeated measures (treatment X time) ANOVA with Holm-Sidak multiple comparison procedures. Values of P < 0.05 were considered statistically significant.
3 Results
3.1 TAK-491 and PIO decreased IS
Ninty-six rats began the protocol, two rats in the TAK 10 mgkg−1d−1 group died during reperfusion. Therefore, a total of 94 rats were included. Body weight and the size of AR were comparable among groups (Table 1). IS, expressed as a percentage of the LV (Table 1), or percentage of the AR (Fig. 1) was significantly smaller in the TAK 1.0 mgkg−1d−1 (40.9 ± 1.7% of the AR), TAK 3.0 mgkg−1d−1 (30.7 ± 1.7%), TAK 10.0 mgkg−1d−1 (8.2 ± 0.6%), PIO 1.0 mgkg−1d−1 alone (43.2 ± 1.2%), PIO 2.5 mgkg−1d−1 alone (40.1 ± 1.0%), PIO 2.5 mgkg−1d−1 + TAK 1.0 mgkg−1d−1 (32.1 ± 0.7%) and PIO 2.5 mgkg−1d−1 +TAK 3.0 mgkg−1d−1 (21.1 ± 1.5%) than in the control group (50.2 ± 1.7% of the AR). TAK 0.3 mgkg−1d−1 had no significant effect. IS was the smallest in the TAK 10.0 mgkg−1d−1 (p < 0.001 versus each of the other groups), followed by PIO+TAK 3.0 mgkg−1d−1. The differences in IS between PIO + TAK 1.0 mgkg−1d−1 and PIO 2.5 mgkg−1d−1 alone (p = 0.008) or TAK 1.0 mgkg−1d−1 alone (p = 0.002) were statistically significant, as were the differences in IS between PIO+TAK 3.0 mgkg−1d−1 and PIO alone (p < 0.001) or TAK 3.0 mgkg−1d−1 alone (p < 0.001). These findings suggested an additive effect.

The effect of TAK and PIO on IS (% of the area at risk). Rats received 4-day pretreatment with TAK (0.3, 1.0, 3.0 or 10.0 mgkg−1d−1), PIO (1.0 or 2.5 mgkg−1d−1), or PIO 2.5 mgkg−1d−1 + TAK (1.0 or 3.0 mgkg−1d−1). Both TAK and PIO dose dependently limited IS. The effect of PIO+TAK combination was additive
Heart rate (HR) and mean blood pressure (MBP) are presented in Figs. 2 and 3, respectively. Overall the results indicated small but statistically significant differences among groups in both HR and MBP, though they indicated a lack of clinical significance. HR was slower in the TAK 10.0 mgkg−1d−1 group than in the other groups.

Mean heart rate (in beats per minute [bpm]) at baseline, before coronary artery occlusion, 25 min of ischemia and after 20 min of reperfusion. Overall, there was a significant treatment effect (p < 0.001) and time effect (p < 0.001). There was a statistically significant interaction between treatment and time (p = 0.038). The difference among the treatment groups is solely due to significantly slower heart rate in the TAK 10.0 mgkg−1d−1 group than in all other groups. We found no significant differences at any time point among other groups

Mean blood pressure (MBP) (mmHg) at baseline, before coronary artery occlusion, after 25 min of ischemia and after 20 min of reperfusion. Overall, there was a significant treatment (p = 0.005) and time (p < 0.001) effects. However, the interaction between treatment and time was not signigicant (p = 0.053). Differences among the treatment groups were solely due to a significant difference in MBP between the TAK 10.0 mgkg−1d−1 and TAK 0.3 mgkg−1d−1 groups at baseline (142 ± 1 vs. 135 ± 3 mmHg); and between TAK 10.0 mgkg−1d−1 (91 ± 2 mmHg) and TAK 1.0 mgkg−1d−1 (86 ± 1 mmHg) or PIO 2.5 mgkg−1d−1 + TAK 3.0 mgkg−1d−1 (86 ± 2 mmHg) groups at reperfusion. No other differences among groups were significant
Echocardiographic assessment of LV dimensions and function 35 days after infarction shows that both TAK 3.0 mgkg−1d−1 and PIO 2.5 mgkg−1d−1 tended to attenuate LV dilatation and improve LV function (Fig. 4). The effect of TAK+PIO tended to be better than TAK or PIO alone; however, the differences did not reach statistical significance.

Echocardiographic assessment of LV dimensions and function 35 days after infarction. LVIDd- LV internal diameter in diastole; LVIDs- LV internal diameter in systole; FS- fractional shortening; LVVd- LV volume in diastole; LVVs- LV volume in systole; LVEF- LV ejection fraction. * p < 0.01 vs. control IR. # p < 0.04 vs. TAK+PIO
3.2 Enzyme activity
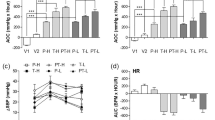
cNOS activity increased after IR in all 4 treatment groups. cNOS significantly increased in the TAK 3.0, PIO and especially in the TAK+PIO group compared to the control group both in hearts not subjected and those who were subjected to IR (Fig. 5a).

Myocardial cNOS (a); ciNOS (b); COX1 (c) and COX2 (d) activity. Rats received 4-day pretreatment with oral water (control), TAK-491 3.0 mgkg−1d−1 (TAK); PIO 2.5 mgkg−1d−1 (PIO); or TAK+PIO. IR- ischemia-reperfusion; NIR- no ischemia reperfusion. * p < 0.01 vs. control NIR; # p < 0.01 vs. TAK+PIO NIR; † p < 0.01 vs. control IR; ‡ p < 0.01 vs. TAK+PIO IR
ciNOS activity was increased in hearts not subjected to IR by PIO. IR increased ciNOS activity in all 4 groups; however, this increase was attenuated by TAK 3.0, PIO and their combination (Fig. 5b).
COX-1 activity was not affected by treatment or IR (Fig. 5c). PIO caused a small, yet significant, increase in COX-2 activity in rats not subjected to IR. IR increased COX-2 activity in all four groups. TAK had no significant effect on the IR induced increase in COX-2 activity, whereas PIO alone or in combination with TAK augmented this increase (Fig. 5d).
3.3 Immunoblotting
IR did not affect total Akt levels. Pretreatment with TAK, PIO and their combination did not affect total Akt levels at 4-hour reperfusion (Fig. 6a&b). IR induced a significant increase in Thr-308 P-Akt, which was further augmented in rats pretreated with PIO, but especially TAK (Fig. 6a&c). The difference in Thr-308 P-Akt levels between the TAK alone and PIO alone was significant (p < 0.001). There was no difference between the TAK alone and the TAK+PIO group; however, the difference between PIO alone and TAK+PIO was statistically significant (p < 0.001). IR also increased myocardial Ser-473 P-Akt levels. Pretreatment with PIO and TAK caused a singificant increase in Ser-473 P-Akt levels compared with the control group, without a significant difference between TAK and PIO. However, Ser-473 P-Akt levels were significantly higher in the TAK+PIO group than in the TAK alone, PIO alone or the control group (Fig. 6a&d).

a. Samples of immunoblots (a) and densitometric analyses of myocardial levels of total Akt (b), Thr-308 P-Akt (c) and Ser-473 P-Akt (d). Rats were treated with vehicle and hearts were explanted without being subjected to IR (Sham), or were pretreated with water (control), PIO 2.5 mgkg−1d−1 (PIO); TAK-491 3.0 mgkg−1d−1 (TAK); or TAK+PIO and were subjected to IR. * p < 0.01 vs. Cont; # p < 0.01 vs. TAK+PIO
IR did not affect total ERK 1/2 levels. Pretreatment with TAK, PIO and their combination did not affect total ERK 1/2 levels at 4-hour reperfusion (Fig. 7a&b). Thr-202/ Thr-204-P-ERK 1/2 levels increased after IR and further increased after pretreatment with PIO, and especially with TAK alone or in combination (Fig. 7a&c). The difference between TAK and PIO was statistically significant (p < 0.001). Pretreatment with TAK+PIO resulted in higher levels of P-ERK 1/2 than treatment with PIO alone (p < 0.001) or TAK alone (p = 0.06).

Samples of immunoblots (a) and densitometric analyses of myocardial levels of total ERK 1/2 (b) and P-ERK 1/2 (c). Rats were treated with vehicle and hearts were explanted without being subjected to IR (Sham), or were pretreated with water (control), PIO 2.5 mgkg−1d−1 (PIO); TAK-491 3.0 mgkg−1d−1 (TAK); or TAK+PIO and were subjected to IR. * p < 0.01 vs. Cont; # p < 0.01 vs. TAK+PIO; & p = 0.06 vs. TAK+PIO
IR did not affect total eNOS levels. Pretreatment with TAK, PIO and their combination did not affect total eNOS levels after 4 h of reperfusion (Fig. 8a). Myocardial Ser-1177 P-eNOS levels were significantly increased by IR. Pretreatment with PIO significantly augmented this increase (Fig. 8b). Pretreatment with TAK resulted in higher levels of P-eNOS compared with those of the control (p < 0.001) or the PIO (p = 0.005) groups. Pretreatment with TAK+PIO significantly heightened levels of P-eNOS compared with pretreatment with PIO alone (p < 0.001) or TAK alone (p < 0.001).

a. Samples of immunoblots and densitometric analyses of myocardial levels of total eNOS (a), Ser-1177 P-eNOS (b), iNOS (c) and COX2 (d). Rats were treated with vehicle and hearts were explanted without being subjected to IR (Sham), or were pretreated with water (control), PIO 2.5 mgkg−1d−1 (PIO); TAK-491 3.0 mgkg−1d−1 (TAK); or TAK+PIO and were subjected to IR. * p < 0.01 vs. Cont; # p < 0.01 vs. TAK+PIO
IR enhanced myocardial iNOS levels. Pretreatment with PIO (p < 0.001), but not TAK (p = 0.818) further increased iNOS levels. Myocardial iNOS levels in the TAK+PIO group were significantly higher than in the control group (p < 0.001), the TAK alone group (p < 0.001), but not the PIO alone group (p = 0.059) (Fig. 8c).
COX2 levels increased after IR. PIO (p < 0.001), but not TAK (p = 0.998) augmented the increase (Fig. 8d). COX2 levels in the TAK+PIO group were significantly higher than in the control group (p < 0.001) and the TAK only group (p < 0.001), but were similar to those seen in the PIO alone group (p = 1.00).
IR increased myocardial levels of P-p38 MAPK. However, TAK, PIO and their combination had no significant effect on P-p38 levels (Fig. 9a&b).

A sample of immunoblot (a) and densitometric analysis (b) of myocardial levels of P-p38. A sample of immunoprecipitations with monoclonal anti-Bax 6A7 antibodies followed by immunoblotting with polyclonal anti-Bax antibodies (c). Rats were treated with vehicle and hearts were explanted without being subjected to IR (Sham), or were pretreated with water (control), PIO 2.5 mgkg−1d−1 (PIO); TAK-491 3.0 mgkg−1d−1 (TAK); or TAK+PIO and were subjected to IR. * p < 0.01 vs. control; # p < 0.01 vs. TAK+PIO. d. TUNEL staining for apoptosis in the border zone. * p < 0.001 vs. control IR; # p < 0.001 vs. TAK+PIO; $ p = 0.010 vs. TAK+PIO
TAK, PIO and their combination attenuated activation of Bax by IR (Fig. 9c). IR increased apoptosis (Fig. 9d). Both TAK and PIO attenuated apoptosis. The effect of TAK+PIO combination was significantly greater than PIO alone and tended to be stronger than TAK alone, suggesting additive effect.
4 Discussion
The major findings of the current study are that pretreatment with TAK-491, a novel highly selective AT1 receptor blocker, and low-dose PIO limited IS in the rat, without significant hemodynamic effects. Moreover, the combination of TAK-491 and PIO had additive effects on myocardial protection. The protective effects seem to be long lasting, as both TAK and PIO tended to attenuate LV remodeling and to preserve LV systolic function, as assessed by echocardiography 35 days after infarction. Both TAK and PIO augmented Akt, ERK 1/2, eNOS phosphorylation and cNOS activity following IR. PIO, but not TAK increased COX2 levels and activity. PIO, but not TAK increased iNOS levels; however, ciNOS activity following IR was significantly lower in the PIO group than in the control group. TAK alone also attenuated the increase in ciNOS activity following IR. P-p38 levels were increased by IR in the control group. Both TAK and PIO attenuated the increase in Bax activation, but did not affect P-p38 levels after IR. Both TAK and PIO and their combination attenuated apoptosis.
Studies using animal models are usually limited to one drug intervention. However, in the clinical setting, patients usually receive several drugs in combination. The combination of ARB with pioglitazone is commonly used in the clinical setting, as both drugs are indicated for diabetic patients. The uniqueness of the present study is that oral ARB can protect the heart and that there is an additive effect with PIO. Another novel aspect is that pretreatment before infarction, without continuation of therapy after infarction, tends to improve cardiac remodeling and preserve function.
It has been suggested that the myocardial protective effects of ARBs against IR injury is mediated by activation of AT2 receptors by angiotensin II, while the deleterious effects of AT1 receptors are blocked by the ARBs [13, 14]. Blocking the AT2 receptors with PD123319 abrogates the IS limiting effects of candesartan [12–14]. However, PD123319 does not block the antiapoptotic effects of candesartan [14]. Valsartan [16] and candesartan [12, 14] increase AT2 receptor protein levels in the myocardial ischemic zone. It was suggested that activation of the AT2 receptors leads to release of bradykinin with downstream activation of protein kinase C (PKC), NOS and eicosanoid release [12, 13, 26]. Indeed, in the present study, TAK augmented eNOS phosphorylation at Ser-1177 and cNOS activity, both at baseline and especially after IR. On the other hand, TAK attenuated the increase in ciNOS activity following IR. Pretreatment with the bradykinin B2-receptor antagonist HOE140 attenuates the IS-limiting effects of candesartan [12, 13].
Akt phosphorylation
kt is part of the Reperfusion Injury Salvage Kinase signaling (RISK) which confers protection against IR injury [27]. In the present study, we observed that PIO, and especially TAK, increased Akt phosphorylation after IR injury. Previous studies have shown that ARBs increase Akt phosphorylation in the hearts [28], leading to eNOS phosphorylation.
Previously we showed that 3-day pretreatment with PIO does not significantly increase myocardial Akt phosphorylation in the rat. However, we formerly studied P-Akt levels in hearts that were not exposed to IR injury [19], whereas in the present study we assessed P-Akt levels after 30 min of ischemia and 4 h of reperfusion.
4.1 ERK 1/2 phosphorylation
ERK 1/2 is also part of the RISK signaling and confers protection against IR injury [27]. In the present study we found that TAK, and to a lesser extent PIO, augmented ERK 1/2 phosphorylation after IR injury. The combination of TAK+PIO resulted in significantly higher levels of P-ERK 1/2, suggesting an additive effect.
Previous studies suggested that ARBs attenuate mechanical strain-induced increases in ERK 1/2 phosphorylation in mesangial cells [29] and in human aortic smooth muscle cells [30]. Valsartan attenuates ERK 1/2 activation in skin fibroblats by blocking angiotensin II-induced transactivation of epidermal growth factor receptors [31]. Losartan prevents upregulation of ERK 1/2 in endothelial cells [32]. Thus, the effect of TAK-491 on the heart after IR injury differs from the previously reported effects of ARBs on ERK 1/2 activation in other models. This observation may represent unique propertiens of TAK-491 or may be related to different class effect of ARBs on the heart exposed to IR injury.
eNOS activation
eNOS is involved in the delayed form of ischemic preconditioning [33] and is essential for the IS-limiting effects of statins [34], but not of PIO [21]. Losartan increases eNOS phosphorylation in the rat heart [10]. In human aortic endothelial cells, valsartan induced eNOS phosphorylation is dependent on PI3K, but not on protein kinase A (PKA), protein kinase C (PKC) or adenosine monophosphate-activated protein kinase (AMPK) [35]. ARBs prevent NOS uncoupling and the production of superoxide, increasing NO availability [36]. The nonspecific NOS inhibitor (N(G)-monomethyl-L-arginine (L-NMMA) and the protein kinase C (PKC) inhibitor (chelerythrine) abrogate the protective effects of candesartan against IR injury in dogs [12].
In our study both TAK and PIO increased eNOS phosphorylation and cNOS activity. There was an additive effect of PIO and TAK, as P-eNOS levels and cNOS activity were significantly higher in the TAK+PIO group than in the TAK alone and PIO alone groups. We have previously shown that PIO increases myocardial Ser-633 and Ser-1177 P-eNOS levels in the wild type and iNOS−/− mice [21], but not in the rat [19]. However, in both previous studies myocardial levels were assessed after 3-day pretreatment without exposing the hearts to ischemia. PIO significantly reduces IS in eNOS−/− mice and in wild type mice treated with L-NAME, suggesting that the protective effect of PIO is not eNOS dependent [21].
4.2 iNOS
Upregulation of iNOS is essential for mediating the IS-limiting effects by the delayed form of ischemic preconditioning [33] and statins [34]. On the other hand, we have previously shown that the IS-limiting effect of PIO is iNOS-independent [21]. In the present study PIO, but not TAK, increased iNOS protein levels in hearts exposed to IR injury. Of interest, myocardial ciNOS activity was attenauted by both TAK and PIO, suggesting that the PIO-induced iNOS is inactive after IR injury.
Losartan inhibits upregulation of iNOS in the cardiomyopathy hamster heart while it increases eNOS phosphorylation [28]. Therefore, it seems that inhibition of iNOS may be a class effect of ARBs.
4.3 COX2
The IS-limiting effects of the delayed form of ischemic preconditioning [37], statins [38] and PIO [19] is dependent on COX2 upregulation. Previously, we have shown that pretreatment with PIO upregulates COX2 expression and enhances COX2 activity [19]. In the present study we confirm that PIO upregulated COX2 expression and activity. In contrast, TAK did not affect COX2 expression and activity. In the present study we did not assess whether COX2 inhibition abrogates the protective effect of TAK , as it does to the protective effects of statins and PIO; however, the fact that COX1 and COX2 activity have not been affected by TAK suggests that the protective effect of TAK is COX-independent.
4.4 p38 mitogen-activated protein kinases (MAPK)
P38 is member of the “death kinases” that increases apoptosis. We are showing that IR injury increased p38 phosphorylation. PIO and TAK had no effect on P-p38 levels after IR. Kumar et al have also shown that losartan does not affect P-p38 levels in isolated rat hearts exposed to ischemia-reperfusion [39], whereas AT2 inhibition by PD123,319 leads to an increase in P-p38 levels [39]. It has been shown that the ARB L-1258,809 attenuated cardiac p38 activation in spontaneously hypertensive heart failure rats [40]. Thus, different effects of ARBs on p38 activation are seen in different experimental models.
4.5 Bax
Bax activation is involved in cell death by means of apoptosis. PIO and TAK attenuated Bax activation, confirming inhibition of apoptosis. Candesartan inhibits apoptosis in hearts exposed to IR injury [14]. Losartan prevents Bax activation in cardiomyocytes exposed to stretch [41]. Thus, previous studies support our findings that ARBs prevent activation of the proapoptotic pathways; however, in our study only activation of Bax was attenuated, whereas p38 phosphorylation was unaffected.
4.6 PIO and myocardial protection
We have previously shown that 3-day pretreatment with oral PIO at a dose of 10 mgkg−1d−1 [19, 21] and 5 mgkg−1d−1 [20] limits IS in rats and mice. In the present study 4-day pretreatment with oral PIO 2.5 mgkg−1d−1 and even 1.0 mgkg−1d−1 significantly reduced IS in the rat. We have previously shown that the protective effects of PIO are dependent on cytosolic phospholipase A2 (cPLA2) and COX-2 activation [19, 21] with subsequent increased production of prostaglandin I2 [19, 21] and 15-epi-lipoxin A4 [42]. In contrast to the protective effects of statins that are eNOS- and iNOS-dependent [34], the IS-limiting effects of PIO are iNOS-independent and only partially dependent on eNOS [21].
Limitations
In the present study we wanted to use medium-term treatment duration. In our previous studies we usually used 3 day pretreatment [19–21, 38]. In a recent study, we extended the treatment with pioglitazone to 14 days and the protective effect was preserved [43]. The present study intended to find the optimal doses and possible additive effects of the two drugs. In future studies we will further explore different treatment periods. In general, the doses of drugs used in rat and mouse models are much higher than the doses used in the clinical setting. It has to be remembered that the affinity of the drugs to their ligands is different in humans and animals. Absorption, pharmacokinetics and pharmacodynamics are also different between humans and small animals. In our previous studies we treated rats and mice with PIO 10 mg/kg/d [19, 21] and 5 mg/kg/d [20, 43]. In the present study we decreased the dose of PIO to 2.5 mg/d and we still show a protective effect. In humans the limiting factor for the dose of ARB is blood pressure. Here we did not observe major effects of the drugs on blood pressure or heart rate. Indeed, the results should not be directly extrapolated to humans and clinical studies are needed to examine the potential protective effects of TAK-491 alone and in combination with PIO (or other drugs) on myocardial (and brain or other organs) protection in patients. In the present study we are showing additive effects when the drugs are combined. Our intention was to show therapeutic effects with relatively low doses that may be more relevant to the clinical setting and not to use the highest doses.
We used 30 min of coronary artery occlusion. Thirty minute coronary artery occlusion is considered standard. Most studies in infarct size in rats and mice are using 30 min of occlusion (some are using up to 40 min). It should be remembered that even the protective effects of ischemic preconditioning are lost when the duration of ischemia is extended [44]. Thus, the protective effects of these drugs with different ischemic times should also be explored.
In conclusion, TAK-491, a selective AT1 receptor blocker and PIO, a PPAR-γ agonist used to treat diabetes mellitus, protect against IR injury and limit myocardial IS. However, it seems that the signaling pathways of both drugs are somewhat different. Although both TAK and PIO augmented Akt, ERK 1/2 and eNOS phosphorylation, as well as cNOS activity following IR; only PIO, but not TAK increased COX2 levels and activity. Both TAK and PIO attenuated the increases in Bax activation, but did not affect P-p38 levels after IR. The effect of TAK+PIO combination was additive. Further studies are needed to assess the signaling pathways involving the protective effects of ARBs against IR injury and the potential favorable effects of combining ARBs with other drugs which are known to have protective effects, such as statins and thiazolidinediones, including PIO.
References
Dahlof B, Devereux RB, Kjeldsen SE, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003.
Lindholm LH, Ibsen H, Dahlof B, et al. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:1004–10.
Young JB, Dunlap ME, Pfeffer MA, et al. Mortality and morbidity reduction with Candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: results of the CHARM low-left ventricular ejection fraction trials. Circulation. 2004;110:2618–26.
Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:E1–E211.
Diaz RJ, Wilson GJ. Selective blockade of AT1 angiotensin II receptors abolishes ischemic preconditioning in isolated rabbit hearts. J Mol Cell Cardiol. 1997;29:129–39.
Liu Y, Tsuchida A, Cohen MV, Downey JM. Pretreatment with angiotensin II activates protein kinase C and limits myocardial infarction in isolated rabbit hearts. J Mol Cell Cardiol. 1995;27:883–92.
Liu YH, Yang XP, Sharov VG, et al. Paracrine systems in the cardioprotective effect of angiotensin-converting enzyme inhibitors on myocardial ischemia/reperfusion injury in rats. Hypertension. 1996;27:7–13.
Sladek T, Sladkova J, Kolar F, et al. The effect of AT1 receptor antagonist on chronic cardiac response to coronary artery ligation in rats. Cardiovasc Res. 1996;31:568–76.
Ozer MK, Sahna E, Birincioglu M, Acet A. Effects of captopril and losartan on myocardial ischemia-reperfusion induced arrhythmias and necrosis in rats. Pharmacol Res. 2002;45:257–63.
Matsuhisa S, Otani H, Okazaki T, et al. Angiotensin II type 1 receptor blocker preserves tolerance to ischemia-reperfusion injury in Dahl salt-sensitive rat heart. Am J Physiol Heart Circ Physiol. 2008;294:H2473–2479.
Zhu BQ, Sievers RE, Browne AE, et al. Comparative effects of aspirin with ACE inhibitor or angiotensin receptor blocker on myocardial infarction and vascular function. J Renin Angiotensin Aldosterone Syst. 2003;4:31–7.
Jugdutt BI, Balghith M. Enhanced regional AT(2)-receptor and PKC(epsilon) expression during cardioprotection induced by AT(1)-receptor blockade after reperfused myocardial infarction. J Renin Angiotensin Aldosterone Syst. 2001;2:134–40.
Jalowy A, Schulz R, Dorge H, Behrends M, Heusch G. Infarct size reduction by AT1-receptor blockade through a signal cascade of AT2-receptor activation, bradykinin and prostaglandins in pigs. J Am Coll Cardiol. 1998;32:1787–96.
Jugdutt BI, Menon V. AT2 receptor and apoptosis during AT1 receptor blockade in reperfused myocardial infarction in the rat. Mol Cell Biochem. 2004;262:203–14.
Chen M, Hamada M, Hiasa G, et al. An angiotensin II type 1 receptor blocker, candesartan, increases myocardial apoptosis in rats with acute ischemia-reperfusion. Hypertens Res. 2001;24:323–9.
Jugdutt BI, Menon V. Valsartan-induced cardioprotection involves angiotensin II type 2 receptor upregulation in dog and rat models of in vivo reperfused myocardial infarction. J Card Fail. 2004;10:74–82.
Sandmann S, Li J, Fritzenkotter C, et al. Differential effects of olmesartan and ramipril on inflammatory response after myocardial infarction in rats. Blood Press. 2006;15:116–28.
Hoshida S, Yamashita N, Kuzuya T, Hori M. Differential effects of long-term renin-angiotensin system blockade on limitation of infarct size in cholesterol-fed rabbits. Atherosclerosis. 2000;149:287–94.
Ye Y, Lin Y, Atar S, et al. Myocardial protection by pioglitazone, atorvastatin, and their combination: mechanisms and possible interactions. Am J Physiol Heart Circ Physiol. 2006;291:H1158–1169.
Ye Y, Lin Y, Perez-Polo JR, Birnbaum Y. Oral glyburide, but not glimepiride, blocks the infarct-size limiting effects of pioglitazone. Cardiovasc Drugs Ther. 2008;22:429–36.
Ye Y, Lin Y, Manickavasagam S, et al. Pioglitazone protects the myocardium against ischemia-reperfusion injury in eNOS and iNOS knockout mice. Am J Physiol Heart Circ Physiol. 2008;295:H2436–2446.
Kido M, Du L, Sullivan CC, et al. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46:2116–24.
Ye Y, Hu Z, Lin Y, Zhang C, Perez-Polo JR. Down-regulation of microRNA-29 by antisense inhibitors and a PPAR-{gamma} agonist protects against myocardial ischemia-reperfusion injury. Cardiovasc Res. 2010.
Murphy KM, Streips UN, Lock RB. Bcl-2 inhibits a Fas-induced conformational change in the Bax N terminus and Bax mitochondrial translocation. J Biol Chem. 2000;275:17225–8.
Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene. 2001;20:7779–86.
Sato M, Engelman RM, Otani H, et al. Myocardial protection by preconditioning of heart with losartan, an angiotensin II type 1-receptor blocker: implication of bradykinin-dependent and bradykinin-independent mechanisms. Circulation. 2000;102:III346–351.
Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217–34.
Matsuhisa S, Otani H, Okazaki T, et al. N-acetylcysteine abolishes the protective effect of losartan against left ventricular remodeling in cardiomyopathy hamster. Antioxid Redox Signal. 2008;10:1999–2008.
Yatabe J, Sanada H, Yatabe MS, et al. Angiotensin II type 1 receptor blocker attenuates the activation of ERK and NADPH oxidase by mechanical strain in mesangial cells in the absence of angiotensin II. Am J Physiol Renal Physiol. 2009;296:F1052–1060.
Iizuka K, Machida T, Kawaguchi H, Hirafuji M. Pulsatile mechanical pressure promotes angiotensin-converting enzyme expression in aortic smooth muscle cells. Cardiovasc Drugs Ther. 2008;22:383–90.
Liu HW, Cheng B, Yu WL, et al. Angiotensin II regulates phosphoinositide 3 kinase/Akt cascade via a negative crosstalk between AT1 and AT2 receptors in skin fibroblasts of human hypertrophic scars. Life Sci. 2006;79:475–83.
Hu C, Dandapat A, Mehta JL. Angiotensin II induces capillary formation from endothelial cells via the LOX-1 dependent redox-sensitive pathway. Hypertension. 2007;50:952–7.
Bolli R, Dawn B, Tang XL, et al. The nitric oxide hypothesis of late preconditioning. Basic Res Cardiol. 1998;93:325–38.
Ye Y, Martinez JD, Perez-Polo RJ, et al. The role of eNOS, iNOS, and NF-kappaB in upregulation and activation of cyclooxygenase-2 and infarct size reduction by atorvastatin. Am J Physiol Heart Circ Physiol. 2008;295:H343–351.
Su KH, Tsai JY, Kou YR, et al. Valsartan regulates the interaction of angiotensin II type 1 receptor and endothelial nitric oxide synthase via Src/PI3K/Akt signalling. Cardiovasc Res. 2009;82:468–75.
Oak JH, Cai H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes. 2007;56:118–26.
Shinmura K, Tang XL, Wang Y, et al. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci USA. 2000;97:10197–202.
Birnbaum Y, Ye Y, Rosanio S, et al. Prostaglandins mediate the cardioprotective effects of atorvastatin against ischemia-reperfusion injury. Cardiovasc Res. 2005;65:345–55.
Kumar D, Menon V, Ford WR, Clanachan AS, Jugdutt BI. Effect of angiotensin II type 2 receptor blockade on activation of mitogen-activated protein kinases after ischemia-reperfusion in isolated working rat hearts. J Cardiovasc Pharmacol Ther. 2003;8:285–96.
Liang Q, Elson AC, Gerdes AM. p38 MAP kinase activity is correlated with angiotensin II type 1 receptor blocker-induced left ventricular reverse remodeling in spontaneously hypertensive heart failure rats. J Card Fail. 2006;12:479–86.
Leri A, Claudio PP, Li Q, et al. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest. 1998;101:1326–42.
Birnbaum Y, Ye Y, Lin Y, et al. Augmentation of myocardial production of 15-epi-lipoxin-a4 by pioglitazone and atorvastatin in the rat. Circulation. 2006;114:929–35.
Ye Y, Keyes KT, Zhang C, et al. The myocardial infarct size limiting effects of sitagliptin is PKA-dependent, whereas the protective effect of pioglitazone is partially dependent on PKA. Am J Physiol Heart Circ Physiol. 2010; ePublished ahead of time.
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36.
Acknowledgement
The study was supported by a grant from Takeda Pharmaceutical Company Limited, Osaka, Japan.
Statement of conflicts of interest
None for all co-authors
Author information
Authors and Affiliations
Corresponding author
Additional information
The study was supported by a grant from Takeda Pharmaceutical Company Limited, Osaka, Japan.
Rights and permissions
About this article
Cite this article
Ye, Y., Keyes, K.T., Zhang, C.F. et al. Additive Effect of TAK-491, a New Angiotensin Receptor Blocker, and Pioglitazone, in Reducing Myocardial Infarct Size. Cardiovasc Drugs Ther 24, 107–120 (2010). https://doi.org/10.1007/s10557-010-6227-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-010-6227-y