
Abstract
The significance of KISS1 goes beyond its original discovery as a metastasis suppressor. Its function as a neuropeptide involved in diverse physiologic processes is more well studied. Enthusiasm regarding KISS1 has cumulated in clinical trials in multiple fields related to reproduction and metabolism. But its cancer therapeutic space is unsettled. This review focuses on collating data from cancer and non-cancer fields in order to understand shared and disparate signaling that might inform clinical development in the cancer therapeutic and biomarker space. Research has focused on amino acid residues 68-121 (kisspeptin 54), binding to the KISS1 receptor and cellular responses. Evidence and counterevidence regarding this canonical pathway require closer look at the covariates so that the incredible potential of KISS1 can be realized.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
1.1 Why care about KISS1?
KISS1 was discovered as a metastasis suppressor in melanoma following microcell-mediated introduction of whole, wild-type chromosome 6 into metastatic melanoma cells followed by subtractive hybridization comparing cells suppressed for metastasis [1]. Early studies identified that KISS1 was highly expressed in the placenta and brain, with lesser expression in the kidney and pancreas, and negligible expression in other tissues [1,2,3,4]. Since invasion of trophoblasts during pregnancy resembles tumor invasion, early speculation was that KISS1 inhibits invasion as the explanation for metastasis suppression. While invasion was inhibited in the majority of cancer cell lines tested, the blockage was not complete, and since metastasis only requires some ability to invade (i.e., even weakly invasive cells can metastasize as long as those cells retain the ability to complete the other steps of the metastatic cascade), other processes were thought to be more relevant. Regardless, the capacity to inhibit metastasis garnered some enthusiasm because of the potential to improve cancer patient outcomes. Subsequently, accumulating clinical evidence in multiple cancer types has reaffirmed KISS1’s relevance in cancer and metastasis, highlighting its prognostic value as well as its therapeutic potential.
In this review, we will summarize what is known about KISS1 from multiple disciplines, focusing on its role in cancer. We will also investigate whether the early promise of KISS1 in cancer therapy has been fulfilled, and if the accumulating data warrant further investment in the cancer therapeutics space. Somewhat surprisingly, given KISS1 was originally defined in the context of cancer, most of the current understanding and clinical promise have been realized in physiology. For this review to put KISS1 into perspective, it is critical to explore what is known about its roles outside of cancer. Those data indeed inform concepts related to KISS1 roles in neoplasia.
Central to KISS1 research for the past quarter century are its crucial roles in reproduction, where KISS1 provided evidence for negative and positive feedback regulators of gonadal hormones. Particularly, KISS1-expressing neurons sit at the apex of the hypothalamic-pituitary-gonadal axis to regulate luteinizing hormone, follicle-stimulating hormone, and consequently two key gonadal hormones, estrogen and testosterone. As for completing a regulatory feedback circuit of the reproduction system, KISS1-expressing neurons express gonadal receptors, and indeed, KISS1 expression in those neurons is regulated by gonadal hormones [5]. Besides being highly expressed in the placenta and selected regions of the brain, KISS1 is expressed in lower levels in the liver, pancreas, adipose, and heart. The broader expression compared to initial studies can be attributed to improved methods and has been correlated with physiologic roles in reproduction (ovulation, fertilization, embryo implantation, placentation, etc.), circadian rhythm, adiposity, kidney development, and bone formation [6, 7]. Besides associating with the aforementioned processes, KISS1 is also a regulator of metabolism [8, 9]. Due to KISS1’s many physiological roles, disruptions of KISS1 are accompanied with pathologic processes, including hypogonadism [10], polycystic ovary syndrome [11, 12], and preeclampsia [13]. Cumulatively to date, twenty-five KISS1-centric clinical trials have been registered on clinicaltrials.gov, targeting reproductive disorders, diabetes, and in vitro fertilization. Ultimately, the research on KISS1 during the last three decades agrees unanimously on its importance in many aspects of medicine.
1.2 How is KISS1 regulated?
To critically evaluate how to fully harness KISS1’s potential, it is imperative that we fully understand how it is regulated and its mechanism of action. Data to address questions of regulation, as expected, come primarily from the endocrinology literature. Furthermore, KISS1 regulation has been determined to occur at both the RNA and protein expression levels in tissue- and cell type-specific manners.
1.2.1 Gene
KISS1 transcription is selectively regulated based on tissue and cell type.
In the hypothalamus, KISS1 is expressed by select subsets of neurons. Depending upon the neuron subpopulation, estradiol either up- or downregulates KISS1 via a classical or nonclassical ERα pathway, respectively [14]. In addition, the classical ERα pathway may be conserved in the uterus where estradiol also upregulates KISS1 expression [15]. In principle, ERα’s classical mechanism of action is exerted through the direct binding of E2-activated ERα to DNA via estrogen responsive elements (ERE). Nonclassical ERα signaling, on the other hand, regulates gene transcription through the binding of ERα to cofactors such as AP1, SP1, NFκB, etc., which is not dependent on the ERE (reviewed in [16]). In addition, sequences upstream of the KISS1 promoter and a 3’ intergenic region downstream of the last exon appear to act as enhancer regions [17, 18]. As in virtually all endocrine systems, KISS1 expression is determined by the combinations of transcription factors, cofactors, and epigenetic machinery present (or absent).
Some of the earliest understanding of KISS1 regulation came from metastatic melanoma studies right after its discovery. As the original quest for a metastasis suppressor gene on chromosome 6 ended up with a gene identified on chromosome 1, KISS1, it stood to reason that KISS1 regulators resided on chromosome 6 (detailed in [19,20,21]). Indeed, subsequent experiments revealed that the essential regulator for the KISS1 in melanoma – the transcription factor CRSP3/DRIP130 – resides on chromosome 6 [21]. CRSP3/DRIP130 also regulates a key cofactor TXNIP/VDUP1 [21]. Subsequently, binding studies using CRSP3, together with TXNIP/VDUP1 and a basal transcription factor SP1, identified SP1 binding sites at nucleotides - 93 to - 58 bp of the KISS1 promoter, the binding results in active gene transcription [22].
In breast cancer cell lines MCF7 and MDA-MB-435, transcriptional activator protein AP-2α binds to SP1 at nucleotides - 288 to - 188 bp in the KISS1 promoter [23]. Using another breast cancer cell line, MDA-MB-231, de Roux’s group suggested that E2 downregulates KISS1 via a nonclassical ERα pathway (like in the hypothalamic subpopulation) but independent of SP1 [24]. As ER happens to be important in KISS1 regulation in the hypothalamus and breast cancer subtypes, an association between hormonal status and KISS1 regulation in breast cancer may exist. Concomitantly, SP1 regulation of KISS1 may also depend on the hormonal status. However, data from two independent ER-negative breast cancer cell lines (MDA-MB-435 and MDA-MB-231) complicate interpretations. It appears that KISS1 regulation in breast cancer cells may depend on additional undetermined features rather than exclusively relying on hormonal status.
Though not directly interacting with the gene region, many other proteins, long noncoding RNA, and miRNA are emerging as important regulators of KISS1 gene expression in diverse cancer types as well (Table 1). Together, these results highlight the complexity and tissue-specific nature of KISS1 regulation.
1.2.2 Protein
As a typical secreted protein, KISS1 has a signaling sequence that targets the nascent protein to the endoplasmic reticulum for transport to the plasma membrane via the Golgi and secretory vesicles. Either at the outer leaflet of the plasma membrane or outside the cell, a proprotein convertase, furin, cleaves full-length KISS1 at dibasic sites into multiple fragments called kisspeptins (KP) [37]. Of note and somewhat unexpected, furin had previously been believed to be catalytic intracellularly [38]. Despite this, the majority of KISS1 research has focused on KISS1/KP after secretion. The most well-studied KP, a 54 amino acid polypeptide, spans residues 68-121 called KP54. Other smaller peptides derived (by still relatively ill-defined mechanisms) from KP54 have been detected and named based upon the number of amino acids: KP14 (aa 108-121), KP13 (aa 109-121), KP10 (aa 112-121). Early data strongly agree that all of these peptides belong to the family of RF-amides [3, 4, 39]. Their C-termini are amidated, which contributes to their binding to the receptor KISS1R which, in turn, triggers multiple signaling cascades (see Sect. 1.3). Because the above-referenced KP equivalently bind to KISS1R, researchers refer to KP-54, KP-14, KP-13, and KP-10 all as kisspeptin for short. As we will discuss below, there are other polypeptides derived from KISS1, which can also legitimately be referred to as KP. We, therefore, recommend that KP be defined by relative position from KISS1 rather than length to avoid confusion. Until a naming consensus is reached, we use the common conventions in this review and will attempt not to be ambiguous about which KP is being discussed.
Besides furin, other enzymes are also associated with KISS1 cellular processing. The most well associated enzymes are the matrix metalloproteinases (MMP), whose expression patterns overlap significantly with KISS1. MMP-16 and MMP-24 are expressed specifically in the brain; MMP-2, MMP-9, and MMP-14 are highly expressed in placenta. These two tissues most highly express KISS1. All of these MMPs (MMP-16, MMP-24, MMP-2, MMP-9, MMP-14) cleave KP at Gly118 and inactivate KP/KISS1R signaling [40].
Though less studied than KP, another fragment of KISS1, named kissorphin (KSO), generated from the cleavage of KP10 at Gly118 by MMP, also has physiologic functions. The 6-residue KSO (aa 112-117) shares sequence with neuropeptide FF (NPFF); can be amidated at the C-terminus; binds and activates the RFamide receptor NPFFR [41]; binds to Alzheimer’s amyloid-β peptide, prion protein, and amylin peptides [42]; and, possibly, possesses an anti-opioid character [43,44,45]. Linkages between KSO and cancer, if any, are not yet clear.
Though preliminary, some posttranslational modifications of KISS1 have also been reported. Yan et al., using thyroid cancer cell lines, suggested that an E3 ubiquitin ligase SMURF1 might be associated with the ubiquitination of KISS1, leading to KISS1 degradation [46]. In two screenings for studying the cellular distribution of phosphorylated proteins, KISS1 appears to be phosphorylated at Ser134 in both Jurkat and MEF cells (https://www.phosphosite.org/siteAction.action?id=11169746). Phosphorylation at Tyr112 in Jurkat cells has also been reported [47]. We found no independent verification of these modifications; however, they posit an intriguing alternative mechanism of KISS1 regulation.
1.3 How does KISS1 mediate cellular responses?
Shortly after the discovery of KISS1, an orphan G protein-coupled receptor which shares significant sequence homology with galanin receptors, GPR54, was identified [48]. In 2001, three labs independently and nearly simultaneously discovered that KP are the ligands of GPR54 [2,3,4]. GPR54 was subsequently named KISS1 receptor (KISS1R).
KISS1R is a 7 transmembrane G protein-coupled receptor. Heterotrimeric G proteins, consisting of subunits α, β, and γ, initiate signals depending upon the α subunit Gαs, Gαi/o, Gαq/11, or Gα12/13 (reviewed in [49]). KISS1R is typically coupled with Gαq/11 but can also associate with another Gαq member, Gα15/16, in hematopoietic organs [3, 50]. Accordingly, KP/KISS1R signaling fits well under a prototypical Gαq model. Briefly, a ligand-activated receptor activates the effector protein PLC-β which, in turn, hydrolyzes PIP2 into two second messengers, IP3 and DAG. IP3 diffuses into the cytosol triggering Ca2+ efflux from the endoplasmic reticulum, while membrane-associated DAG activates PKC. KP/KISS1R signaling in specific cell types diverges from here. The main route for downstream PKC signal is through MAP kinases (ERK1/2 and p38-related pathways) [2,3,4, 51,52,53,54].
KISS1R also reportedly activates another small G protein, RhoA [55, 56], transactivates EGFR [57], and associates with β-arrestin [58]. There exists crosstalk with KISS1/KISS1R signaling and other cancer-associated signaling pathways as well, including EGFR [57], CXCL12/CXCR4 [59,60,61], TNFα [55], NFκB [62], PI3K [62], and TGFβ [51]. Therefore, downstream pathways of KISS1R signaling are numerous and have the potential to affect multiple cellular processing and phenotypes. When coupled with knowledge that KISS1 and KISS1R are differentially expressed and differentially regulated in a cell type-specific manner, one must be careful not to extrapolate findings from one cell type to another.
The list of cellular responses of KP/KISS1R signaling continues to expand. Each of the pathways in the previous paragraph regulate cancer-associated phenotypes, such as migration and invasion [52,53,54,55, 61,62,63,64], stress fiber formation [3], proliferation [53, 62], cell cycle arrest [65], apoptosis [62, 65, 66], autophagy [36, 66, 67], and angiogenesis [56, 68]. Therefore, each represents a viable explanation for how KISS1/KISS1R signaling mediates metastasis suppression.
To further understand KISS1 mechanisms of action, several labs have overexpressed KISS1. Yan et al. overexpressed KISS1 and observed repression of NF-κB translocation to the nucleus which, in turn, reduced MMP9 expression in HT1080 cells [69]. Complicating interpretation, many studies (including our own), express KISS1 in cells which do not have detectable expression of KISS1R. Yet, re-expression or over-expression of KISS1 results in phenotypic changes, including stabilizing the master of mitochondrial biogenesis PGC1α, inhibition of AMPK, and downregulation of PPARα [70, 71]. Likewise, Jiang et al. demonstrated that KISS1 suppresses metastasis in ovarian and prostate cancer cells that do not express KISS1R through PKCα [72].
While intriguing, challenges still exist in ascribing particular signaling cascades to the anti-metastatic functions of KISS1. These challenges include the following: (i) many reports utilize non-metastatic cells from multiple tissue origins; (ii) some studies either over-express KISS1R or use cancer cell lines in which the KISS1R is not expressed; and (iii) drug (i.e., KP or KP mimic) concentration and exposure time vary widely. Taken together, these results present a highly complex situation in which canonical KP/KISS1R signaling is called into question as the exclusive mechanism by which KISS1 mediates functions.
2 The relationship of KISS1 and KISS1R
Whereas KISS1 and KISS1R function to regulate many aspects of development (see [6, 73, 74] for comprehensive review), from a cancer perspective and possibly normal physiological perspective, it is intriguing to critically evaluate the discrepancies observed in which KISS1 and KISS1R may be independent of each other. Knockout models for Kiss1 and Kiss1r in mice provided an early indication for potential independent roles, as whereas mice from either knockout background do not undergo normal sexual maturation resulting in infertility, Kiss1-/- are less severely affected than Kiss1r-/- mice [75]. Some potential explanations for the observed differences are as follows: (i) unknown, yet independent functional roles for both Kiss1 and Kiss1r, (ii) genetic polymorphism(s) that subtly affect Kiss1 or Kiss1r function or penetrance, or (iii) an incomplete knockout of Kiss1 (either from a technical limitation or unknown biology such as transmission of kisspeptin from the placenta) [75,76,77]. While no data yet exists to back up the last two hypotheses, accumulating evidence support the hypothesis that KISS1 and KISS1R can function independently. Therefore, this section aims to dissect the multidisciplinary evidence for independent functional roles for the ligand KISS1 and the receptor KISS1R.
Before elaborating upon putative alternative functions, it is important to recognize that nature has done some of the experiments for us. Critically, germline mutations of Kiss1 or Kiss1r have been observed in patients [10, 78,79,80,81,82,83], but also in multiple species. In the majority of cases, hypogonadism or reproductive deficiencies have been observed. However, the severity of the pathologies is variable and obviously affected by polygenic signaling and covariates.
2.1 Evolutionary history
KISS1 phylogenetics collectively show that, throughout evolution, KISS1/KISS1R biology contributes to genetic fitness in species rather than conferring an essential unique biological function [84, 85]. For example, kisspeptin appears to be dispensable for reproduction in teleost while necessary in placental mammals. In addition, kiss and kiss receptor genes are missing altogether in chickens. Furthermore, phylogenetics offers insights into the relationships between KISS1 and KISS1R throughout evolution.
It stands to reason that if KISS1 and KISS1R function together exclusively, they should coevolve. But do they coevolve? Synteny analysis has identified 3 paralogs of KISS (Kiss1, 2, 3) and 4 paralogs of KISSR (KissR1, 2, 3, 4) in vertebrates and 2 paralogs of KISS and 2 paralogs of KISSR in mammals. Importantly, the annotated number does not indicate a one-to-one pairing relationship between the ligand and receptor (e.g., kiss1 pairs with kissR1); rather, it is based on the order of their discovery. Interestingly, early research led to the hypothesis that there is a conservation of kiss/kissR pair, i.e., in select species the pairing appeared to match (in primates, rodents, and cattle, kiss1 and kissR1; in platypus, kiss1 and 2 and kissR1 and 2; in lizard, kiss2 and kissR2 [85]).
Accumulating evidence challenges the coevolution hypothesis. First, in vitro studies show that both kiss1 and kiss2 can activate both kissR1 and kissR2. In other words, there is no unique selectivity for the pairing. Second, later studies - using more complete genome databases and expanded species analyses - showed that some species have more kiss receptors than kiss (ligand) genes (e.g., in spotted gar, kissR1–4 and kiss1 and 2; in European eel, kissR1–3 and kiss1 and 2; in coelacanth, kissR1–4 and kiss1–3 [86]). Third, the presence of a pseudo-kiss2 gene (translated into KP10 that is nonamidated, inactive) was reported in primates including human [87]. While challenging the coevolution hypothesis, the above evidence suggests that a high degree of conservation between the paralogs circumvents the need for two paralogs to coexist. Pasquier et al. suggest that this may be attributed to differential physiological roles, which may include tissue specificity, differential regulation, and/or differential mechanisms of action, e.g., differential regulation in the hypothalamic subpopulations [86]. In that same line, alternative splicing of different isoforms of KISS1R has been identified in a modern teleost species, implicating differential tissue expression [88].
Since the discovery of its pairing to GPR54, KISS1 has long been classified under the RF-amide peptide family based on its RF-amide motif (other members include ligands NPFF, QRFP, NPVF, PrRP). Due to the diversity in KISS paralogs, they form their own branch in the RF-amide peptide family. Different ligand branches within the family promiscuously bind with receptors in other branches. Accordingly, in vitro studies show that KP binds to NPFFR1 (GPR147) and NPFFR2 (GPR74) [89, 90]. However, the classification under the RF-amide peptide was recently questioned partly because KISS-KISSR evolutionary history is distinct from other members in the family. Instead, assuming coevolution with their cognate receptor, KISS may deserve their own group called KISS/galanin/spexin family based on their cognate receptor (KISS1R is mostly homologous to galanin receptor) [91].
Altogether, despite inconclusive data, phylogenetic studies suggest that other receptors for KISS1 exist and the action mode of KP/KISS1R is tissue-specific. Of note, phylogenetics also provide useful data of which researchers should be aware, such as existent isoforms of genes and the necessity for appropriate model animal selection.
2.2 Differential effect of KP10 in mouse and humans
Alignment of KISS1 protein sequences across species has identified that the majority of KISS1 (precursor) amino acid sequence is highly variable. Despite this variability, KP10 is a highly conserved domain in primates, rodents, cattle, and zebrafish. Despite sequence conservation, KP10 exhibits differential effects within a given species, as illustrated by studies in the pancreas and placenta below, which suggests a genetic conservation, but a physiological divergence.
In the studies of KISS1 regulating pancreatic insulin production, both stimulation vs inhibition have been observed. Initially, discrepancies were attributed to differences in experimental models (whole pancreas vs cells, perfused vs static tissue culture), forms of KISS1 (KP54, KP13, KP10), and species (mouse, rat, human, monkey, pig). Subsequently, Song and colleagues concluded the discrepancy arose from the spectrum of KISS1 concentration among research groups [92]. At nM concentrations, KISS1 inhibits glucose-stimulated insulin secretion, while at μM concentrations, KISS1 stimulates insulin secretion in mouse pancreas islets in both perfused or static cultures as well as in an in vivo mouse model [92]. The conflict seemed to be resolved until a recent trial of KP10 at nM administration in 19 healthy men concluded KP10 stimulated insulin secretion [93]. Interestingly, Lyubimov et al. showed that human KP10 has higher affinity for NPFF2R than murine KP10, resulting in slightly less than 20-fold differential EC50 [90]. Their results highlight how different pathways may be activated when utilizing reagents which are not from the same species. Also, the findings illustrate some level of promiscuity for ligand-receptor binding in KISS1 signaling.
Lastly, the conserved physiology of KISS1 in placentation and pregnancy between mice and humans has been questioned as well. Whereas compelling evidence suggests that KISS1 plays significant roles in regulating human placentation, Kiss1-/- mice still delivered litters that were not significantly different from Kiss1WT [94]. Taken together, these results imply that important considerations need to be taken into account when translating KISS1 findings from model systems to humans.
2.3 Constitutive receptor activity of KISS1R
Direct evidence supporting KISS1-independent functions of KISS1R stem from studies showing KISS1R desensitization [95] via intracellular internalization of KISS1R [58]. This observation implicates a constitutive receptor activity [96]. Briefly, after prolonged KP exposure, GRK2 rapidly uncouples KISS1R from Gαq/11 (desensitization) and facilitates KISS1R binding to β-arrestin. β-arrestin then sequesters membrane KISS1R via clathrin-coated vesicles. A small portion of “used” KISS1R undergoes degradation, while the rest is recycled back to the cellular membrane. Even in the absence of KP, KISS1R internalization (regardless of Gαq/11-coupling) displays dynamic turnover, with a high degree of internalization (~ 60–70% of the total receptors) [58]. This helps to maintain a sufficient pool of signaling-competent KISS1R on the cell surface. Under chronic KP stimulation, provided that the cytosolic Ca2+ pool can sustain the Gαq/11-coupled-KISS1R pathway and that KISS1R is retained on the cell surface, signaling continues. The proposed mechanism fits well in the case of KISS1R-expressing neurons, which quickly respond to cues without new cycles of transcription or translation. Here, an interesting observation is that the internalized KISS1R may trigger signaling on its own without KP stimulation (constitutive receptor activity) [58]. Subsequently, Zajac et al. showed that KISS1R is directly associated with EGFR and stimulation of ER-negative breast cancer cells with EGF can regulate the endocytosis of both receptors, regardless of KP10 treatment [57]. Moreover, knocking-down KISS1R in an MDA-MB-231 variant cell line that does not express KISS1 [Note: other variants of MDA-MB-231 express KISS1 and KISS1R.] reduces cell migration, even with no KP treatment [97]. Thus, it appears that the mode of action of KISS1R probably expands beyond the prototypical Gαq-coupled receptor.
In addition, MMTV-PyMT/Kiss1r+/- mice develop tumors later than MMTV-PyMT/Kiss1rWT mice. Particularly, subcutaneously implanting primary MMTV-PyMT/Kiss1r+/- cancer cells into immunocompromised mice shows reduced primary tumor growth, suggesting KISS1R has a role in tumorigenicity [98]. This could be explained by the tumorigenicity promoting role of KP/KISS1R but also could implicate the involvement of KISS1R in cancer, with or without KISS1. These transplantation experiments could have just as easily been done in syngeneic FVB mice which has an intact immune system and could be more amenable to dissecting any purported immune functions. Unpublished data from our group show KISS1R in macrophage populations, implicating an immune paracrine crosstalk in addition to autocrine or endocrine functions (Ben Beck, Warren Denning and Danny Welch, unpublished observations).
2.4 KISS1 function independent of KISS1R
Strong supporting evidence for an independent function of KISS1 comes from Kiss1r-/- mice studies where KP shows subtle effects. First, KP at μM concentrations stimulates insulin secretion in response to glucose in Kiss1r-/- mice [92]. Second, KP still regulates neuronal excitability in Kiss1r-/- mice. Similar excitation is observed when activating NPFFR1, suggesting that KP effect may be exerted through NPFFR1 instead of Kiss1r [99]. A role for KISS1 that is independent of both KISS1R and NPFFR is also a possibility as shown by a study in neurotoxicity by Chilumuri and colleagues [100]. Particularly, knocking-down KISS1 in human neuronal cells shows increased amyloid toxicity. In contrast, KISS1 overexpression induces neuroprotection. Intriguingly, their initial hypothesis that the neuroprotective effect is exerted through either KP or KSO has been experimentally refuted, as administering antagonists of either receptors, KISS1R and NPFFR1, shows the same effect. Taken together, the data do not preclude an as yet unidentified receptor as well.
3 Why study KISS1 in cancer?
3.1 Clinical evidence of KISS1 relevance in metastasis
Most clinical evidence supports, or is at least consistent with, KISS1 metastasis suppressor roles as observed in preclinical models, i.e., expression is lost as tumors progress toward metastasis and/or increased expression is associated with better prognosis (Table 2). Data vary depending upon whether KISS1 is measured at the protein or RNA level, mostly likely because protein and RNA expression do not directly correlate [133, 134].
Clinical data in some cancer types provide contradictory evidence, most notably in liver, breast, and thyroid cancers. A common denominator for these cancer types is that the primary sites are highly hormonally active but direct connections have not yet been established. Considering that KISS1 is regulated (both negatively or positively) by estradiol depending on the hypothalamic subpopulation neurons and that KISS1 is widely associated with other hormones (e.g., insulin, leptin, prolactin, etc., all of which have been described to associate with cancer to some extent), it stands to reason that tumor hormonal status and the secondary microenvironmental physiology could influence the capacity of KISS1 to suppress metastasis. Moshmi Bhattacharya’s group has most extensively explored such relationships in different breast cancer cell lines. Overall, they find that presence of ERα in luminal subtypes is associated with KP/KISS1R suppression of invasion and metastasis using MCF7 [59]. In contrast, ERα-negative cells exhibit promotion of metastatic phenotypes in MCF10A [97], Hs578T, and MDA-MB-231 cells [135, 136]. The situation is not entirely clear, however. Using MDA-MB-231, other groups find KISS1/KISS1R anti-metastatic roles [67, 137]. The discrepancy may arise from distinctive epigenetics of the cell lines. Liu’s group proposes that in breast cancer, KP/KISS1R signaling has dual roles: to initially promote tumorigenesis and then to suppress the invasion in the early stage of metastasis [137]. Also, the different laboratories studied metastasis formation in different tissues (i.e., the lung and brain, respectively). Perhaps KISS1/KISS1R effects on metastasis have organ-specific effects on tumor cells. Besides molecular mechanisms studied in breast cancer, these unexpected observations that KISS1 promotes metastasis in liver and thyroid cancer have not been followed up.
Also, although molecular studies have focused on KISS1/KISS1R underlying cancer suppression, many clinical studies do not take KISS1R into account. Interestingly, in those that do, KISS1 and KISS1R expression levels do not correlate. The following section will address these mechanisms of KISS1 loss in more detail.
3.1.1 Epigenetic silencing or downregulation of KISS1-regulating transcription factors
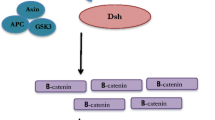
The KISS1 gene was discovered based on the clinical observation that the deletion of long arm of chromosome 6 (6q) occurs in > 80% of late-stage cases of metastatic melanoma. As discussed above, the long arm of chromosome 6 consists of the positive transcription factor CRSP3/DRIP130 that regulates KISS1 promoting transcription factors TXNIP/VDUP1 (on chromosome 1q) and disruption of the cascade can lead to KISS1 suppression [21]. An alternative route for losing KISS1 expression in melanoma is hypermethylation of another positive transcription factor TCF21 [138].
In bladder cancers, loss of KISS1 expression occurs through hypermethylation in the gene promoter [101]. Mechanistically, the overexpression of UHRF1 increases methylation of CpG in the KISS1 promoter repressing its expression [30]. Likewise, hypermethylation was described in colorectal cancer [108]; however, conflicting evidence exists as another study presented an inconclusive role of hypermethylation in colorectal cancer [139]. Lastly, hypermethylation in the KISS1 promoter was also described but not associated with the downregulation of KISS1 mRNA in pancreatic ductal adenocarcinoma [140].
Overall, the loss of expression of KISS1 due to epigenetic silencing aligns well with what is commonly observed in most other metastasis suppressors (i.e., there are relatively few mutations observed, but gene expression is silenced [141]). The predominance of epigenetic changes in cancer does not preclude mutations or structural modifications of KISS1 or KISS1R in other pathogenic states.
3.1.2 Single-nucleotide polymorphism (SNP)
In the latest update of the human genome (GRCh37.p13 (Dec 2019)), the KISS1 gene region includes 2014 SNP. Among them, the most clinically significant, rs587777835, results in an inactivating KISS1 mutation and, ultimately, hypogonadotropic hypogonadism [10]. There are several studies of KISS1 SNP association in cancer. In the study of breast cancer in Mexican populations, Quevedo et al. specifically chose to focus on 2 SNP, rs12998, and rs5780218 and reported that the latter correlates with higher risk for developing breast cancer [142]. Collectively, however, the majority of KISS1 SNP studies do not find a statistically significant association between SNPs or mutations of KISS1 with cancer development or disease prognosis [143, 144]. Instead, the loss of KISS1 expression is more commonly explained via epigenetic silencing or downregulation of the transcription factors discussed above. Nevertheless, several studies of KISS1 SNP in cancer offer intriguing implications, provided that study cohorts are statistically sufficient, and the biology of SNP is characterized. To illustrate these implications, 2 cases are discussed below.
Dova et al. used carcinoma of unknown primary tumor samples and found that 49 out of 50 tumor samples presented wildtype KISS1, similar to KISS1 in the peripheral blood lymphocytes of healthy controls. In contrast, only 1/50 tumor samples displayed a point substitution in the last exon, resulting in P81R KISS1 [143]. Intriguingly, P81R KISS1 was independently reported by Pentheroudakis et al. as well [144]. In the latter study, the mutation resulting in P81R was detected in the cell lines MCF7 and A549 and in 5/50 breast adenocarcinomas samples (3/5 present germline mutation). Regarding the phenotype, P81R KISS1 tumors have less KISS1 immunoreactivity, 20% vs 50% in wildtype, and account for higher rate of axillary node involvement, 80% vs 55% in wildtype. Although both studies conclude no significant association between P81R KISS1 mutation and disease, P81R KISS1 detection in two independent studies (ranged from 2 to 10% and 2 out of 3 studied cancer cell lines) suggests that the mutation could be significant in a larger cohort.
The study by Brunet et al. is especially interesting [145]. Particularly, rs71745629 KISS1 was associated with prolonged latency of metastatic colorectal cancer. In the studied cohort (N=172), although colorectal cancer patients with the KISS1 rs71745629, T/* genotype do not have better overall survival, they do have a significantly better progression-free survival, 12 months vs 4 months for those with the homozygous T/T genotype. Mechanistically, KISS1 rs71745629, T/* genotype results in the deletion of adenosine 417 (417delA) in the terminal exon of KISS1 gene; this creates a frameshift and a downstream STOP codon, translated into a 145-aa KISS1 protein. In contrast, in the homozygous T/T genotype (417A), the protein is 138 aa long. This implicates an isoform of KISS1 protein more likely to suppress metastasis. Of note, the 145-aa isoform was reported in the discovery of KISS1 as a metastasis suppressor in 1996 [1]. Through multiple updates of the genome reference consortium database, the 138-aa KISS1 seems to be more prevalent. Nonetheless, multiple intriguing questions arise from this study, including how the genotype affects KISS1 production and whether the isoforms have different effects on cellular response.
3.2 A model to study cancer dormancy
3.2.1 The clinical relevance of cancer dormancy and where KISS1 fits in
Metastasis is the major cause of cancer-related deaths. After treatment of primary cancer, despite being considered disease-free, a substantial cohort of cancer patients relapse in a type-specific manner. The time between the disease-free announcement and relapse is called metastatic latency. Particularly, long metastatic latency (years), or metastatic dormancy, has been clinically observed in breast, prostate, melanoma, renal, lung, and head and neck cancer [146]. Metastatic latency represents a promising window of opportunity to screen, intervene, and prevent a relapse [147]. Metastatic latency can vary significantly in patients. Further identification of the mechanisms that promote residual cells dormancy may provide the necessary framework for the development of novel therapeutics to prevent progressive disease [148,149,150].
Cancer dormancy research has evolved from captivating interest to recent mechanistic studies, and we will hopefully see its clinical applications in the future [151]. Molecular pathways in cancer dormancy have been compiled extensively in the last decade [146, 152]. However, challenges remain which include a lack of robust study models and limited study material for statistical analyses. We predict that KISS1 can help fill in this gap. The direct evidence comes from experiments in which introducing KISS1 gene in highly metastatic cancer cells keeps them dormant in secondary site [72, 153, 154]. Though currently there is no direct data for the molecular mechanism underlying this observation, accumulating data of KISS1 both in physiology and cancer signaling highly overlap with the molecular pathways described in cancer dormancy (Table 3). Whereas the predominant approach in the study of KISS1 in cancer is to either utilize endogenously expressing KISS1R or overexpressed KISS1 cancer cells, in our model showing KISS1-induced dormancy, cancer cells do not express KISS1R. Altogether, KISS1-induced dormancy is likely attributed to multiple molecular players in both cancer cells and the tumor microenvironment.
3.2.2 in vitro models do not recapitulate KISS1 dormancy effects
Metastasis is a stepwise process in which a single step is necessary but insufficient to lead to the end point: secondary outgrowth [173]. Pinning down the exact step(s) in which a metastasis suppressor is involved will inform development as a therapeutic. Unfortunately, in vitro studies have not always led to unequivocal definition of KISS1’s mechanism of action. For example, KISS1-expressing cancer cells can form primary tumors, circulate, and seed in the lung, but do not grow out. In other words, KISS1 suppresses metastasis at the last step, the outgrowth of cancer cells at the secondary site. To study the secondary outgrowth in lung, an ex vivo pulmonary metastasis assay (PUMA) has been proposed and shown to be an appropriate model [174]. Accordingly, Young et al. utilized the PUMA with the goal to model the metastasis suppressor effect of KISS1 and study the underlying mechanism [175]. GFP-labeled, KISS1-expressing cancer cells were injected into the tail vein; 20 min later when cells are lodged in the lung capillaries, lungs were harvested, cut into small sections, and ex vivo cultured up to 3 weeks. After 3 weeks, whereas modest fluorescent puncta were detected in vivo, the fluorescent signal increased dramatically in the PuMA lung. Something essential for KISS1 to suppress outgrowth may have been altered in the PUMA. The result once again emphasizes that in vivo models most faithfully recapitulate the metastasis process.
3.3 KISS1 potential clinical application for cancer: diagnosis, prognosis, and therapy
3.3.1 Diagnosis
Dotterweich et al. demonstrated the use of KP10 conjugated with a fluorophore for the diagnosis of multiple myeloma (malignant plasma cells homing in the bone marrow). Conventional detection methods for myeloma are MRI and measurement of excess serum or urinary IgG; however, downsides exist regarding the accessibility and specificity. Experiments show that when myeloma cells homing to the bone marrow interacts with mesenchymal and osteoprogenitor cells, these stromal cells significantly upregulate KISS1R. Using fluorophore-conjugated KP10, the group further showed that the bone where tumor is injected fluoresces compared to no signal observation in the un-injected bones, suggesting the specific binding of fluorophore-conjugated KP10 to tumor site. Subsequent mechanistic studies will further the exciting potential of this application [176].
3.3.2 Prognosis
Clinical evidence (Table 2) consolidates KISS1’s relevance in disease progression toward metastasis. Accordingly, many studies propose the prognostic value for KISS1. The promise of KISS1 as a biomarker for predicting metastasis or survival is context dependent. Additionally, most studies have measured KISS1 expression within the primary tumor and lacked parallel measurement in metastases. As discussed previously, loss of expression in what is likely to be a minority population of metastatic cells within a primary tumor is suboptimal as an expression biomarker. Furthermore, realistically, a single gene is implausible to be a prognostic tool for such a complex multifactorial disease as cancer (though there are exceptions, e.g., CML with the involvement of Philadelphia chromosome, rare cancers where single genes can confer malignancy). As we further categorize cancer into molecular subtypes for precision medicine, the combinations of multiple genes including KISS1 may be useful; nevertheless, taking all variants into account is a statistical and experimental challenge.
3.3.3 Therapy
The relevance of KISS1 to cancer dormancy not only provides a study model for cancer dormancy but also presents a potential therapy to intervene in metastasis (discussed in [19]). As KISS1 is typically downregulated in cancer, it stands to reason that finding a way to re-express or add back a metastasis suppressor will have significant therapeutic potential in preventing metastatic outgrowth. Firstly, KISS1’s metastasis suppressor effect is exerted after its secretion, which omits complications of the cellular membrane barrier that impedes many drugs to access the cell genome or other intracellular targets. Secondly, KISS1 is endogenous, thus theoretically less or non-immunogenic. In addition, its limited side effects in clinical trials also demonstrate a strong safety profile. Ultimately, characterizing the mechanism by which KISS1 suppresses metastasis is a must. Immunotherapy breakthroughs have recently revolutionized the cancer therapeutic space. While there is no direct association of KISS1, the fact that KISS1R is more relatively ubiquitously expressed in lymph nodes suggests that KISS1R may have a yet to be defined role in the immune system. In the meantime, early therapeutic progress related to KISS1 is encouraging.
Empirically, natural compounds have demonstrated anticancer effects. Though it is challenging to pin down the exact mechanisms underlying the biological effects of natural compounds, many research groups employ this drug discovery approach. Honokiol, a small biphenolic compound extracted from magnolia bark, exerts its anticancer effect through diverse molecular pathways essential for cancer [177]. Interestingly, a microarray of renal cell carcinoma treated with honokiol (40 μM) for 24 hours identified KISS1 as the top upregulated gene and KISS1R as the third most upregulated gene; further validation and knockdown study confirmed honokiol inhibited renal cancer cell invasion partly via the upregulation of KISS1 [178]. Research on the anticancer effect of honokiol has continued characterizing the detailed pathways and provides intriguing suggestive evidence for further development [177].
After the seminal case reports in 2003 identifying a clinical phenotype (hypogonadism) due to impaired KISS1/KISS1R signaling [76, 179], KP/KISS1R garnered great research interest and thrived beyond cancer, moving to physiology fields. The recognition of the great potential of a short 10-aa peptide as a drug candidate was illustrated by attempts of multiple research groups to generate the stable synthetic mimetic-KP10 peptides [180,181,182]; the culmination is a drug called TAK-448 (Takeda Pharmaceuticals) put on clinical trial phase I (NCT02381288) for the effect of downregulating testosterone in healthy and prostate cancer males. This study demonstrated that TAK-448, an agonist of KISS1R, is tolerable and can effectively reduce PSA in cancer patients; however, the effect was not robust and did not go to the next phase for cancer trials [183]. Unfortunately, challenges exist in measuring effective therapeutic indices for anti-metastatic drugs, and thus further development of biomarkers and criteria for measuring efficacy are desperately needed. Follow-up studies of KP-10 in combination with cytotoxic therapies, KP-10 prevention of relapse, and/or KP-10 mediated immune activation/regulation may provide additional opportunities for the advancement of KISS1 cancer therapeutics.
4 Concluding remarks
The review attempts to integrate KISS1 data from multiple fields to make sense of the biology of KISS1 with the goal to realize its clinical potential in metastatic cancer. Unfortunately, what was once thought of as a straightforward exercise has been more difficult than initially expected. Nonetheless, some clear lessons have been learned. Critically, KISS1 is a central node in signaling where its upstream and downstream vary depending on tissue and cell type. As conflicting data arise, researchers should be aware of alternative hypotheses besides the long-standing presumptions and be stringent in including covariates such as status of KISS1, KISS1R, ERα, and polymorphisms.
The existence of, and detection of, KSO shows that additional, similarly sized KISS1-derived polypeptides exist. Many of those peptides do not share functions (e.g., KSO does not interact with nor activate KISS1R); therefore, we recommend a naming convention in which all KISS1-derived peptides (termed kisspeptins) are defined by position, not size. Doing so will reduce confusion in the future. Still, the promise of KISS1 as a metastasis suppressor which could improve cancer patient outcomes remains.
Abbreviations
- DAG:
-
Diacylglycerol
- E2 :
-
Estrogen
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular signal-regulated kinase
- FSH:
-
Follicle-stimulating hormone
- GPCR:
-
G protein-coupled receptor
- IP3:
-
Inositol trisphosphate
- LH:
-
Luteinizing hormone
- MAPK:
-
Mitogen activate protein kinase
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIP2:
-
Phosphatidylinositol (4,5)-bisphosphate
- PLC:
-
Phospholipase C
- SNP:
-
Single-nucleotide polymorphism
- TNBC:
-
Triple-negative breast cancer
References
Lee, J.-H., Doumen, D. J., & Welch, D. R. (1996). Cloning of a novel gene, KiSS-1, which is responsible for metastasis suppression in chromosome 6/human melanoma hybrid cells. PNAS, 37, 531.
Ohtaki, T., Shintani, Y., Honda, S., Matsumoto, H., Hori, A., Kanehashi, K., et al. (2001). Metastasis suppressor gene KiSS1 encodes peptide ligand of a G-protein-coupled receptor. Nature, 411(6837), 613–617.
Kotani, M., Detheux, M., Vandenbogaerde, A., Communi, D., Vanderwinden, J. M., Le Poul, E., et al. (2001). The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. The Journal of Biological Chemistry, 276(37), 34631–34636.
Muir, A. I., Chamberlain, L., Elshourbagy, N. A., Michalovich, D., Moore, D. J., Calamari, A., et al. (2001). AXOR12, a novel human G protein-coupled receptor, activated by the peptide KiSS-1. The Journal of Biological Chemistry, 276(31), 28969–28975.
Uenoyama, Y., Inoue, N., Maeda, K. I., & Tsukamura, H. (2018). The roles of kisspeptin in the mechanism underlying reproductive functions in mammals. The Journal of Reproduction and Development, 64(6), 469–476.
Bhattacharya, M., & Babwah, A. V. (2015). Kisspeptin: beyond the brain. Endocrinology, 156(4), 1218–1227.
Brommage, R., Liu, J., Hansen, G. M., Kirkpatrick, L. L., Potter, D. G., Sands, A. T., et al. (2014). High-throughput screening of mouse gene knockouts identifies established and novel skeletal phenotypes. Bone Res, 2, 14034.
Wolfe, A., & Hussain, M. A. (2018). The emerging role(s) for kisspeptin in metabolism in mammals. Front Endocrinol (Lausanne), 9(184), 184.
Dudek, M., Ziarniak, K., & Sliwowska, J. H. (2018). Kisspeptin and metabolism: the brain and beyond. Front Endocrinol (Lausanne), 9(145), 145.
Topaloglu, A. K., Tello, J. A., Kotan, L. D., Ozbek, M. N., Yilmaz, M. B., Erdogan, S., et al. (2012). Inactivating KISS1 mutation and hypogonadotropic hypogonadism. The New England Journal of Medicine, 366(7), 629–635.
Tang, R., Ding, X., & Zhu, J. (2019). Kisspeptin and polycystic ovary syndrome. Front Endocrinol (Lausanne), 10, 298.
Witchel, S. F., & Tena-Sempere, M. (2013). The Kiss1 system and polycystic ovary syndrome: lessons from physiology and putative pathophysiologic implications. Fertility and Sterility, 100(1), 12–22.
Hu, K. L., Zhao, H., Yu, Y., & Li, R. (2019). Kisspeptin as a potential biomarker throughout pregnancy. European Journal of Obstetrics, Gynecology, and Reproductive Biology, 240, 261–266.
Gottsch, M. L., Navarro, V. M., Zhao, Z., Glidewell-Kenney, C., Weiss, J., Jameson, J. L., et al. (2009). Regulation of kiss1 and dynorphin gene expression in the murine brain by classical and nonclassical estrogen receptor pathways. The Journal of Neuroscience, 29(29), 9390–9395.
Zhang, P., Tang, M., Zhong, T., Lin, Y., Zong, T., Zhong, C., et al. (2014). Expression and function of kisspeptin during mouse decidualization. PLoS One, 9(5), e97647.
Dahlman-Wright, K., Cavailles, V., Fuqua, S. A., Jordan, V. C., Katzenellenbogen, J. A., Korach, K. S., et al. (2006). International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacological Reviews, 58(4), 773–781.
Tomikawa, J., Uenoyama, Y., Ozawa, M., Fukanuma, T., Takase, K., Goto, T., et al. (2012). Epigenetic regulation of Kiss1 gene expression mediating estrogen-positive feedback action in the mouse brain. Proceedings of the National Academy of Sciences of the United States of America, 109(20), E1294–E1301.
Uenoyama, Y., Tomikawa, J., Inoue, N., Goto, T., Minabe, S., Ieda, N., et al. (2016). Molecular and epigenetic mechanism regulating hypothalamic Kiss1 gene expression in mammals. Neuroendocrinology, 103(6), 640–649.
Beck, B. H., & Welch, D. R. (2010). The KISS1 metastasis suppressor: a good night kiss for disseminated cancer cells. European Journal of Cancer, 46(7), 1283–1289.
Harms, J. F., Welch, D. R., & Miele, M. E. (2003). KISS1 metastasis suppression and emergent pathways. Clinical & Experimental Metastasis, 20(1), 11–18.
Goldberg, S. F., Miele, M. E., Hatta, N., Takata, M., Paquette-Straub, C., Freedman, L. P., et al. (2003). Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Research, 63(2), 432–440.
Mitchell, D. C., Stafford, L. J., Li, D., Bar-Eli, M., & Liu, M. (2007). Transcriptional regulation of KiSS-1 gene expression in metastatic melanoma by specificity protein-1 and its coactivator DRIP-130. Oncogene, 26(12), 1739–1747.
Mitchell, D. C., Abdelrahim, M., Weng, J. S., Stafford, L. J., Safe, S., Bar-Eli, M., et al. (2006). Regulation of KiSS-1 metastasis suppressor gene expression in breast cancer cells by direct interaction of transcription factors activator protein-2a and specificity protein-1. JBC, 281(1), 51–58.
Huijbregts, L., & de Roux, N. (2010). KISS1 is down-regulated by 17 beta-estradiol in MDA-MB-231 cells through a nonclassical mechanism and loss of ribonucleic acid polymerase II binding at the proximal promoter. Endocrinology, 151(8), 3764–3772.
Teng, Y., Liu, M. Y., & Cowell, J. K. (2011). Functional interrelationship between the WASF3 and KISS1 metastasis-associated genes in breast cancer cells. International Journal of Cancer, 129(12), 2825–2835.
Kim, T. H., & Cho, S. G. (2017). Melatonin-induced KiSS1 expression inhibits triple-negative breast cancer cell invasiveness. Oncology Letters, 14(2), 2511–2516.
Mitra, A., Fillmore, R. A., Metge, B. J., Rajesh, M., Xi, Y., King, J., et al. (2008). Large isoform of MRJ (DNAJB6) reduces malignant activity of breast cancer. Breast Cancer Research, 10(2), R22.
Dissanayake, S. K., Wade, M., Johnson, C. E., O'Connell, M. P., Leotlela, P. D., French, A. D., et al. (2007). The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. Journal of Biological Chemistry, 282(23), 17259–17271.
Deng, G., Zheng, X., Jiang, P., Chen, K., Wang, X., Jiang, K., et al. (2017). Notch1 suppresses prostate cancer cell invasion via the metastasis-associated 1-KiSS-1 metastasis-suppressor pathway. Oncology Letters, 14(4), 4477–4482.
Zhang, Y., Huang, Z., Zhu, Z., Zheng, X., Liu, J., Han, Z., et al. (2014). Upregulated UHRF1 promotes bladder cancer cell invasion by epigenetic silencing of KiSS1. PLoS One, 9(10), e104252.
Shen, Z. L., Wang, B., Jiang, K. W., Ye, C. X., Cheng, C., Yan, Y. C., et al. (2016). Downregulation of miR-199b is associated with distant metastasis in colorectal cancer via activation of SIRT1 and inhibition of CREB/KISS1 signaling. Oncotarget, 7(23), 35092–35105.
Liu, G., Zhao, X., Zhou, J., Cheng, X., Ye, Z., & Ji, Z. (2018). LncRNA TP73-AS1 promotes cell proliferation and inhibits cell apoptosis in clear cell renal cell carcinoma through repressing KISS1 expression and inactivation of PI3K/Akt/mTOR signaling pathway. Cellular Physiology and Biochemistry, 48(1), 371–384.
Liu, C., Wang, L., Li, Y. W., Cui, Y. S., Wang, Y. Q., & Liu, S. (2019). Long noncoding RNA LUCAT1 promotes migration and invasion of prostate cancer cells by inhibiting KISS1 expression. European Review for Medical and Pharmacological Sciences, 23(8), 3277–3283.
Qiu, J.J., Lin, X.J., Tang, X.Y., Zheng, T.T., Zhang, X.Y., & Hua, K.Q. (2019). Long noncoding RNA TC0101441 induces epithelial-mesenchymal transition in epithelial ovarian cancer metastasis by downregulating KiSS1. International Journal of Cancer, 0(ja). https://doi.org/10.1002/ijc.32692.
Zhang, Y. X., Cui, H. X., Liu, L., & Yi, G. K. (2019). Long non-coding RNA MNX1-AS1 promoted osteosarcoma proliferation and invasion via inhibiting KISS1. European Review for Medical and Pharmacological Sciences, 23(14), 6045–6052.
Kaverina, N., Borovjagin, A. V., Kadagidze, Z., Baryshnikov, A., Baryshnikova, M., Malin, D., et al. (2017). Astrocytes promote progression of breast cancer metastases to the brain via a KISS1-mediated autophagy. Autophagy, 13(11), 1905–1923.
Harihar, S., Pounds, K. M., Iwakuma, T., Seidah, N. G., & Welch, D. R. (2014). Furin is the major proprotein convertase required for KISS1-to-Kisspeptin processing. PLoS One, 9(1), e84958.
Thomas, G. (2002). Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nature Reviews. Molecular Cell Biology, 3(10), 753–766.
Findeisen, M., Rathmann, D., & Beck-Sickinger, A. G. (2011). RFamide peptides: structure, function, mechanisms and pharmaceutical potential. Pharmaceuticals, 4(9), 1248–1280.
Takino, T., Koshikawa, N., Miyamori, H., Tanaka, M., Sasaki, T., Okada, Y., et al. (2003). Cleavage of metastasis suppressor gene product KiSS-1 protein/metastin by matrix metalloproteinases. Oncogene, 22(30), 4617–4626.
Milton, N. G. (2012). in vitro activities of Kissorphin, a novel hexapeptide KiSS-1 derivative, in neuronal cells. Journal of Amino Acids, 2012, 691463.
Milton, N. G., Chilumuri, A., Rocha-Ferreira, E., Nercessian, A. N., & Ashioti, M. (2012). Kisspeptin prevention of amyloid-beta peptide neurotoxicity in vitro. ACS Chemical Neuroscience, 3(9), 706–719.
Gibula-Bruzda, E., Marszalek-Grabska, M., Gawel, K., Trzcinska, R., Silberring, J., & Kotlinska, J. H. (2017). The new kisspeptin derivative - kissorphin (KSO) - attenuates acute hyperlocomotion and sensitization induced by ethanol and morphine in mice. Alcohol, 64, 45–53.
Gibula-Tarlowska, E., Kedzierska, E., Piechura, K., Silberring, J., & Kotlinska, J. H. (2019). The influence of a new derivate of kisspeptin-10 - kissorphin (KSO) on the rewarding effects of morphine in the conditioned place preference (CPP) test in male rats. Behavioural Brain Research, 372, 112043.
Gibula-Tarlowska, E., Grochecki, P., Silberring, J., & Kotlinska, J. H. (2019). The kisspeptin derivative kissorphin reduces the acquisition, expression, and reinstatement of ethanol-induced conditioned place preference in rats. Alcohol, 81, 11–19.
Yan, C., Su, H., Song, X., Cao, H., Kong, L., & Cui, W. (2018). Smad ubiquitination regulatory factor 1 (Smurf1) promotes thyroid cancer cell proliferation and migration via ubiquitin-dependent degradation of Kisspeptin-1. Cellular Physiology and Biochemistry, 49(5), 2047–2059.
Brill, L. M., Salomon, A. R., Ficarro, S. B., Mukherji, M., Stettler-Gill, M., & Peters, E. C. (2004). Robust phosphoproteomic profiling of tyrosine phosphorylation sites from human T cells using immobilized metal affinity chromatography and tandem mass spectrometry. Analytical Chemistry, 76(10), 2763–2772.
Lee, D. K., Nguyen, T., O'Neill, G. P., Cheng, R., Liu, Y., Howard, A. D., et al. (1999). Discovery of a receptor related to the galanin receptors. FEBS Letters, 446(1), 103–107.
Wettschureck, N., & Offermanns, S. (2005). Mammalian G proteins and their cell type specific functions. Physiological Reviews, 85(4), 1159–1204.
Wacker, J. L., Feller, D. B., Tang, X. B., Defino, M. C., Namkung, Y., Lyssand, J. S., et al. (2008). Disease-causing mutation in GPR54 reveals the importance of the second intracellular loop for class A G-protein-coupled receptor function. The Journal of Biological Chemistry, 283(45), 31068–31078.
Tian, J., Al-Odaini, A. A., Wang, Y., Korah, J., Dai, M., Xiao, L., et al. (2018). KiSS1 gene as a novel mediator of TGFbeta-mediated cell invasion in triple negative breast cancer. Cellular Signalling, 42, 1–10.
Francis, V. A., Abera, A. B., Matjila, M., Millar, R. P., & Katz, A. A. (2014). Kisspeptin regulation of genes involved in cell invasion and angiogenesis in first trimester human trophoblast cells. PLoS One, 9(6), e99680.
Zhang, Y., Tang, Y. J., Li, Z. H., Pan, F., Huang, K., & Xu, G. H. (2013). KiSS1 inhibits growth and invasion of osteosarcoma cells through inhibition of the MAPK pathway. European Journal of Histochemistry, 57(4), 199–204.
Noonan, M. M., Dragan, M., Mehta, M. M., Hess, D. A., Brackstone, M., Tuck, A. B., et al. (2018). The matrix protein Fibulin-3 promotes KISS1R induced triple negative breast cancer cell invasion. Oncotarget, 9(53), 30034–30052.
Cho, S. G., Li, D. L., Stafford, L. J., Luo, J., Rodriguez-Villanueva, M., Wang, Y., et al. (2009). KiSS1 suppresses TNFa-induced breast cancer cell invasion via an inhibition of RhoA-mediated NFkB activation. Journal of Cellular Biochemistry, 107(6), 1139–1149.
Cho, S. G., Yi, Z., Pang, X., Yi, T., Wang, Y., Luo, J., et al. (2009). Kisspeptin-10, a KISS1-derived decapeptide, inhibits tumor angiogenesis by suppressing Sp1-mediated VEGF expression and FAK/Rho GTPase activation. Cancer Research, 69(17), 7062–7070.
Zajac, M., Law, J., Cvetkovic, D. D., Pampillo, M., McColl, L., Pape, C., et al. (2011). GPR54 (KISS1R) transactivates EGFR to promote breast cancer cell invasiveness. PLoS One, 6(6), e21599.
Pampillo, M., Camuso, N., Taylor, J. E., Szereszewski, J. M., Ahow, M. R., Zajac, M., et al. (2009). Regulation of GPR54 signaling by GRK2 and {beta}-arrestin. Molecular Endocrinology, 23(12), 2060–2074.
Olbrich, T., Ziegler, E., Turk, G., Schubert, A., Emons, G., & Grundker, C. (2010). Kisspeptin-10 inhibits bone-directed migration of GPR54-positive breast cancer cells: evidence for a dose-window effect. Gynecologic Oncology, 119(3), 571–578.
Navenot, J. M., Wang, Z., Chopin, M., Fujii, N., & Peiper, S. C. (2005). Kisspeptin-10-induced signaling of GPR54 negatively regulates chemotactic responses mediated by CXCR4: a potential mechanism for the metastasis suppressor activity of kisspeptins. Cancer Research, 65(22), 10450–10456.
Schmidt, E., Haase, M., Ziegler, E., Emons, G., & Grundker, C. (2014). Kisspeptin-10 inhibits stromal-derived factor 1-induced invasion of human endometrial cancer cells. International Journal of Gynecological Cancer, 24(2), 210–217.
Chen, S., Chen, W., Zhang, X., Lin, S., & Chen, Z. (2016). Overexpression of KiSS-1 reduces colorectal cancer cell invasion by downregulating MMP-9 via blocking PI3K/Akt/NF-kappaB signal pathway. International Journal of Oncology, 48(4), 1391–1398.
Bilban, M., Ghaffari-Tabrizi, N., Hintermann, E., Bauer, S., Molzer, S., Zoratti, C., et al. (2004). Kisspeptin-10, a KiSS-1/metastin-derived decapeptide, is a physiological invasion inhibitor of primary human trophoblasts. Journal of Cell Science, 117(8), 1319–1328.
Tian, J., Al-Odaini, A. A., Wang, Y., Korah, J., Dai, M., Xiao, L., et al. (2017). KiSS1 gene as a novel mediator of TGFbeta-mediated cell invasion in triple negative breast cancer. Cellular Signalling, 42, 1–10.
Becker, J. A., Mirjolet, J. F., Bernard, J., Burgeon, E., Simons, M. J., Vassart, G., et al. (2005). Activation of GPR54 promotes cell cycle arrest and apoptosis of human tumor cells through a specific transcriptional program not shared by other G(q)-coupled receptors. BBRC, 326(3), 677–686.
Yin, Y., Tang, L., & Shi, L. (2017). The metastasis suppressor gene KISS-1 regulates osteosarcoma apoptosis and autophagy processes. Molecular Medicine Reports, 15(3), 1286–1290.
Ulasov, I. V., Borovjagin, A. V., Timashev, P., Cristofanili, M., & Welch, D. R. (2019). KISS1 in breast cancer progression and autophagy. Cancer Metastasis Reviews, 38(3), 493–506.
Ramaesh, T., Logie, J. J., Roseweir, A. K., Millar, R. P., Walker, B. R., Hadoke, P. W., et al. (2010). Kisspeptin-10 inhibits angiogenesis in human placental vessels ex vivo and endothelial cells in vitro. Endocrinology, 151(12), 5927–5934.
Yan, C. H., Wang, H., & Boyd, D. D. (2001). KiSS-1 represses 92-kDa type IV collagenase expression by down- regulating NFkB binding to the promoter as a consequence of IkBa-induced block of p65/p50 nuclear translocation. The Journal of Biological Chemistry, 276(2), 1164–1172.
Liu, W., Beck, B. H., Vaidya, K. S., Nash, K. T., Feeley, K. P., Ballinger, S. W., et al. (2014). Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Research, 74(3), 954–963.
Manley, S. J., Liu, W., & Welch, D. R. (2017). The KISS1 metastasis suppressor appears to reverse the Warburg effect by shifting from glycolysis to mitochondrial beta-oxidation. Journal of Molecular Medicine (Berlin, Germany), 95(9), 951–963.
Jiang, Y., Berk, M., Singh, L. S., Tan, H., Yin, L., Powell, C. T., et al. (2005). KiSS1 suppresses metastasis in human ovarian cancer via inhibition of protein kinase C alpha. Clinical & Experimental Metastasis, 22(5), 369–376.
Uenoyama, Y., Pheng, V., Tsukamura, H., & Maeda, K. I. (2016). The roles of kisspeptin revisited: inside and outside the hypothalamus. The Journal of Reproduction and Development, 62(6), 537–545.
Oakley, A. E., Clifton, D. K., & Steiner, R. A. (2009). Kisspeptin signaling in the brain. Endocrine Reviews, 30(6), 713–743.
Lapatto, R., Pallais, J. C., Zhang, D., Chan, Y. M., Mahan, A., Cerrato, F., et al. (2007). Kiss1-/- mice exhibit more variable hypogonadism than Gpr54-/- mice. Endocrinology, 148(10), 4927–4936.
Seminara, S. B., Messager, S., Chatzidaki, E. E., Thresher, R. R., Acierno, J. S., Shagoury, J. K., et al. (2003). The GPR54 gene as a regulator of puberty. New England Journal of Medicine, 349(17), 1614–U1618.
Chan, Y. M., Broder-Fingert, S., Wong, K. M., & Seminara, S. B. (2009). Kisspeptin/Gpr54-independent gonadotrophin-releasing hormone activity in Kiss1 and Gpr54 mutant mice. Journal of Neuroendocrinology, 21(12), 1015–1023.
Tenenbaum-Rakover, Y., Commenges-Ducos, M., Iovane, A., Aumas, C., Admoni, O., & de Roux, N. (2007). Neuroendocrine phenotype analysis in five patients with isolated hypogonadotropic hypogonadism due to a L102P inactivating mutation of GPR54. The Journal of Clinical Endocrinology and Metabolism, 92(3), 1137–1144.
Silveira, L. G., Noel, S. D., Silveira-Neto, A. P., Abreu, A. P., Brito, V. N., Santos, M. G., et al. (2010). Mutations of the KISS1 gene in disorders of puberty. The Journal of Clinical Endocrinology and Metabolism, 95(5), 2276–2280.
Silveira, L. F., Santos, M. A., Brito, V. N., Silveira-Neto, A. P., Mendonca, B. B., & Latronico, A. C. (2008). Two KISS1 mutations associated with gonadotropin-dependent precocious puberty. Hormone Research, 70, 20–20.
Semple, R. K., Achermann, J. C., Ellery, J., Farooqi, I. S., Karet, F. E., Stanhope, R. G., et al. (2004). Two novel missense mutations in GPR54 in a patient with hypogonadotropic hypogonadism. The Journal of Clinical Endocrinology and Metabolism, 90(3), 1849–1855.
Pallais, J. C., Bo-Abbas, Y., Pitteloud, N., Crowley Jr., W. F., & Seminara, S. B. (2006). Neuroendocrine, gonadal, placental, and obstetric phenotypes in patients with IHH and mutations in the G-protein coupled receptor, GPR54. Molecular and Cellular Endocrinology, 254-255, 70–77.
Nimri, R., Lebenthal, Y., Lazar, L., Chevrier, L., Phillip, M., Bar, M., et al. (2011). A novel loss-of-function mutation in GPR54/KISS1R leads to hypogonadotropic hypogonadism in a highly consanguineous family. The Journal of Clinical Endocrinology and Metabolism, 96(3), E536–E545.
Pasquier, J., Kamech, N., Lafont, A.G., Vaudry, H., Rousseau, K., & Dufour, S. (2014). Molecular evolution and structure-activity relationships of kisspeptins and their receptors. Journal of Molecular Endocrinology, 52, T101–117.
Kanda, S., & Oka, Y. (2013). Structure, synthesis, and phylogeny of kisspeptin and its receptor. Advances in Experimental Medicine and Biology, 784, 9–26.
Pasquier, J., Kamech, N., Lafont, A. G., Vaudry, H., Rousseau, K., & Dufour, S. (2014). Molecular evolution of GPCRs: kisspeptin/kisspeptin receptors. Journal of Molecular Endocrinology, 52(3), T101–T117.
Osugi, T., Ohtaki, N., Sunakawa, Y., Son, Y. L., Ohkubo, M., Iigo, M., et al. (2013). Molecular evolution of kiss2 genes and peptides in vertebrates. Endocrinology, 154(11), 4270–4280.
Mechaly, A. S., Vinas, J., & Piferrer, F. (2009). Identification of two isoforms of the Kisspeptin-1 receptor (kiss1r) generated by alternative splicing in a modern teleost, the Senegalese sole (Solea senegalensis). Biology of Reproduction, 80(1), 60–69.
Oishi, S., Misu, R., Tomita, K., Setsuda, S., Masuda, R., Ohno, H., et al. (2011). Activation of neuropeptide FF receptors by kisspeptin receptor ligands. ACS Medicinal Chemistry Letters, 2(1), 53–57.
Lyubimov, Y., Engstrom, M., Wurster, S., Savola, J. M., Korpi, E. R., & Panula, P. (2010). Human kisspeptins activate neuropeptide FF2 receptor. Neuroscience, 170(1), 117–122.
Yun, S., Kim, D. K., Furlong, M., Hwang, J. I., Vaudry, H., & Seong, J. Y. (2014). Does Kisspeptin belong to the proposed RF-amide peptide family? Frontiers in Endocrinology (Lausanne), 5, 134.
Song, W. J., Mondal, P., Wolfe, A., Alonso, L. C., Stamateris, R., Ong, B. W., et al. (2014). Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metabolism, 19(4), 667–681.
Izzi-Engbeaya, C., Comninos, A. N., Clarke, S. A., Jomard, A., Yang, L., Jones, S., et al. (2018). The effects of kisspeptin on beta-cell function, serum metabolites and appetite in humans. Diabetes, Obesity and Metabolism, 20(12), 2800–2810.
Babwah, A. V. (2015). Uterine and placental KISS1 regulate pregnancy: what we know and the challenges that lie ahead. Reproduction, 150(4), R121–R128.
Millar, R. P., & Babwah, A. V. (2015). KISS1R: hallmarks of an effective regulator of the neuroendocrine axis. Neuroendocrinology, 101(3), 193–210.
Smit, M. J., Vischer, H. F., Bakker, R. A., Jongejan, A., Timmerman, H., Pardo, L., et al. (2007). Pharmacogenomic and structural analysis of constitutive g protein-coupled receptor activity. Annual Review of Pharmacology and Toxicology, 47, 53–87.
Goertzen, C. G., Dragan, M., Turley, E., Babwah, A. V., & Bhattacharya, M. (2016). KISS1R signaling promotes invadopodia formation in human breast cancer cell via beta-arrestin2/ERK. Cellular Signalling, 28(3), 165–176.
Cho, S. G., Wang, Y., Rodriguez, M., Tan, K., Zhang, W., Luo, J., et al. (2011). Haploinsufficiency in the prometastasis Kiss1 receptor Gpr54 delays breast tumor initiation, progression, and lung metastasis. Cancer Research, 71(20), 6535–6546.
Liu, X., & Herbison, A. (2015). Kisspeptin regulation of arcuate neuron excitability in kisspeptin receptor knockout mice. Endocrinology, 156(5), 1815–1827.
Chilumuri, A., & Milton, N. G. (2013). The role of neurotransmitters in protection against amyloid- beta toxicity by KiSS-1 overexpression in SH-SY5Y neurons. ISRN Neuroscience, 2013, 253210.
Cebrian, V., Fierro, M., Orenes-Pinero, E., Grau, L., Moya, P., Ecke, T., et al. (2011). KISS1 methylation and expression as tumor stratification biomarkers and clinical outcome prognosticators for bladder cancer patients. American Journal of Pathology, 179(2), 540–546.
Sanchez-Carbayo, M., Capodieci, P., & Cordon-Cardo, C. (2003). Tumor suppressor role of KiSS-1 in bladder cancer – Loss of KiSS-1 expression is associated with bladder cancer progression and clinical outcome. AJP, 162(2), 609–617.
Stark, A. M., Tongers, K., Maass, N., Mehdorn, H. M., & Held-Feindt, J. (2005). Reduced metastasis-suppressor gene mRNA-expression in breast cancer brain metastases. Journal of Cancer Research and Clinical Oncology, 131(3), 191–198.
Mooez, S., Malik, F. A., Kayani, M. A., Rashid, R., Zahid, A., & Khan, A. (2011). Expressional alterations and transcript isoforms of metastasis suppressor genes (KAI1 and KiSS1) in breast cancer patients. Asian Pacific Journal of Cancer Prevention, 12(10), 2785–2791.
Kostadima, L., Pentheroudakis, G., & Pavlidis, N. (2007). The missing kiss of life: transcriptional activity of the metastasis suppressor gene KiSS1 in early breast cancer. Anticancer Research, 27(4B), 2499–2504.
Ulasov, I. V., Kaverina, N. V., Pytel, P., Thaci, B., Liu, F., Hurst, D. R., et al. (2012). Clinical significance of KISS1 protein expression for brain invasion and metastasis. Cancer, 118(8), 2096–2105.
Xie, F., Yang, H., Wang, S., Zhou, B., Tong, F., Yang, D., et al. (2012). A logistic regression model for predicting axillary lymph node metastases in early breast carcinoma patients. Sensors (Basel), 12(7), 9936–9950.
Chen, S. Q., Chen, Z. H., Lin, S. Y., Dai, Q. B., Fu, L. X., & Chen, R. Q. (2014). KISS1 methylation and expression as predictors of disease progression in colorectal cancer patients. World Journal of Gastroenterology, 20(29), 10071–10081.
Wang, X. Q., Fang, P. F., Zhang, C., Xu, Y. Y., Song, X. B., Liang, J., et al. (2019). Low KISS1 expression predicts poor prognosis for patients with colorectal cancer: a meta-analysis. Clinical and Experimental Pharmacology & Physiology, 46(7), 625–634.
Huo, X., Zhang, L., & Li, T. (2018). Analysis of the association of the expression of KiSS-1 in colorectal cancer tissues with the pathology and prognosis. Oncology Letters, 15(3), 3056–3060.
Jiang, T., Zhang, S. L., Lin, B., Meng, L. R., & Gao, H. (2005). Expression and clinical significance of KISS-1 and GPR54 mRNA in endometrial carcinoma. Zhonghua Zhong Liu Za Zhi, 27(4), 229–231.
Ikeguchi, M., Yamaguchi, K., & Kaibara, N. (2004). Clinical significance of the loss of KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in esophageal squamous cell carcinoma. Clinical Cancer Research, 10(4), 1379–1383.
Guan-Zhen, Y., Ying, C., Can-Rong, N., Guo-Dong, W., Jian-Xin, Q., & Jie-Jun, W. (2007). Reduced protein expression of metastasis-related genes (nm23, KISS1, KAI1 and p53) in lymph node and liver metastases of gastric cancer. International Journal of Experimental Pathology, 88(3), 175–183.
Dhar, D. K., Naora, H., Kubota, H., Maruyama, R., Yoshimura, H., Tonomoto, Y., et al. (2004). Downregulation of KiSS-1 expression is responsible for tumor invasion and worse prognosis in gastric carcinoma. International Journal of Cancer, 111(6), 868–872.
Shengbing, Z., Feng, L. J., Bin, W., Lingyun, G., & Aimin, H. (2009). Expression of KiSS-1 gene and its role in invasion and metastasis of human hepatocellular carcinoma. Anat Rec (Hoboken), 292(8), 1128–1134.
Hou, Y. K., Wang, Y., Cong, W. M., & Wu, M. C. (2007). [Expression of tumor metastasis-suppressor gene KiSS-1 and matrix metalloproteinase-9 in portal vein tumor thrombus of hepatocellular carcinoma]. Ai Zheng. Aizheng. Chinese Journal of Cancer, 26(6), 591–595.
Song, W. W., Gui, A. P., Li, W., Chen, H. S., & Li, J. M. (2017). Expressions of HIF-1alpha and KISS-1 in patients with liver cancer and correlation analysis. European Review for Medical and Pharmacological Sciences, 21(18), 4058–4063.
Zheng, S., Chang, Y., Hodges, K. B., Sun, Y., Ma, X., Xue, Y., et al. (2010). Expression of KISS1 and MMP-9 in non-small cell lung cancer and their relations to metastasis and survival. Anticancer Research, 30(3), 713–718.
Sun, Y. B., & Xu, S. (2013). Expression of KISS1 and KISS1R (GPR54) may be used as favorable prognostic markers for patients with non-small cell lung cancer. International Journal of Oncology, 43(2), 521–530.
Shirasaki, F., Takata, M., Hatta, N., & Takehara, K. (2001). Loss of expression of the metastasis suppressor gene KiSS1 during melanoma progression and its association with LOH of chromosome 6q16.3-q23. Cancer Research, 61(20), 7422–7425.
Prentice, L. M., Klausen, C., Kalloger, S., Kobel, M., McKinney, S., Santos, J. L., et al. (2007). Kisspeptin and GPR54 immunoreactivity in a cohort of 518 patients defines favourable prognosis and clear cell subtype in ovarian carcinoma. BMC Medicine, 5, 33.
Hata, K., Dhar, D. K., Watanabe, Y., Nakai, H., & Hoshiai, H. (2007). Expression of metastin and a G-protein-coupled receptor (AXOR12) in epithelial ovarian cancer. European Journal of Cancer, 43(9), 1452–1459.
Yu, L., Zhu, B., Wu, S., Zhou, L., Song, W., Gong, X., et al. (2017). Evaluation of the correlation of vasculogenic mimicry, ALDH1, KiSS-1, and MACC1 in the prediction of metastasis and prognosis in ovarian carcinoma. Diagnostic Pathology, 12(1), 23.
Masui, T., Doi, R., Mori, T., Toyoda, E., Koizumi, M., Kami, K., et al. (2004). Metastin and its variant forms suppress migration of pancreatic cancer cells. Biochemical and Biophysical Research Communications, 315(1), 85–92.
Nagai, K., Doi, R., Katagiri, F., Ito, T., Kida, A., Koizumi, M., et al. (2009). Prognostic value of metastin expression in human pancreatic cancer. Journal of Experimental & Clinical Cancer Research, 28, 9.
Marot, D., Bieche, I., Aumas, C., Esselin, S., Bouquet, C., Vacher, S., et al. (2007). High tumoral levels of Kiss1 and G-protein-coupled receptor 54 expression are correlated with poor prognosis of estrogen receptor-positive breast tumors. Endocrine-Related Cancer, 14(3), 691–702.
Martin, T. A., Watkins, G., & Jiang, W. G. (2005). KiSS-1 expression in human breast cancer. Clinical & Experimental Metastasis, 22(6), 503–511.
Ikeguchi, M., Hirooka, Y., & Kaibara, N. (2003). Quantitative reverse transcriptase polymerase chain reaction analysis for KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in hepatocellular carcinoma. Journal of Cancer Research and Clinical Oncology, 129(9), 531–535.
Schmid, K., Wang, X., Haitel, A., Sieghart, W., Peck-Radosavljevic, M., Bodingbauer, M., et al. (2007). KiSS-1 overexpression as an independent prognostic marker in hepatocellular carcinoma: an immunohistochemical study. Virchows Archiv, 450(2), 143–149.
Chen, H., Chen, P. S., Lin, F. F., Chen, S. Y., & Lin, J. H. (2019). KISS1 protein expression is associated with worse prognosis in osteosarcoma patients: a long-term follow-up study. Translational Cancer Research, 8(5), 1756–1762.
Ringel, M. D., Hardy, E., Bernet, V. J., Burch, H. B., Schuppert, F., Burman, K. D., et al. (2002). Metastin receptor is overexpressed in papillary thyroid cancer and activates MAP kinase in thyroid cancer cells. Journal of Clinical Endocrinology and Metabolism, 87(5), 2399–2402.
Karapanagiotou, E. M., Dilana, K. D., Gkiozos, I., Gratsias, I., Tsimpoukis, S., Polyzos, A., et al. (2011). Metastin is not involved in metastatic potential of non-small cell lung cancer. Medical Oncology, 28(2), 559–564.
Ziegler, E., Olbrich, T., Emons, G., & Grundker, C. (2013). Antiproliferative effects of kisspeptin10 depend on artificial GPR54 (KISS1R) expression levels. Oncology Reports, 29(2), 549–554.
Tan, K., Cho, S. G., Luo, W., Yi, T., Wu, X., Siwko, S., et al. (2014). KiSS1-induced GPR54 signaling inhibits breast cancer cell migration and epithelial-mesenchymal transition via protein kinase D1. Current Molecular Medicine, 14(5), 652–662.
Cvetkovic, D., Babwah, A. V., & Bhattacharya, M. (2013). Kisspeptin/KISS1R system in breast cancer. Journal of Cancer, 4(8), 653–661.
Guzman, S., Brackstone, M., Wondisford, F., Babwah, A. V., & Bhattacharya, M. (2019). KISS1/KISS1R and breast cancer: metastasis promoter. Seminars in Reproductive Medicine, 37(4), 197–206.
Cho, S. G., Li, D., Tan, K., Siwko, S. K., & Liu, M. (2012). KiSS1 and its G-protein-coupled receptor GPR54 in cancer development and metastasis. Cancer Metastasis Reviews, 31(3-4), 585–591.
Arab, K., Smith, L. T., Gast, A., Weichenhan, D., Huang, J. P., Claus, R., et al. (2011). Epigenetic deregulation of TCF21 inhibits metastasis suppressor KISS1 in metastatic melanoma. Carcinogenesis, 32(10), 1467–1473.
Mousavi Ardehaie, R., Hashemzadeh, S., Behrouz Sharif, S., Ghojazadeh, M., Teimoori-Toolabi, L., & Sakhinia, E. (2017). Aberrant methylated EDNRB can act as a potential diagnostic biomarker in sporadic colorectal cancer while KISS1 is controversial. Bioengineered, 8(5), 555–564.
Mardin, W. A., Haier, J., & Mees, S. T. (2013). Epigenetic regulation and role of metastasis suppressor genes in pancreatic ductal adenocarcinoma. BMC Cancer, 13, 264.
Hurst, D. R., & Welch, D. R. (2011). Metastasis suppressor genes at the interface between the environment and tumor cell growth. International Review of Cell and Molecular Biology, 286, 107–180.
Quevedo, E. G., Aguilar, G. M., Aguilar, L. A., Rubio, S. A., Martinez, S. E., Rodriguez, I. P., et al. (2015). Polymorphisms rs12998 and rs5780218 in KiSS1 suppressor metastasis gene in Mexican patients with breast cancer. Disease Markers, 2015, 365845.
Dova, L., Golfinopoulos, V., Pentheroudakis, G., Georgiou, I., & Pavlidis, N. (2008). Systemic dissemination in cancer of unknown primary is independent of mutational inactivation of the KiSS-1 metastasis-suppressor gene. Pathology Oncology Research, 14(3), 239–241.
Pentheroudakis, G., Kostadima, L., Dova, L., Georgiou, I., Tzavaras, T., Vartholomatos, G., et al. (2010). A twisted kiss: in vitro and in vivo evidence of genetic variation and suppressed transcription of the metastasis-suppressor gene KiSS1 in early breast cancer. Neoplasma, 57(1), 47–54.
Pare-Brunet, L., Sebio, A., Salazar, J., Berenguer-Llergo, A., Rio, E., Barnadas, A., et al. (2015). Genetic variations in the VEGF pathway as prognostic factors in metastatic colorectal cancer patients treated with oxaliplatin-based chemotherapy. The Pharmacogenomics Journal, 15(5), 397–404.
Kim, K., Marquez-Palencia, M., & Malladi, S. (2019). Metastatic latency, a veiled threat. Frontiers in Immunology, 10, 1836.
Recasens, A., & Munoz, L. (2019). Targeting cancer cell dormancy. Trends in Pharmacological Sciences, 40(2), 128–141.
Klein, C. A. (2013). Selection and adaptation during metastatic cancer progression. Nature, 501(7467), 365–372.
Werner-Klein, M., & Klein, C. A. (2019). Therapy resistance beyond cellular dormancy. Nature Cell Biology, 21(2), 117–119.
Polzer, B., & Klein, C. A. (2013). Metastasis awakening: the challenges of targeting minimal residual cancer. Nature Medicine, 19(3), 274–275.
Sosa, M. S., Bragado, P., & Aguirre-Ghiso, J. A. (2014). Mechanisms of disseminated cancer cell dormancy: an awakening field. Nature Reviews. Cancer, 14(9), 611–622.
Aguirre-Ghiso, J. A. (2007). Models, mechanisms and clinical evidence for cancer dormancy. Nature Reviews. Cancer, 7(11), 834–846.
McNally, L. R., Welch, D. R., Beck, B. H., Stafford, L. J., Long, J. W., Sellers, J. C., et al. (2010). KISS1 over-expression suppresses metastasis of pancreatic adenocarcinoma in a xenograft mouse model. Clinical & Experimental Metastasis, 27(8), 591–600.
Nash, K. T., Phadke, P. A., Navenot, J.-M., Hurst, D. R., Accavitti-Loper, M. A., Sztul, E., et al. (2007). KISS1 metastasis suppressor secretion, multiple organ metastasis suppression, and maintenance of tumor dormancy. JNCI, 99(4), 309–321.
Corno, C., & Perego, P. (2019). KiSS1 in regulation of metastasis and response to antitumor drugs. Drug Resistance Updates, 42, 12–21.
Aguirre-Ghiso, J. A., & Sosa, M. S. (2018). Emerging topics on disseminated cancer cell dormancy and the paradigm of metastasis. Annual Review of Cancer Biology, 2(1), 377–393.
Aguirre Ghiso, J. A. (2002). Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene, 21(16), 2513–2524.
Yi, T. F., Tan, K. R., Cho, S. G., Wang, Y., Luo, J., Zhang, W. Z., et al. (2010). Regulation of embryonic kidney branching morphogenesis and glomerular development by KISS1 Receptor (Gpr54) through NFAT2-and Sp1-mediated Bmp7 expression. Journal of Biological Chemistry, 285(23), 17811–17820.
Bragado, P., Estrada, Y., Parikh, F., Krause, S., Capobianco, C., Farina, H. G., et al. (2013). TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nature Cell Biology, 15(11), 1351–1361.
Kobayashi, A., Okuda, H., Xing, F., Pandey, P. R., Watabe, M., Hirota, S., et al. (2011). Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. Journal of Experimental Medicine, 208(13), 2641–2655.
Song, G. Q., & Zhao, Y. (2016). Kisspeptin 10 inhibits the Warburg effect in breast cancer through the Smad signaling pathway: both in vitro and in vivo. American Journal of Translational Research, 8(1), 188–195.
Johnson, R. W., Finger, E. C., Olcina, M. M., Vilalta, M., Aguilera, T., Miao, Y., et al. (2016). Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nature Cell Biology, 18(10), 1078–1089.
Kim, T. H., & Cho, S. G. (2017). Kisspeptin inhibits cancer growth and metastasis via activation of EIF2AK2. Molecular Medicine Reports, 16(5), 7585–7590.
Taylor, J., Pampillo, M., Bhattacharya, M., & Babwah, A. V. (2014). Kisspeptin/KISS1R signaling potentiates extravillous trophoblast adhesion to type-I collagen in a PKC- and ERK1/2-dependent manner. Molecular Reproduction and Development, 81(1), 42–54.
Kim, G. L., Dhillon, S. S., & Belsham, D. D. (2010). Kisspeptin directly regulates neuropeptide Y synthesis and secretion via the ERK1/2 and p38 mitogen-activated protein kinase signaling pathways in NPY-secreting hypothalamic neurons. Endocrinology, 151(10), 5038–5047.
Chen, J., Fu, R., Cui, Y., Pan, J., Li, Y., Zhang, X., et al. (2014). LIM-homeodomain transcription factor Isl-1 mediates kisspeptin's effect on insulin secretion in mice. Molecular Endocrinology, 28(8), 1276–1290.
Navenot, J. M., Fujii, N., & Peiper, S. C. (2009). KiSS1 metastasis suppressor gene product induces suppression of tyrosine kinase receptor signaling to Akt, tumor necrosis factor family ligand expression, and apoptosis. Molecular Pharmacology, 75(5), 1074–1083.
Stathatos, N., Bourdeau, I., Espinosa, A. V., Saji, M., Vasko, V. V., Burman, K. D., et al. (2005). KiSS-1/G protein-coupled receptor 54 metastasis suppressor pathway increases myocyte-enriched calcineurin interacting protein 1 expression and chronically inhibits calcineurin activity. The Journal of Clinical Endocrinology and Metabolism, 90(9), 5432–5440.
Platonov, M. E., Borovjagin, A. V., Kaverina, N., Xiao, T., Kadagidze, Z., Lesniak, M., et al. (2018). KISS1 tumor suppressor restricts angiogenesis of breast cancer brain metastases and sensitizes them to oncolytic virotherapy in vitro. Cancer Letters, 417, 75–88.
Chen, S., Su, X., Gao, J., Han, H., Chen, Z., & Lin, S. (2015). [Suppression of Kiss-1 gene inhibits HCT116 human colorectal carcinoma cell migration in vitro via nuclear factor-kappaB signaling pathway]. Nan Fang Yi Ke Da Xue Xue Bao. Journal of Southern Medical University, 35(11), 1643–1648.
Gorbunova, O. L., & Shirshev, S. V. (2014). The role of kisspeptin in immune tolerance formation during pregnancy. Doklady Biological Sciences, 457(1), 258–260.
Huang, C., Wang, H. Y., Wang, M. E., Hsu, M. C., Wu, Y. S., Jiang, Y. F., et al. (2019). Kisspeptin-activated autophagy independently suppresses non-glucose-stimulated insulin secretion from pancreatic beta-cells. Scientific Reports, 9(1), 17451.
Welch, D. R., & Hurst, D. R. (2019). Defining the hallmarks of metastasis. Cancer Research, 79(12), 3011–3027.
Mendoza, A., Hong, S. H., Osborne, T., Khan, M. A., Campbell, K., Briggs, J., et al. (2010). Modeling metastasis biology and therapy in real time in the mouse lung. The Journal of Clinical Investigation, 120(8), 2979–2988.
Young, E. D., Strom, K., Tsue, A. F., Usset, J. L., MacPherson, S., McGuire, J. T., et al. (2018). Automated quantitative image analysis for ex vivo metastasis assays reveals differing lung composition requirements for metastasis suppression by KISS1. Clinical & Experimental Metastasis, 35(1-2), 77–86.
Dotterweich, J., Tower, R. J., Brandl, A., Muller, M., Hofbauer, L. C., Beilhack, A., et al. (2016). The KISS1 receptor as an in vivo microenvironment imaging biomarker of multiple myeloma bone disease. PLoS One, 11(5), e0155087.
Ong, C. P., Lee, W. L., Tang, Y. Q., & Yap, W. H. (2019). Honokiol: a review of its anticancer potential and mechanisms. Cancers (Basel), 12(1), 48.
Cheng, S., Castillo, V., Eliaz, I., & Sliva, D. (2015). Honokiol suppresses metastasis of renal cell carcinoma by targeting KISS1/KISS1R signaling. International Journal of Oncology, 46(6), 2293–2298.
deRoux, N., Genin, E., Carel, J. C., Matsuda, F., Chaussain, J. L., & Milgrom, E. (2003). Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. PNAS, 100(19), 10972–10976.
Curtis, A. E., Cooke, J. H., Baxter, J. E., Parkinson, J. R., Bataveljic, A., Ghatei, M. A., et al. (2010). A kisspeptin-10 analog with greater in vivo bioactivity than kisspeptin-10. American Journal of Physiology. Endocrinology and Metabolism, 298(2), E296–E303.
Asami, T., Nishizawa, N., Ishibashi, Y., Nishibori, K., Horikoshi, Y., Matsumoto, H., et al. (2012). Trypsin resistance of a decapeptide KISS1R agonist containing an Nomega-methylarginine substitution. Bioorganic & Medicinal Chemistry Letters, 22(20), 6328–6332.
Niida, A., Wang, Z., Tomita, K., Oishi, S., Tamamura, H., Otaka, A., et al. (2006). Design and synthesis of downsized metastin (45-54) analogs with maintenance of high GPR54 agonistic activity. Bioorganic & Medicinal Chemistry Letters, 16(1), 134–137.
MacLean, D. B., Matsui, H., Suri, A., Neuwirth, R., & Colombel, M. (2014). Sustained exposure to the investigational Kisspeptin analog, TAK-448, down-regulates testosterone into the castration range in healthy males and in patients with prostate cancer: results from two phase 1 studies. The Journal of Clinical Endocrinology and Metabolism, 99(8), E1445–E1453.
Acknowledgements
Work done in the authors’ labs was funded primarily by the National Foundation for Cancer Research; Susan G. Komen for the Cure (SAC110037); METAvivor Research and Support, Inc.; and USPHS-National Institutes of Health (Grant No. CA87728; CA134981); National Cancer Institute P30-CA168524 (DRW) and National Institutes of Health GM103418 and the KUMC Biomedical Research Training Program (TL); and the Hall Family Professorship in Molecular Medicine. We apologize to any authors whose work was omitted due to article guidelines. We are also grateful for helpful comments and suggestions from Thomas Beadnell, Rosalyn Zimmermann, and Adam Scheid.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Informed Consent
This review summarizes only published results from clinical studies. To the best of the authors’ knowledge, all studies were performed in compliance with applicable human subject protection policies, guidelines, and laws.
Animal Studies
This review summarizes only published results utilizing animals for experimental studies. All work from the authors’ laboratories was approved by relevant Institutional Animal Care and Use Committees. To the best of the authors’ knowledge, all other studies were performed in compliance with applicable policies, guidelines, and laws regarding humane housing, handling, and treatment of research animals.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ly, T., Harihar, S. & Welch, D.R. KISS1 in metastatic cancer research and treatment: potential and paradoxes. Cancer Metastasis Rev 39, 739–754 (2020). https://doi.org/10.1007/s10555-020-09868-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-020-09868-9