
The article deals with the investigation of the ethylene-butane natural gases mixture that is promising for practical application. A unit with a small volume cell created to obtain data on phase equilibria and thermal properties of binary mixtures of little investigated and rare substances using several experimental methods was presented. It is shown that it was possible to use non-standard measuring equipment to obtain sufficiently reliable results in a wide range of temperatures, pressures and compositions. Application of a new method to determine two-component gas mixtures concentration was demonstrated. The method of experimentation was described as well. The experimental results of ethylene–butane mixture thermal properties and vapor-liquid equilibrium that have expanded our knowledge in relation to this two-component natural refrigerant behavior in the low-temperature region and at high dilutions were tabulated. The comparison of the experimental and calculated data showed the possibility of applying the three-parameter cubic equation of state to simulate the thermodynamic properties of the ethylene–butane mixture without using test data of the mixture.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Modern low-parametric equations of state make it possible with limited initial information to predict the thermodynamic properties of substances for solving a number of technical problems [1]. However, in order to adequately describe the thermodynamic properties of the working fluid and to verify the quality of its model, experimental data are required in the temperature and pressure range typical for the production process.
The object of the study was a binary ethylene–butane (C2H4–C4H10) mixture of natural gases, which is a promising two-component working fluid of vapor-compression refrigeration systems to provide a cooling level of 190 K [2]. An analysis of published sources showed that the ethylene–butane system was experimentally studied mainly in the region of critical temperatures [3,4,5].
The limited field of research of this mixture and the promise of its use as a working fluid make it expedient to experimentally study its thermal properties at lower temperatures (173 to 273 K).
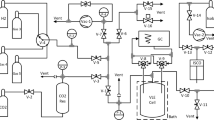
Experimental study. Equipment. The experimental setup (Fig. 1) allows to investigate the vapor-liquid equilibrium in a wide range of pressures (0.01 to 15 MPa), temperatures (77 to 450 K) and concentrations (from 0.0001 mol/mol). The measuring cell of the apparatus is a metal cylindrical vessel 17 with a volume of 15 cm3 with a magnetic stirrer, placed in a stabilizing copper block of the cryostat 18.

Experimental setup diagram: 1) vacuum pump; 2, 25) thermocouple pressure transducer; 3) gauge tank; 4) expansion tank; 5) nitrogen trap (cryogenic pump); 6, 11, 23) pressure transducer DD2.5; 7) pressure transducer D100; 8) thermocompressor (cryopump); 9, 24) gauge cylinder; 10) mass converter S-05; 12, 14, 16) pressure transducer D16; 13) cylinders with clean components; 15) separation vessel and mercury displacer; 17) equilibrium chamber; 18) cryostat; 19) mercury collection; 20) nitrogen-oil separator; 21) deadweight tester; 22) cylinder with liquid; 26) cylinder with CGS (calibration gas mixture); 27) nitrogen cylinder.
The design of the measuring cell is shown in Fig. 2. The vessel 2 is an austenitic chromium-nickel stainless steel (Kh18H9T) 18 × 1.5 mm tube with spherical bottoms, enclosed in a copper sheath 3 and placed in a copper temperature stabilizer 2 with sectional heaters 7, 21, 23 bifilarly wound around its cylindrical surface. The temperature is maintained and equalized by height by analog temperature controllers. The temperature sensor is a copper resistance thermometer 20, the unbalance signal source is a copper-constantan differential thermocouple with junctions 17, 26.

Diagram of the cryostat with the measuring cell: 1) liquid nitrogen supply tube; 2) corrosion-resistant steel vessel; 3) copper sheath; 4) spiral stirrer; 5, 9) upper and lower coolers; 6) platinum resistance thermometer; 7, 21, 23) heaters; 8) copper shells; 10) funnel cryogenic agent supply; 11, 13) valves; 12) vacuum pump; 14) magnet pull; 15) guide; 16) cooled screen; 18, 27) covers; 17, 19, 26) points of installation of differential thermocouple junctions; 20) copper resistance thermometer; 22) magnet; 24) heat stabilizer; 25) thermal bridge.
The heat-stabilizing block is cooled by two copper coils 5, 9 (shown in Fig. 2) bifilarly wound around the copper shell 8 (top and bottom). In evaporator coils, liquid nitrogen is bubbled out from the Dewar flask. The flow rate of the evacuated vapors of the cryogen is regulated by means of the valves 11, 13. The heater 7 is used for heating and temperature maintenance. The axial gradient of the block is eliminated with an accuracy of ± 0.01 K by heaters 21, 23.
For better thermal stabilization of the equilibrium chamber, copper thermal bridges 25 are provided to reduce the heat inflow through the filling and analysis lines, the magnet 22 pull 14 of the stirrer 4 (see Fig. 2). The use of copper cooled lids 18, 27 helps to reduce the radiant and convective components of heat inflows. Cylindrical copper thermal bridge 25 provides adjustable compensation of heat influx and cooling of the copper block in the temperature range of 150–250 K. The correspondence of the temperature of the stabilizing block to the temperature of the equilibrium chamber with an error of ±0.01 K is estimated from the EMF of the differential thermocouple with junctions at 17, 19, periodically measured by a voltmeter.
The temperature of the block is determined using a universal digital voltmeter V7-78/1 and a platinum resistance thermometer PTSV-2-1, calibrated in ITS-90 with an error of ±0.01 K. The measured temperature corresponds to the cell temperature with an error not exceeding ±0.03 K.
The pressure in the cell is calculated from the electric signal of the strain gauge D16 14 (see Fig. 1), measured by the same voltmeter. The strain gauges DD2.5, D16, D100 (6, 23, 12, 16, 7), used for measurement and mutual monitoring, are calibrated in accordance with the deadweight testers MP6, MP60, MP600 class 0.05.
The total composition of the mixture is calculated from the mass values of the components, obtained with an error of ±1 mg from the difference in mass of the respective filling cylinders 9 (see Fig. 1) weighed on the analytical balance ADV-200 before and after filling the substance into the cell. Small or fractional amounts of the dissolved component are determined with an error of ±1% by the volumetric method from the pressure of the substance, determined using the transducer 6, in a temperature-controlled filling line of a constant volume. The density of the saturated liquid phase is calculated from the values of the mass of the components and the known volume of the cell at the boiling point with an error of ±0.2%.
With the created device (Fig. 3), the molar concentration of the volatile component of the binary gas mixture can be determined (based on the gravimetric static method [6]) with an error of 0.001–0.01 mol/mol.

Diagram of the device for determining the concentration of two-component gas mixtures by the gravimetric static method: 1, 2) cylinders with clean components; 3) pressure transducer; 4) support pier; 5) thermostat; 6) gauge tank; 7) counterweight; 8) mass tensor; 9) digital voltmeter; 10, 11) sources of constant voltage; 12) voltage comparator; 13) filling valve; 14) vacuum pump; 15) cylinder with a stirred mixture of components.
The device (see Fig. 3) makes it possible to make measurements of a gas sample with a mass of 5–50 mg at a pressure of 0.005–0.1 MPa. For this, all cylinders and tubing are evacuated through the nitrogen trap 5 by the pump 1 with a thermocouple pressure transducer 2 (see Fig. 1). The vapor phase sample is carried to the tubing and, if necessary, to the expansion vessel 4. The sample is then heated to ambient temperature and fed to the temperature-controlled measuring constant volume 3, where the values of the corresponding quantities are determined from the strain 10 and pressure 11 gauges.
The molar concentration of gas mixture components is calculated from the results of measurements obtained in the temperature-controlled constant volume (Tmix = T1 = T2, Vmix = V1 = V2):
where pmix, p1, p2 represent the absolute pressure of the mixture and its components; mmix, m1, m2 represent the mass of the gas mixture and its components; and U is the measurement signal proportional to the indicated values.
Experiment methodology. The pre-evacuated cell was filled to 95–99% with the first component (less volatile) from the measuring cylinder. The vapor volume Vv ≈ 0.03–0.3 ml was set by overheating of the saturated liquid followed by the release of its excess amount, by determining the density at the point of inflection on the temperature–pressure relationship curve and the subsequent cooling to the initial state. The amount of the substance remaining in the cell was determined by weighing the measuring cylinder before and after filling. The second component (more volatile) was added from another measuring cylinder. Small amounts of the component, the precise weighing of which on the analytical balance was difficult, were determined from the pressure in the filling line (the dependence of the mass of the gas on the pressure in the line was obtained previously by experiment). The gas was dosed via the fi lling capillary to the lower part of the cell. The gas flow rate was determined using a digital voltmeter from the pressure drop in the filling line and was set according to the nature of the pressure change in the cell. The sensitivity of the change in the measuring signal of the pressure transducer ΔUp = 1 μV corresponds to the change in pressure in the lines Δp = l2ΔUp = 14 Pa and to the change in the total composition N ≈ 0.1 ppm. Equilibrium in this case was established within 20–60 min. Pressure for the next 10 min remained unchanged within δp = ±0.001 MPa and was recorded by the measuring system. Then the refueling of the component and the subsequent measurement were carried out.
After filling the cell with the second component in several stages, its residues in the lines were frozen into a measuring cylinder, the weight of the charged substance and the total molar concentration of the mixture were determined by weight, and the calculated intermediate masses and compositions were corrected.
Further studies were carried out by a static method, measuring the compositions of the equilibrium liquid and vapor phases by the gravimetric static technique. Before sampling the liquid phase, the capillaries were blown into a preliminary measuring cylinder of the filling lines, and then evacuated. Then, the sample was heat-stabilized and fed into an evacuated measuring volume, where its mass and pressure were determined at the beginning and end of the process. After evacuation of the measuring volumes, the sample was re-sampled with subsequent measurements, the measurement cycle was repeated until stable results were obtained within the error of determining the composition of the gas mixture (generally, two or three attempts were sufficient).
The flow rate for sampling the vapor phase was chosen so that the total pressure did not decrease by more than 1%, in order to avoid a significant disruption of the composition. For purging the capillary, it was necessary to have 2–5 ml of the vapor phase. 5–15 ml were used for the measurements. With such a quantity of the selected vapor phase, the equilibrium total pressure changed with the corresponding concentration of the liquid phase. The reference pressure in this case was the arithmetic mean equilibrium pressure measured at the beginning and at the end of the last sampling.
The vapor phase with a low content of the volatile component was sampled while equally replenished (flow rate G = 2–10 mg/h) through the lower filling capillary.
In studies over a wide range of compositions, a predetermined amount of the component was determined only by weighing, and refilling with the second component was carried out after measuring the equilibrium pressure at all temperature levels. When calculating the concentration of components in a liquid, their content in the vapor phase was taken into account.
The testing of the installation in a wide range of temperatures, pressures and densities was carried out by measuring the thermal properties of pure substances N2 (99.995%), C2H4 (99.88%), C4H10 (99.95%), C4F8 (99.99%) on the vapor pressure curve and in the single-phase region. The compositions of the equilibrium phases were measured on isotherms: 89.68 K for the nitrogen–neon mixture; 310.95 K for the nitrogen–butane mixture and compared with the corresponding literature data. The consistency of the measurement results with true values confirmed reliability of the measuring techniques.
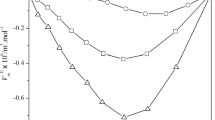
Results of simulation and experiment. The solubility of gaseous ethylene in liquid butane was studied at 283.15, 303.15, 322.04, 348.15 K in a range of compositions, respectively, 0.0006–0.76, 0.0003–0.76, 0.0001–0.78, 0.52 mol/mol by the static method, where the content was specified and the concentration by weight of the charged components was determined (Table 1, Fig. 4).

The presented calculated data are consistent with the results presented by Rainwater and Lynch [3] in the critical region and diverge at low pressures, approaching the values presented by Efremova and Sorina [4].
The low-temperature range on the boiling and condensation curves, supplementing studies in the critical region, was extended by measurements at temperatures of 173.15, 193.15, 213.15, 233.15, 253.15, and 273 K (Table 2) by the static method. The total composition of the mixture was set. The concentration in the liquid was determined from the masses of the charged components and using the volumetric method, and the concentration of the components in the vapor was determined only by the volumetric method (gravimetric static method). The homogeneity of the liquid phase was confirmed up to the melting curve.
The characteristics of the ethylene–butane system in the single-phase region are represented as p–T dependences on liquid isochores of mixtures of constant compositions (Table 3). Confidence limits for the determination of temperatures were Δ0.95T = 0.02–0.05 K; compositions – \( {\tilde{\varDelta}}_{0.95}x=0.1-8\%,\kern0.5em {\tilde{\varDelta}}_{0.95}y=2-6\%; \) specific volumes of liquid – \( {\tilde{\varDelta}}_{0.95}{v}^{\prime }=0.2-0.8\% \) and pressure – \( {\tilde{\varDelta}}_{0.95}p=0.1-0.5\%. \)
Simulation of the thermodynamic properties of the ethylene–butane mixture was carried out using a three-parameter cubic equation of state (TCEOS), which for the ith component of the mixture is written in the form [7, 8]:
where v is the molar volume; R is the gas constant; b i , c i are the parameters of the TCEOS for the ith component; and a i (T) is the temperature function for the ith component.
Algorithms for calculating the vapor-liquid equilibrium and the solubility of gas in a liquid are given in [9, 10]. To compare the experimental and published data on the solubility of this mixture, graphs were constructed (see Fig. 4). The results of the comparison testify to the possibility of using the TCEOS for thermodynamic calculations of process units operating on ethylene–butane mixture.
Conclusions
-
1.
The developed gravimetric static method for measuring the concentration of two-component gas mixtures allows, with an accuracy proportional to the mass of the sample and the difference in the molecular masses of the components, to determine their content in the entire concentration range at pressures an order of magnitude lower than the atmospheric pressure after measuring the masses of samples of pure starting materials only once.
-
2.
The obtained experimental data on the phase equilibrium for the ethylene–butane system can be used in the development of refrigeration devices and methods for the highest degree of purification of substances.
-
3.
Approbation of the three-parameter cubic equation of state gives grounds for its use in modeling the thermodynamic properties of the ethylene–butane mixture.
References
A. V. Trotsenko, “Modeling thermodynamic properties of little-researched substances,” Tekh. Gazy, No. 3, 33–39 (2002).
V. A. Naer and A. V. Rozhentsev, “Influence of the mass of the charge of a mixture of refrigerants on the performance characteristics of a low-temperature refrigeration device,” Kholod. Tekhn. Tekhnol., No. 1, 28–39 (2008).
J. C. Rainwater and J. J. Lynch, “A non-linear correlation of high pressure vapor-liquid equilibrium data for ethylene + butane showing inconsistencies in experimental compositions,” Int. J. Thermophys., No. 15, 1231–1239 (1994).
G. D. Efremova and G. A. Sorina, “Phase and volume relations in the ethylene–butane system,” Khim. Prom., No. 6, 503–510 (1960).
B. Williams, PhD Thesis, Vol. 6. Pt. 1, Department of Chemical and Materials Engineering, University of Michigan, Dechema, Michigan, (1949).
Patent 71993 Ukraine, V. N. Valyakin, A. V. Trotsenko, and A. V. Valyakina, IPC G01N 9/02, G01N 27/407, “Gravimetric static method for determining the concentration of two-component gas mixtures,” Byull., No. 15 (2012).
A. V. Trotsenko, “Equation of state of technical gases,” Tekh. Gazy, No. 2, 57–62 (2002).
A. V. Trotsenko and A. V. Valyakina, “Modeling of thermodynamic properties of working fluids on the basis of three-parameter cubic equations of state,” Kholod. Tekhn. Tekhnol., No. 2, 38–42 (2007).
A. V. Trotsenko and A. V. Valyakina, “Generalized algorithm for calculating vapor-liquid equilibrium of pure substances on the basis of cubic equations of state,” Kholod. Tekhn. Tekhnol., No. 1, 80–84 (2011).
A. V. Trotsenko and A. V. Valyakina, “Modeling the processes of gas solubility in liquids for the working fluids of cryogenic and refrigerating machines,” Kholod. Tekhn., No. 8, 24–29 (2016).
Author information
Authors and Affiliations
Additional information
Translated from Khimicheskoe i Neftegazovoe Mashinostroenie, No. 12, pp. 15–21, December, 2017.
Rights and permissions
About this article

Cite this article
Bondarenko, V.L., Valyakina, A.V., Borisenko, A.V. et al. Vapor-Liquid Equilibrium of the Ethylene–Butane Mixture. Chem Petrol Eng 53, 778–787 (2018). https://doi.org/10.1007/s10556-018-0421-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10556-018-0421-3