
Abstract
In the pediatric population, brain tumors represent the most commonly diagnosed solid neoplasms and the leading cause of cancer-related deaths globally. They include low-grade gliomas (LGGs), medulloblastomas (MBs), and other embryonal, ependymal, and neuroectodermal tumors. The mainstay of treatment for most brain tumors includes surgical intervention, radiation therapy, and chemotherapy. However, resistance to conventional therapy is widespread, which contributes to the high mortality rates reported and lack of improvement in patient survival despite advancement in therapeutic research. This has been attributed to the presence of a subpopulation of cells, known as cancer stem cells (CSCs), which reside within the tumor bulk and maintain self-renewal and recurrence potential of the tumor. An emerging promising approach that enables identifying novel therapeutic strategies to target CSCs and overcome therapy resistance is drug repurposing or repositioning. This is based on using previously approved drugs with known pharmacokinetic and pharmacodynamic characteristics for indications other than their traditional ones, like cancer. In this review, we provide a synopsis of the drug repurposing methodologies that have been used in pediatric brain tumors, and we argue how this selective compilation of approaches, with a focus on CSC targeting, could elevate drug repurposing to the next level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Brain tumors are the most common solid neoplasms and the leading cause of cancer-related deaths among children worldwide [1,2,3,4]. They are a group of neoplasms, each with its own pathophysiology, prognosis, and treatment, that arise from different types of cells within the central nervous system (CNS). Low-grade gliomas (LGGs) are by far the most commonly diagnosed brain tumors in the pediatric age group, followed by medulloblastomas (MBs) and other embryonal, ependymal, and neuroectodermal tumors [5].
Treatment of most brain tumors requires a multimodality approach that includes surgical intervention, radiation therapy (RT), and chemotherapy. Unlike many other forms of cancer, treatment of brain tumors has proved to be particularly challenging due to their location, which renders them beyond the reach of neurosurgeons and inoperable. In addition, chemotherapeutic agents are unable to traverse the blood-brain barrier (BBB) system due to its selective permeability [6]. Those tumors also harbor unique genetic and epigenetic characteristics that make them resistant to various conventional chemotherapeutic agents [7,8,9]. Moreover, several studies demonstrated that therapy resistance could be attributed to the presence of a subpopulation of cells residing within the tumor bulk, known as cancer stem cells (CSCs) [10,11,12]. Indeed, it is becoming apparent that CSCs play a crucial role in the failure to respond to conventional therapy and subsequent, recurrence of different brain tumors [13]. Hence, it is crucial—with the unassertive improvements in the outcomes of patients during the past few years—to look for new treatment strategies that focus on targeting CSCs via a computational-based drug repurposing approach.
The idea of drug repurposing—by using previously approved drugs for new indications other than their traditional use—has recently gained considerable popularity in different fields of medicine, especially cancer [14, 15]. This attractive approach is anticipated to provide novel avenues to overcome conventional therapy resistance and eradicate the highly malignant brain CSCs. In tumors such as breast cancer, for example, genomics-based tools have been used to identify Food and Drug Administration (FDA)-approved drugs and repurpose them to treat patients through targeting CSCs within the tumor [16]. In addition, data-driven computational methods were utilized in glioblastoma multiforme (GBM) to identify compounds and drugs that can be repositioned to inhibit GBM CSCs [17]. Such synthetic drugs could present promising molecular weapons against cancer, and henceforth predict a bright future for efficient management of lethal cancers such as brain tumors.
2 Repurposing approved drugs in pediatric brain tumors
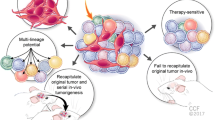
Advancements in analyzing the genetic and epigenetic alterations in tumor cells in the past few years drove the discovery of novel molecular signatures and therapeutic targets implicated in oncogenesis, including pediatric tumors, which in turn instigated remarkable improvement in the survival rates of patients [18]. According to the Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute, there was a conspicuous increase in the 5-year relative survival rate for all childhood cancer combined from 61.7% in 1975–1977 to 81.4% in 1999–2006, except for gliomas [19]. Particularly, children with metastatic diseases and brain tumors have not shown significant improvement in their survival over the past three decades [18]. This necessitates seeking a different approach to identify more effective drugs for pediatric brain tumors that mainly target signaling pathways of stemness and thus block tumor growth and metastasis. Consequently, drug repurposing could be the promising next-generation strategy to pursue in cancer research [20]. By combining genetic evidence with suitable in vitro/in vivo phenotypic analyses, FDA-approved drugs—with known pharmacokinetics/pharmacodynamics and drug-drug interaction parameters—could be repurposed to serve as potential candidates to treat brain tumors among children [14]. However, those candidate compounds must have a number of unique features in order to be efficient in the case of pediatric brain tumors, including (1) ability to traverse the BBB, (2) safe for infants and children with no serious adverse effects, (3) FDA approved, (4) considerably less costly and less time-intensive, and (5) proven efficient and safe against brain CSCs in preclinical models (Figs. 1 and 2).

Schematic diagram showing how repurposed drugs targeting cancer stem cells (CSCs) promote tumor regression. While majority of cancer cells within a tumor are affected by conventional therapies leading to shrinkage of the tumor, surviving CSCs may resist these types of therapeutic agents prompting cancer recurrence with time. This renders the need for novel targeted therapeutic approaches for “repurposed drugs” directed against CSCs to elicit complete tumor regression and prevent recurrence

Drug repurposing to target cancer stem cells (CSCs) in pediatric brain tumors. Using genomics-based tools, computational approaches, and in vitro/in vivo analyses, many Food and Drug Administration (FDA)-approved drugs could be identified and repurposed to specifically target CSCs in patients with pediatric brain tumors
3 Pediatric glioma
Gliomas are the most common brain tumors in children [21]. These tumors are divided into two categories; the low-grade glioma (LGG) and the high-grade glioma (HGG), where the former accounts for 50% of all primary pediatric CNS tumors [22] while the latter accounts for only 8% [23]. Surgical resection is the mainstay of management for gliomas in both children and adults, followed by conventional chemotherapy, mainly Temozolomide (TMZ), or radiation therapy [22, 23]. Although these conventional therapies might improve the quality of life, they do not always increase patients’ survival [24]. Under the current treatments, LGG shows much higher survival rates than HGG, where patients diagnosed with glioblastoma (astrocytoma grade IV), the most aggressive form of glioma, have an overall survival of several months after diagnosis despite major advances in translational research and therapeutic interventions [24,25,26]. This has been attributed, in part, to the incomplete eradication of the CSC subpopulation present within the heterogeneous tumor bulk, which is held responsible for fueling tumor regrowth and self-renewal, leading to tumor progression and malignant recurrence [27, 28].
New studies are redirecting the use of pre-approved existing drugs, which have been used clinically for nonneoplastic pathologies, alone or combined with conventional therapies to increase their efficacy [29]. This is the case for several solid tumors as it is for glioma, where a set of drugs is under investigation for targeting cancer cells, and in particular the CSC subpopulation, to increase survival of glioma patients [29] (Table 1).
3.1 Antidiabetic drugs
Metformin and its analog phenformin, two biguanide drugs used to treat type 2 diabetes mellitus, were found to be effective on glioma cell lines leading to reduced migration [44] and cell death through the generation of reactive oxygen species (ROS) [30]. When compared to metformin, phenformin was able to significantly decrease the proliferation of glioblastoma stem cells in vitro and to reduce the expression of stem cell markers such as SOX2, OCT4 and CD44. Phenformin was also shown to prolong the survival of xenografted mice by inhibiting angiogenesis within the tumor and inducing apoptosis of tumor cells in vivo, especially when combined with TMZ [31].
3.2 Antihyperlipidemic drugs
Another category of repurposed therapeutic drugs for targeting glioma (specifically the HGG) includes statins. These lipid-lowering drugs are traditionally used to reduce heart attacks and strokes, therefore decreasing the mortality of patients with cardiovascular diseases and lowering pleiotropic effects [45]. In cancer, simvastatin was shown to reduce proliferation and induce apoptosis of the C6 glioma cell line via increased phosphorylation of c-jun by activating the JNK pathway [32]. Other statins were shown to induce cytotoxicity, reduce migration and invasion, and cause apoptosis of glioblastoma cell lines mainly through manipulating TGF-β activity, a common target for statins in both glioma and cardiovascular diseases [33]. However, when combined with chemotherapy, and specifically TMZ, statins sensitized glioblastoma cell lines to this treatment, therefore increasing the efficiency of standard of care therapy in targeting this tumor [46].
3.3 Antihypertensive drugs
Angiotensin I converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs)—known for lowering blood pressure in hypertensive patients and treating congestive heart failure [47, 48]—have been involved in targeting solid tumors [34]. In glioblastoma, studies revealed numerous ways by which ACEIs and ARBs can efficiently decrease tumorigenic properties. It was proven that angiotensin and angiotensinogen receptors I and II signaling promote the release of several cytokines and proinflammatory factors favoring tumor development and aggressiveness [49,50,51]. Interestingly, ACEIs and ARBs played a critical role in modifying the tumor microenvironment by reducing hypoxia, enhancing T cell infiltration, and ameliorating the antitumor immunity. In addition, these drugs were able to enhance the delivery of conventional therapies to the destined sites by increased BBB penetrance, hence prolonging the survival of glioblastoma patients [34]. These data support the notion that targeting the renin-angiotensin system by ACEIs and ARBs, improves cancer treatment via activating immune-stimulatory pathways to enhance cancer immunotherapy [34].
3.4 GSK3-β inhibitors
TMZ is the most effective FDA-approved chemotherapeutic treatment for glioma, especially when combined with radiation therapy [52]. However, during treatment, cancer cells might become resistant, a phenomenon attributed to the CSC population residing within the tumor bulk [10, 53]. Thus, shortly after treatment, glioma, in particular HGG, becomes refractory to both chemotherapy and radiation therapy, and the survival is drastically decreased. Therefore, studies have focused on improving glioblastoma prognosis mainly by targeting the CSC population and by enhancing the outcome of conventional treatments [35]. To fulfill these purposes, several molecular studies were conducted to better understand the mechanisms underlying aggressiveness and therapy resistance in glioblastoma.
One of the most critical findings states that GSK3-β, a serine-threonine kinase, is overexpressed in glioblastoma, yet its role remains controversial as it promotes cancer progression in some tissues and suppresses tumors in others [54]. In glioblastoma, knocking down GSK3-β has been shown to promote cellular apoptosis [55]. In light of these data, new strategies have emerged to inhibit GSK3-β, especially after the pathologic link of some neurodegenerative diseases to GSK3-β, such as Alzheimer’s disease (AD) [56]. This fortunate finding has led to the commencement of several clinical trials using GSK3-β inhibitors to treat AD and progressive supranuclear palsy [57, 58]. Hence, it was worth repurposing GSK3-β inhibitors to treat glioblastoma, thereby decreasing drug costs and increasing treatment success rates. Several drugs have been tested for this purpose, out of which kenpaullone was found to inhibit stemness properties by reducing the number and size of cultured glioblastoma spheres [35]. In vivo studies also showed that combinatorial treatment of kenpaullone with TMZ prolonged survival of mice compared to sole treatment with TMZ as the penetrance of kenpaullone across the BBB enhances when administered with TMZ [35]. Lithium chloride, another GSK3-β inhibitor and well-known drug for the treatment of bipolar disorder, was shown to inhibit cellular proliferation and invasion of glioblastoma spheres in vitro [36, 37].
Moreover, several studies reported more potent antitumor effects of other GSK3-β inhibitors in targeting CSCs as alternatives to lithium due to its potential toxicities in humans when administered at high concentrations. Those inhibitors include indirubins and their derivatives which reduce the phosphorylation of β-Catenin, a GSK3-β substrate, thereby decreasing migration and increasing survival [36], and CHIR99021 which increases the permeability of endothelial cells, allowing therapeutic drugs to traverse the BBB and reach the tumor site [36]. More recently, a new drug named tideglusib that irreversibly inhibits GSK3-β was placed on clinical trials for AD [57, 59]. This same drug is referred to as an “orphan drug” for AD [60]. Tideglusib is still under investigation for its role in treating glioblastoma and other nervous system tumors. In one study, tideglusib was found to significantly reduce the sphere forming ability of glioblastoma cells and to sensitize cancer cells to TMZ chemotherapy by inhibiting GSK3-β [61]. A study from our group also revealed that tideglusib is an effective in vitro treatment for neuroblastoma by significantly reducing cell proliferation, viability, and migration of SK-N-SH and SH-SY5Y cells, and inhibiting the neurosphere forming ability of SH-SY5Y cells [62].
3.5 NSAIDs
Nonsteroidal anti-inflammatory drugs (NSAIDs) are used as antipyretics, anti-inflammatory, and analgesic agents. They are arachidonic acid pathway inhibitors primarily targeting cyclooxegenase-2 (Cox-2) enzyme, hence decreasing the synthesis of various prostaglandins implicated in a wide range of diseases such as cancer, inflammation, cardiovascular disease, and hypertension [63]. Prostaglandin E2 (PGE2), through EP receptor signaling, enhances tumor cell proliferation and angiogenesis and inhibits apoptosis and immune responses [63, 64].
Ibuprofen and diclofenac, which are usually used as analgesics, have been shown to reduce cell proliferation and migration of glioblastoma cell lines [38, 39]. While both reduced signal transducer and activator of transcription 3 (STAT3) phosphorylation, diclofenac further reduced extracellular lactate, c-myc expression, and lactate dehydrogenase (LDH-A) activity. As such, diclofenac may give rise to decreased lactate-mediated immunosuppression in gliomas. The COX-2 inhibitor celecoxib reduced tumor edema and exhibited an anti-angiogenic effect on glioblastoma [40, 65].
3.6 Antipsychotic drugs
Antipsychotic drugs such as thioridazine [41] and trifluoperazine [42] were tested as anticancerous agents as well. Thioridazine prevented the occurrence of adaptive metabolic alterations associated with TMZ resistance and was able to impair autophagy by inhibiting the fusion between lysosomes and autophagosomes, thereby increasing the chemosensitivity of glioblastoma to TMZ [41]. In vivo studies showed that an analog of trifluoperazine, 3 dc, reduced the size of xenografted brain tumors in a mouse model of glioblastoma and increased intracellular calcium levels with a selective effect on glioblastoma cells over normal neural cells [42].
3.7 Antidepressants
Antidepressants were also part of the repurposing strategy. One such drug, fluvoxamine, is able to disrupt actin polymerization, inhibit FAK and AKT/mTOR signaling, and subsequently inhibit glioblastoma migration and invasion in vitro. Similarly, an in vivo model demonstrated the anti-invasive effect of fluvoxamine, where it confined the CSC (CD133+ staining) dissemination to the area where the tumor was located compared to the untreated group of mice, where the CSC population was more widespread [43].
3.8 Antineoplastic drugs
Another category of repurposed drugs for treating glioma lies in the cancer field itself. In an attempt to reduce the long-term toxicity caused by radiotherapy in pediatric glioma patients, a study showed that combining rapamycin, an mTOR signaling pathway inhibitor [66], and erlotinib, an EGFR tyrosine kinase inhibitor [67], both used to treat lung cancer, was found to be well tolerated in children with LGGs and some patients exhibited prolonged disease stabilization [68].
4 Medulloblastoma
Medulloblastoma (MB) is a highly aggressive malignant tumor arising from the CNS primitive neuroectodermal cells, mainly originating from the cerebellum or posterior fossa [69]. It is by far the most common malignant tumor of the CNS in children, consisting of about 20% of the total brain tumors in this patient population, with a prevalence of around 5 per 1000,000 children younger than 15 years [70, 71].
There are distinct molecular classifications of MB, as demonstrated by Pomeroy et al. [72], Thompson et al. [73], Kool et al. [74], and E. Cho et al. [75]. These molecular subgroups differ in their demographics, transcriptomes, somatic genetic events, and clinical outcomes. Variations in the number, composition, and nature of the subgroups between studies brought about a consensus conference in Boston in the fall of 2010, where the discussants agreed on four main transcriptional subgroups of MB named MBWNT, MBSHH, MBGroup 3, and MBGroup 4, clearly distinct in terms of demographics, histology, DNA copy-number aberrations, and clinical outcome [76].
The standard of care therapy for MB encompasses maximal safe resection followed by radiation therapy and chemotherapy [77]. However, MB harbors a population of CSCs that are resistant to the multimodal therapy used and are thought to be responsible for the consequent increase in tumor progression, metastasis, and recurrence [78, 79]. Moreover, the tumorigenic interplay that exists between the neighboring signaling cascades, the epidermal growth factor receptor (EGFR), and platelet-derived growth factor receptor (PDGFR), previously reported in MB cells [80, 81], may explain the aggressive mechanisms these cells utilize to evade therapy. However, many drugs used in the treatment of chronic and nonneoplastic diseases have been repurposed to target molecular pathways that are involved in the pathogenesis of MB, as shown below (Table 2).
4.1 Cardiac glycosides
Cardiac glycosides, such as digoxin, digitoxin, and ouabain, are naturally derived steroid-like compounds widely used in the treatment of congestive heart failure and cardiac arrhythmias. They act by binding to and inhibiting sodium-potassium ATPase (Na+/K+-ATPase) [95, 96]. The initial interest in cardiac glycosides was for use in cardiac disease, yet several studies were conducted thereafter to assess the use of these medications in the treatment of cancer [97].
Among the receptor tyrosine kinases, the EGFR family is known to regulate growth, differentiation, motility, and survival of fibroblasts and epithelial cells. It triggers various downstream signaling cascades, including the Erk1/2 (MAPK) and phosphoinositide-3 (PI3)-kinase/Akt pathways. EGFR family is also expressed in neurons of the hippocampus, cerebellum, and cerebral cortex in addition to other regions of the CNS [82, 98, 99]. Linking EGFR with MB, co-expression of c-erbB2/HER2 and c-erbB4/HER2 subtypes of the EGFR family had been significantly associated with the advanced metastatic disease and poor clinical outcome [100,101,102]. D. Wolle et al. revealed a crosstalk between Na, K-ATPase and EGFR signaling, where treatment of DOAY MB cells with ouabain, a cardiac glycoside, inhibited EGF-induced downstream activation of Erk1/2 and Akt but activated Erk1/2 and Akt independently from EGFR. Aside from that, ouabain inhibited p21 Ras activation, hindered EGF-mediated formation of actin stress fibers, prevented EGF-induced FAK phosphorylation, and hence DAOY cell motility, hypothetically through stress signaling activation [82].
Huang et al. studied the in vitro effects of 5 members of the cardiac glycoside family (proscillaridin A, digoxin, lanatoside C, digitoxigenin, and digoxigenin) on MED8A (MBGroup 3) and D283 (MBGroup 4) cells [103]. Both cell lines showed a significant decrease in viability, with ascending doses of all cardiac glycoside family members. In addition, digoxin was found to prolong survival in orthotopic PDX models. Huang et al. also reported better survival among mice treated with digoxin and radiation therapy compared to mice treated with radiation therapy only [83].
4.2 Antihyperlipidemic drugs
Statins, antihyperlipidemic drugs that inhibit the HMG-CoA reductase enzyme of cholesterol biosynthesis, have also been used to target MB [104, 105]. HMG-CoA inhibition prevents the formation of isoprenoid intermediates that are essential for post-translational modification (prenylation) of intracellular signaling proteins, including the G-proteins Ras and Rho [105, 106]. Interestingly, inhibiting Ras and Rho prenylation by statins alters numerous downstream signaling pathways that are implicated in cancer, such as the PI3K/Akt/mTOR and MAPK/ERK pathways [107, 108].
Takwi et al. studied the interplay between statins, microRNAs (miRNAs) and c-Myc in vitro [84]. Lovastatin was found to induce miR-33b (miRNA precursor whose gene is located at 17p11.2, a genetic locus identified to be lost in patients with MB [109]) expression, which in turn negatively regulated c-Myc expression, and extended survival and reduced growth of xenografted DAOY cells in mice [84].
Another studied target for statins is the Hedgehog (Hh) signaling pathway. Hh pathway is an essential pathway to maintain the activity of CSCs population in various tumors [110]. PTCH1 gene mutation, which is reported in 8–10% of sporadic MB cases, leads to ligand-independent constitutive activation of the Hh pathway [110, 111]. Hh signaling pathway effector Gli 1 was associated with BclII overexpression, and both were present in areas of decreased apoptosis in nodular MB. Lovastatin treatment of DAOY MB cells was associated with upregulation of pro-apoptotic genes with reduction in BclII mRNA and protein levels. Significant apoptosis was seen with lovastatin monotherapy of DAOY cells and co-treatment with cyclopamine, an Hh antagonist [85, 86]. In another study by Sheikholeslami et al., the effect of Simvastatin on 3 MB cell lines (DAOY, D283, D231) was investigated [87]. Simvastatin induced apoptotic cell death in the treated cell lines compared to untreated controls by activation of intrinsic and extrinsic apoptosis pathways. Rescue assays reversed simvastatin induced cell death, variably among the three studied MB cell lines. Simvastatin also decreased Mcl-1 and Bcl2 expression, both of which are anti-apoptotic proteins that regulate apoptosis [87].
4.3 Antihelminthic drugs
Mebendazole is a benzimidazole antihelminthic drug used to treat parasitic infestations via inhibiting microtubule formation within the parasites [112, 113]. This drug was repurposed to study its effect on MB. Bai et al. revealed that mebendazole inhibited vascular endothelial growth factor receptor 2 (VEGFR-2) in cultured HUVECs in vitro and in MB xenografts. Notably, VEGFR-2 is known to induce angiogenesis, enhance vascular permeability, and maintain CSCs [114]. Tumor sections from mebendazole-treated mice also showed a decrease in the angiogenic activity within the tumor with no effect on the microvasculature within normal brain tissue. Furthermore, results showed improved survival by 150% and slowed tumor growth [88]. In a second study, Bai et al. demonstrated the brain penetration and efficacy of the 3 mebendazole polymorphs A, B, and C [115]. Mebendazole polymorph C exhibited greater brain tissue penetrance in mice leading to a significant concentration and high brain to plasma ratio (B/P ratio: 0.82). Combining mebendazole with elacridar, a third generation P-glycoprotein inhibitor, the latter marginally increased mebendazole cytotoxicity in vitro on GL261 mouse glioma cells. However, co-administration of mebendazole polymorph C and elacridar had no effect on mebendazole brain concentration. Interestingly, the combination of the two drugs improved survival in GL261 glioma and D425 MB models [115].
Larsen et al. also advocated the anticancer effect of mebendazole as a target for SHH signaling pathway [89]. Knowing that the primary cilia is an essential component for SHH signal transduction [116], binding of mebendazole to human tubulin and subsequent inhibition of primary cilia assembly was an important mechanism by which mebendazole exerted its anti-SHH signaling pathway effect. Mebendazole-treated DAOY MB cells showed decreased expression of GLI-1, a downstream effector of SHH signaling pathway. Mebendazole in vitro studies also revealed a decrease in DAOY cells proliferation and viability with biochemical and morphological evidence of apoptosis. In vivo studies in mice xenografted with DAOY cells exhibited a prolongation in median survival with decreased expression of GLI1 and PTCH1 expression and reduced tumor cell proliferation.
4.4 HDL nanoparticles
As described above, cholesterol signaling plays an essential role in tumor growth and CSCs population maintenance. Bell et al. studied the effect of HDL nanoparticles (HDL NPs) in SHH-MB that express scavenger receptor type B-1 (SCARB1), being as HDL NPs receptor. SCARB1 showed to be highly expressed in SHH MB subtype and associated with poor prognosis. Co-expression of other Hh pathway genes such as GLI2, GLI3, and HHIP was also seen in MB patients with high SCARB1 expression. Upon HDL NPs binding to SCARB1, there was an increase in cholesterol efflux with a decrease in total cholesterol in SHH driven MB and Ewing sarcoma cells. HDL NPs also disrupted MB and Ewing sarcoma cellular viability. Importantly, CSC frequency was also reduced in HDL NPs-treated MB and Ewing sarcoma-treated cells, reflected by a decrease in ALDEFLUOR positive (ALDH+) cells and reduction in 3-D sphere formation in CSC medium [90].
4.5 Antiretroviral drugs
Abacavir is a nucleoside antiretroviral drug that acts by inhibiting the reverse transcriptase enzyme. It has been approved for the treatment of HIV infection with good tolerability and favorable safety profile [117]. Abacavir was also studied as a potential inhibitor of human telomerase activity, a ribonucleoprotein complex that maintains telomere length at the end of chromosomes, including CSCs [118], thereby contributing to tumorigenesis and cellular immortalization [119, 120]. Increased telomerase activity was reported in MB, rendering it as a potential target in the treatment of MB [121]. A study by Rossi et al. demonstrated the effect of Abacavir on MB cell lines [91], revealing decreased cellular proliferation with cellular accumulation at G2/M phase of the cell cycle. In addition, Abacavir-treated cells exhibited a significant increase in cell death, inhibition of telomerase activity, and downregulation of hTERT mRNA. Abacavir also induced cellular differentiation, rendering tumor cells responsive to normal growth regulatory signals and sensitive to chemotherapeutic agents [122]. The Abacavir-induced inhibition of telomerase activity resulted in telomere induced senescence, which is considered one of the tumor suppressive mechanisms that mediate the antitumor effects of chemotherapeutic agents [123].
4.6 NSAIDs
As previously described, NSAIDs are potent COX inhibitors capable of reducing the synthesis of various prostaglandins. Baryawno et al. evidenced the expression of Cox-2, microsomal prostaglandin E synthase-1 (mPGES-1), and Prostanoid EP receptors in MB primary tumors and cell lines [92]. He demonstrated the effect of diclofenac (Cox1/Cox2 inhibitor) and celecoxib (Cox2 inhibitor) on MB. In vitro studies on 9 MB cell lines revealed a dose and time-dependent decrease in cell viability of MB cells, associated with the induction of caspase-dependent apoptosis [92]. In vivo studies on mice carrying xenografted D283 cells treated with either diclofenac or celecoxib suppressed tumor growth by stimulating apoptosis and inhibiting angiogenesis [92].
STAT3 activation plays a crucial role in the progression of many tumors, including brain tumors, by enhancing cancer cell growth, invasion, and metastasis while hindering apoptosis [124, 125]. STAT3-related pathways are constitutively activated in MB-derived CD133/Nestin double-positive cells (MB-DPs) and regulate cancer stem-like properties in MB-DPs [93]. MB-DPs treated with celecoxib demonstrated downregulation of STAT3 gene, phosphorylated STAT3, STAT3-related protein, JAK2, Bcl2, and c-Myc, suppressing their stem-like gene properties [93]. Moreover, celecoxib potentiated the effect of ionizing radiation in both in vivo and in vitro studies [93, 126].
A study by Eslin et al. revealed the effect of the NSAID tolfenamic acid on MB cell lines and mouse xenografted tumor models, targeting specificity protein 1 (Sp1) and survivin [94]. Sp1 is a transcription factor that regulates genes involved in cellular proliferation, differentiation and growth [127], including survivin, an inhibitor of apoptosis protein that regulates apoptosis and cellular mitosis in cancer cells [128]. Overexpression of Sp1 and survivin has been associated with unfavorable prognosis in cancer [129]. Tolfenamic acid showed a decrease in cellular proliferation in DAOY and D283 MB cell lines in a time and dose-dependent manner and induced apoptosis in both MB cell lines with increased expression of the pro-apoptotic c-PARP [94]. Tolfenamic acid also caused downregulation of Sp1 and survivin expression in both cell lines [94]. In vivo studies on nude mice xenografted with D283 MB cells and treated with tolfenamic acid revealed reduced tumor growth and decreased expression of Sp1 and survivin in mice tumor tissues [94]. Similar observations were demonstrated in another study by Patil et al. that reported synergistic effects of tolfenamic acid and vincristine on inhibition of MB cellular growth, increased apoptosis and cell cycle arrest, and decreased survivin expression [130].
5 Neuroblastoma
Neuroblastoma (NB) is the most common extra-cranial solid tumor in the pediatric population, accounting for almost 15% of cancer-related deaths in this group [131]. The current standard of care for NB is a combination of chemotherapy, radiotherapy, and surgical intervention depending on the staging of the tumor. Unfortunately, despite major advancements in treatment options, therapeutic resistance, often manifested by compensatory activation of tumorigenic pathways [132], especially in NB CSCs [133], leads to malignant recurrence [131], and hence, alternative treatment modalities must be sought. Drug repurposing is indeed a promising strategy that allows existing drugs with known pharmacokinetic and pharmacodynamic profiles to be evaluated in treating cancer such as NB. A multitude of drugs have been identified to have in vitro and in vivo effects on NB (Table 3).
5.1 Antimicrobials
Ketoconazole, an antifungal drug and CYP3A4 inhibitor, was shown to increase intra-tumoral levels of the NB chemotherapeutic agent fenretinide, subsequently enhancing its cytotoxicity and inhibiting NB tumor growth in vivo [134]. Another antihelminthic drug, flubendazole, was found to destabilize microtubules, induce p53-dependent apoptosis, and increase necrotic areas within UKF-NB-3 and UKF-NB-3rCDDP1000 (human NB cell lines derived from bone marrow metastasis) NB tumors [135]. Tigecycline, a widely used antibiotic, was also studied in NB revealing decreased cell proliferation and tumor growth by disrupting PI3K/Akt pathway and inducing G1-phase cell cycle arrest [136].
Di Zanni et al. proved that chloroquine (CQ), an antimalarial drug, inhibits IMR32 NB cell proliferation through PHOX2B gene downregulation in the early phase of tumorigenesis [137]. CQ also appears to increase the antitumor activity of RTKi by inhibiting autophagy, a potential escape mechanism used by NB cells when treated with RTKi [138]. In a case report, nifurtimox, an antiprotozoal drug used in Chagas disease, caused remission of NB in a child with concomitant NB and Chagas [139]. In other studies, this same drug was found to inhibit NB cell growth in vitro and in vivo via ROS formation, inhibition of Akt phosphorylation [140], and upregulation of downstream GSK3β [142]. It also decreased NMYC expression and glucose utilization [141]. DFMO is another antiprotozoal repurposed as a potential treatment of NB. It was found to block polyamine synthesis by inhibiting ornithine decarboxylase, which is implicated in NB and to inhibit MYCN driven protein translation, leading to depletion of thymidine pools and disruption of the tumor microenvironment (TME) [143]. Interestingly, it also caused dysregulation of CSCs and glycolytic metabolism via the LIN28/Let-7 pathway, specifically in the setting of elevated MYCN levels [144]. In fact, DFMO appears to be more effective against NB cells when combined with celecoxib [143].
5.2 Drugs used in musculoskeletal diseases
Larsson et al. demonstrated that diclofenac NSAID reduces PGE2 secreted by cancer-associated fibroblast (CAFs) in the immunosuppressant TME of chemotherapy-resistant 11q-deleted NB [145]. Sulfasalazine, a drug used in rheumatoid arthritis, hindered NB cell proliferation by decreasing tetrahydrobiopterin (BH4) levels through the inhibition of sepiapterin reductase (SPR) [146]. Interestingly, TL-118 anti-angiogenic drug combination of cyclophosphamide, diclofenac, sulfasalazine, and cimetidine displayed synergy with gemcitabine through the former’s anti-angiogenic activity and the latter’s cytotoxic effect [147]. Among drugs used in gout, probenecid was found to be effective in NB cell treatment, especially when combined with cisplatin leading to reduced expression of the drug efflux transporters, multidrug resistance-associated proteins (MRPs) and to a diminished population of CSCs within the tumor [148].
Cinacalcet may be a promising drug for NB because it upregulated and stimulated the calcium-sensing receptors (CaSR) that are normally downregulated in MYCN NB tumors. In addition, its capacity to upregulate the expression of cancer-testis antigens (CTAs) warrant it as a potential target for immunotherapy [149]. Zoledronate, another drug that could be repurposed for NB treatment functions by blocking the mevalonate pathway, which enhances the cytolytic activity of γδ T cells in NB cells (including CSCs) [150].
5.3 Antihyperlipidemic drugs
Simvastatin, an inhibitor of mevalonate synthesis that has been repurposed to target gliomas and MBs, has also been evaluated as a potential drug for NB treatment. This antihyperlipidemic medication induced apoptosis of NB cells via impaired prenylation of small Rho GTPases, which are implicated in tumor proliferation and migration [151]. Another drug indicated for hyperlipidemia, fenofibrate, was found to increase oxidative stress in NB cells by enhancing production of the ROS scavenger TXNIP [152].
5.4 Antidiabetic drugs
Metformin is another promising agent for NB [153,154,155,156,157,158], particularly if used in combination with conventional chemotherapeutic agents [155]. It induced apoptosis by disrupting both Rho GTPase/MAPK [154] and Akt/mTOR pathways [153, 155]. Our group showed that metformin, an activator of the AMPK pathway, reduced NB and glioblastoma sphere formation and cell migration and invasiveness via decreasing MMP-2 production [156]. Moreover, Binlateh et al. recently showed that metformin promotes NB cell differentiation by increasing ROS levels, and subsequent disinhibition of Sox6 by cdk5, as well as dephosphorylating Erk1/2, and phosphorylating Akt [157]. Thiazolidinediones (TZDs), such as ciglitazone, pioglitazone, troglitazone, and rosiglitazone, were also found to inhibit NB cells in vitro by activating PPAR gamma. Rosiglitazone and pioglitazone specifically inhibited cell adhesion and migration, stimulated differentiation, and decreased tumor growth in vivo [158].
5.5 Drugs used in cardiovascular diseases
Propranolol, a beta-blocker drug used in cardiovascular diseases, increased apoptosis of NB cells by acting on β2 receptors to induce expression of p53 and its paralogue TAp73. It appears to be more effective when combined with SN-38 and celecoxib [159]. In addition, the anti-anginal drug, perhexiline maleate, downregulated the expression of ABC transporters, key players in chemoresistance, via NDM29. It enhanced the effect of cisplatin, decreased sphere formation—hence targeting the CSCs population, and enhanced cellular differentiation [160].
5.6 Drugs used in neurological disorders
Valproic acid (VPA) has been well documented to target NB cells via epigenetic modifications mediated by HDAC inhibition [161,162,163,164, 170,171,172,173,174] and has displayed synergy with etoposide, cisplatin [163], ellipticine [170], and celecoxib [171]. Gu et al. showed that VPA induces cell cycle arrest in MYCN NB cell lines LA1-55n and NBL-W-N by “rescuing tumor suppressor genes” [161]. Shah and colleagues showed that besides its HDAC inhibitory functions, VPA also downregulates Akt/Survivin and induces staurosporine (STS) mediated cell cycle arrest [162]. Moreover, VPA inhibits NB cell proliferation by downregulating MKK7/Raf1, inhibiting subsequent Fra-1/c-Jun dimer formation and, AP-1 dimer activation [172]. Recently, a study showed that VPA inhibits aerobic glycolysis in NB cells by downregulating E2F, the transcription factor that controls GPI and PGK1 glycolysis genes [164]. Also, VPA abolished the stimulatory effect of BDNF seen in aggressive MYCN NB tumors by suppressing TrkB via RUNX3 upregulation [173]. However, a drawback to VPA is its ability to induce CD133 expression in UKF-NB-3 cells displaying pluripotency biomarkers Nanog, Oct-4 and Sox2, higher proliferation, increased sphere formation, and lower sensitivity to cytostatic drugs [174]. When acetazolamide was combined with MS-275, a HDAC inhibitor, there was reduced proliferation and migration of tumor cells, particularly SP cells, and decreased expression of these markers [165]. Moreover, mTOR and Akt inhibitors have been documented to reduce the number of CD133+ cells [169], signifying the importance of different drug combinations as a promising therapeutic intervention that warrants further investigation.
Fingolimod, a drug used in refractory multiple sclerosis, inhibited TRMP7 channels, which are associated with a higher risk of mortality in NB. Their inhibition decreased intracellular calcium levels and compromised calcium-dependent signaling pathways. This drug might be a strong candidate for repurposing as it sensitizes drug-resistant NB cells to doxorubicin and other chemotherapeutics [175]. Bilir et al. elaborated that through inhibition of CSCs, lithium chloride (LiCl) and clomipramine (CLMP) are able to potentiate the activity of vinorelbine (VNR) on SH-SY5Y NB cells and that the combination of lithium chloride with VNR was associated with lower levels of the chemoresistance biomarker midkine (MK) [166].
5.7 Antineoplastic drugs
Vorinostat, another HDAC inhibitor, increased the chemosensitivity of N-MYC amplified NB cells and reduced stemness [167]. Ponatinib is also another drug that induced PARP mediated apoptosis and inhibited proliferation, colony formation, and invasion of NB cells. Ponatinib treatment of CHP-134 and IMR-32 spheroids impaired migration without affecting spheroid size. This effect had been attributed to the inhibition of the mTOR pathway and its downstream players [168]. This is similar to what our group has previously documented, whereby Triciribine (Akt inhibitor) and Rapamycin (mTOR inhibitor) dysregulated the oncogenic PI3K/Akt/mTOR/S6K1 pathway and inhibit CSCs in both NB and glioblastoma [169].
6 Other pediatric brain tumors
6.1 Pinealoma
Pineal parenchymal tumors (PPTs) are aggressive brain tumors that are usually treated with chemoradiotherapy and surgery [176]. Due to their rarity, the literature is scant. A case report showed that the combination of the HDAC inhibitor vorinostat and retinoic acid exhibited complete remission of a pineoblastoma [177].
6.2 Ependymoma
Ependymomas usually arise in the brain (children) or spinal cord (adults) and rarely metastasize outside the central nervous system. The standard of care therapy involves surgical resection with or without the need for further radio- or chemotherapy depending on the tumor infiltration into the neighboring healthy tissue [178]. In an in vivo study, Nimmervoll et al. showed that gemcitabine can be repurposed to be used in ependymoma as gemcitabine infusions following tumor resection and radiotherapy doubled the median survival of mice and led to 50% cure [179].
7 Conclusion
Repurposing of previously approved drugs and bypassing the time-consuming toxicology/safety pharmacology testing in drug development steps carries a ray of hope in tackling extensive drug resistance in pediatric brain tumors to help better manage these diseases of childhood. However, and as previously mentioned, the candidate compounds must be brain penetrants, safe for infants and children, FDA approved, and previously tried in preclinical models with proven efficiency against CSCs. In this review, we propose that a multidisciplinary approach might be required to set up the landscape for a successful computational-based drug repurposing in pediatric brain tumors as well as other cancer types. Such an approach must take into consideration a group of parameters, such as cancer bioinformatics data, sub-networks of biomolecules and/or pathways controlled by each drug, drug pharmacokinetic/pharmacodynamic characteristics, related preclinical studies conducted, and drug-sensitive cell types within the tumor among others.
References
Pollack, I. F., & Jakacki, R. I. (2011). Childhood brain tumors: epidemiology, current management and future directions. Nature Reviews Neurology, 7(9), 495–506. https://doi.org/10.1038/nrneurol.2011.110.
Pollack, I. F. (1994). Brain tumors in children. New England Journal of Medicine, 331(22), 1500–1507. https://doi.org/10.1056/nejm199412013312207.
Siegel, R. L., Miller, K. D., & Jemal, A. (2019). Cancer statistics, 2019. CA: a Cancer Journal for Clinicians, 69(1), 7–34. https://doi.org/10.3322/caac.21551.
Smith, M. A., & Reaman, G. H. (2015). Remaining challenges in childhood cancer and newer targeted therapeutics. Pediatric Clinics of North America, 62(1), 301–312. https://doi.org/10.1016/j.pcl.2014.09.018.
Jones, D. T. W., Kieran, M. W., Bouffet, E., Alexandrescu, S., Bandopadhayay, P., Bornhorst, M., et al. (2018). Pediatric low-grade gliomas: next biologically driven steps. Neuro-Oncology, 20(2), 160–173. https://doi.org/10.1093/neuonc/nox141.
Aldape, K., Brindle, K. M., Chesler, L., Chopra, R., Gajjar, A., Gilbert, M. R., et al. (2019). Challenges to curing primary brain tumours. Nature Reviews Clinical Oncology, 16(8), 509–520. https://doi.org/10.1038/s41571-019-0177-5.
Mackay, A., Burford, A., Carvalho, D., Izquierdo, E., Fazal-Salom, J., Taylor, K. R., et al. (2017). Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell, 32(4), 520–537.e525. https://doi.org/10.1016/j.ccell.2017.08.017.
Quail, D. F., & Joyce, J. A. (2017). The microenvironmental landscape of brain tumors. Cancer Cell, 31(3), 326–341. https://doi.org/10.1016/j.ccell.2017.02.009.
Gilbertson, R. J. (2011). Mapping cancer origins. Cell, 145(1), 25–29. https://doi.org/10.1016/j.cell.2011.03.019.
Abou-Antoun, T. J., Hale, J. S., Lathia, J. D., & Dombrowski, S. M. (2017). Brain cancer stem cells in adults and children: cell biology and therapeutic implications. Neurotherapeutics, 14(2), 372–384. https://doi.org/10.1007/s13311-017-0524-0.
Bahmad, H. F., Chamaa, F., Assi, S., Chalhoub, R. M., Abou-Antoun, T., & Abou-Kheir, W. (2019). Cancer stem cells in neuroblastoma: expanding the therapeutic frontier. Frontiers in Molecular Neuroscience, 12, 131. https://doi.org/10.3389/fnmol.2019.00131.
Bahmad, H. F., & Poppiti, R. J. (submitted). Medulloblastoma cancer stem cells: molecular signatures and therapeutic targets. Journal of Clinical Pathology.
Lathia, J. D. (2013). Cancer stem cells: moving past the controversy. CNS Oncol, 2(6), 465–467. https://doi.org/10.2217/cns.13.42.
Nowak-Sliwinska, P., Scapozza, L., & Altaba, A. R. I. (2019). Drug repurposing in oncology: Compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochimica et biophysica acta. Reviews on cancer, 1871(2), 434–454, https://doi.org/10.1016/j.bbcan.2019.04.005.
Hernandez, J. J., Pryszlak, M., Smith, L., Yanchus, C., Kurji, N., Shahani, V. M., et al. (2017). Giving drugs a second chance: overcoming regulatory and financial hurdles in repurposing approved drugs as Cancer therapeutics. Frontiers in Oncology, 7, 273. https://doi.org/10.3389/fonc.2017.00273.
Bhat-Nakshatri, P., Goswami, C. P., Badve, S., Sledge Jr., G. W., & Nakshatri, H. (2013). Identification of FDA-approved drugs targeting breast cancer stem cells along with biomarkers of sensitivity. Scientific Reports, 3, 2530. https://doi.org/10.1038/srep02530.
Tan, S. K., Jermakowicz, A., Mookhtiar, A. K., Nemeroff, C. B., Schürer, S. C., & Ayad, N. G. (2018). Drug repositioning in glioblastoma: a pathway perspective. [review]. Front Pharmacol, 9(218). https://doi.org/10.3389/fphar.2018.00218.
Pui, C.-H., Gajjar, A. J., Kane, J. R., Qaddoumi, I. A., & Pappo, A. S. (2011). Challenging issues in pediatric oncology. Nature Reviews Clinical Oncology, 8(9), 540–549. https://doi.org/10.1038/nrclinonc.2011.95.
National Cancer Institute. (2010). Surveillance, epidemiology and end results (pp. 1975–2007). SEER Cancer Statistics Review: Previous Version http://seer.cancer.gov/csr/.
Corsello, S. M., Bittker, J. A., Liu, Z., Gould, J., McCarren, P., Hirschman, J. E., et al. (2017). The drug repurposing hub: a next-generation drug library and information resource. Nature Medicine, 23(4), 405–408. https://doi.org/10.1038/nm.4306.
Minturn, J. E., & Fisher, M. J. (2013). Gliomas in children. Current Treatment Options in Neurology, 15(3), 316–327. https://doi.org/10.1007/s11940-013-0225-x.
Sievert, A. J., & Fisher, M. J. (2009). Pediatric low-grade gliomas. Journal of Child Neurology, 24(11), 1397–1408. https://doi.org/10.1177/0883073809342005.
El-Ayadi, M., Ansari, M., Sturm, D., Gielen, G. H., Warmuth-Metz, M., Kramm, C. M., et al. (2017). High-grade glioma in very young children: a rare and particular patient population. Oncotarget, 8(38), 64564–64578. https://doi.org/10.18632/oncotarget.18478.
Sturm, D., Pfister, S. M., & Jones, D. T. W. (2017). Pediatric gliomas: current concepts on diagnosis, biology, and clinical management. Journal of Clinical Oncology, 35(21), 2370–2377. https://doi.org/10.1200/JCO.2017.73.0242.
Miyashita, K., Kawakami, K., Nakada, M., Mai, W., Shakoori, A., Fujisawa, H., et al. (2009). Potential therapeutic effect of glycogen synthase kinase 3beta inhibition against human glioblastoma. Clinical Cancer Research, 15(3), 887–897. https://doi.org/10.1158/1078-0432.CCR-08-0760.
Nam, J. Y., & de Groot, J. F. (2017). Treatment of glioblastoma. Journal of Oncology Practice/ American Society of Clinical Oncology, 13(10), 629–638. https://doi.org/10.1200/JOP.2017.025536.
Xu, H. S., Qin, X. L., Zong, H. L., He, X. G., & Cao, L. (2017). Cancer stem cell markers in glioblastoma—an update. European Review for Medical and Pharmacological Sciences, 21(14), 3207–3211.
Singh, S. K., Clarke, I. D., Hide, T., & Dirks, P. B. (2004). Cancer stem cells in nervous system tumors. Oncogene, 23(43), 7267–7273. https://doi.org/10.1038/sj.onc.1207946.
Abbruzzese, C., Matteoni, S., Signore, M., Cardone, L., Nath, K., Glickson, J. D., et al. (2017). Drug repurposing for the treatment of glioblastoma multiforme. Journal of Experimental & Clinical Cancer Research, 36(1), 169. https://doi.org/10.1186/s13046-017-0642-x.
Wang, Y., Meng, Y., Zhang, S., Wu, H., Yang, D., Nie, C., et al. (2018). Phenformin and metformin inhibit growth and migration of LN229 glioma cells in vitro and in vivo. Onco Targets Ther, 11, 6039–6048. https://doi.org/10.2147/OTT.S168981.
Jiang, W., Finniss, S., Cazacu, S., Xiang, C., Brodie, Z., Mikkelsen, T., et al. (2016). Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget, 7(35), 56456–56470. https://doi.org/10.18632/oncotarget.10919.
Koyuturk, M., Ersoz, M., & Altiok, N. (2004). Simvastatin induces proliferation inhibition and apoptosis in C6 glioma cells via c-jun N-terminal kinase. Neuroscience Letters, 370(2–3), 212–217. https://doi.org/10.1016/j.neulet.2004.08.020.
Xiao, A., Brenneman, B., Floyd, D., Comeau, L., Spurio, K., Olmez, I., et al. (2019). Statins affect human glioblastoma and other cancers through TGF-beta inhibition. Oncotarget, 10(18), 1716–1728. https://doi.org/10.18632/oncotarget.26733.
Pinter, M., & Jain, R. K. (2017). Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Science Translational Medicine, 9(410). https://doi.org/10.1126/scitranslmed.aan5616.
Kitabayashi, T., Dong, Y., Furuta, T., Sabit, H., Jiapaer, S., Zhang, J., et al. (2019). Identification of GSK3β inhibitor kenpaullone as a temozolomide enhancer against glioblastoma. Scientific Reports, 9(1), 10049. https://doi.org/10.1038/s41598-019-46454-8.
Handley, M. V. (2015). GSK-3 inhibitors in glioblastoma therapy: mechanisms of action. BOSTON UNIVERSITY.
Nowicki, M. O., Dmitrieva, N., Stein, A. M., Cutter, J. L., Godlewski, J., Saeki, Y., et al. (2008). Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro-Oncology, 10(5), 690–699. https://doi.org/10.1215/15228517-2008-041.
Chirasani, S. R., Leukel, P., Gottfried, E., Hochrein, J., Stadler, K., Neumann, B., et al. (2013). Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. International Journal of Cancer, 132(4), 843–853. https://doi.org/10.1002/ijc.27712.
Leidgens, V., Seliger, C., Jachnik, B., Welz, T., Leukel, P., Vollmann-Zwerenz, A., et al. (2015). Ibuprofen and diclofenac restrict migration and proliferation of human glioma cells by distinct molecular mechanisms. PLoS One, 10(10), e0140613. https://doi.org/10.1371/journal.pone.0140613.
Tuettenberg, J., Grobholz, R., Korn, T., Wenz, F., Erber, R., & Vajkoczy, P. (2005). Continuous low-dose chemotherapy plus inhibition of cyclooxygenase-2 as an antiangiogenic therapy of glioblastoma multiforme. Journal of Cancer Research and Clinical Oncology, 131(1), 31–40. https://doi.org/10.1007/s00432-004-0620-5.
Johannessen, T. C., Hasan-Olive, M. M., Zhu, H., Denisova, O., Grudic, A., Latif, M. A., et al. (2019). Thioridazine inhibits autophagy and sensitizes glioblastoma cells to temozolomide. International Journal of Cancer, 144(7), 1735–1745. https://doi.org/10.1002/ijc.31912.
Kang, S., Lee, J. M., Jeon, B., Elkamhawy, A., Paik, S., Hong, J., et al. (2018). Repositioning of the antipsychotic trifluoperazine: synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. European Journal of Medicinal Chemistry, 151, 186–198. https://doi.org/10.1016/j.ejmech.2018.03.055.
Hayashi, K., Michiue, H., Yamada, H., Takata, K., Nakayama, H., Wei, F. Y., et al. (2016). Fluvoxamine, an anti-depressant, inhibits human glioblastoma invasion by disrupting actin polymerization. Scientific Reports, 6, 23372. https://doi.org/10.1038/srep23372.
Al Hassan, M., Fakhoury, I., El Masri, Z., Ghazale, N., Dennaoui, R., El Atat, O., et al. (2018). Metformin treatment inhibits motility and invasion of glioblastoma cancer cells. Analytical Cellular Pathology, 2018, 9. https://doi.org/10.1155/2018/5917470.
Oesterle, A., Laufs, U., & Liao, J. K. (2017). Pleiotropic effects of statins on the cardiovascular system. Circulation Research, 120(1), 229–243. https://doi.org/10.1161/CIRCRESAHA.116.308537.
Shojaei, S., Alizadeh, J., Thliveris, J., Koleini, N., Kardami, E., Hatch, G. M., et al. (2018). Statins: a new approach to combat temozolomide chemoresistance in glioblastoma. Journal of Investigative Medicine, 66(8), 1083–1087. https://doi.org/10.1136/jim-2018-000874.
Rasmussen, E. R., Pottegard, A., Bygum, A., von Buchwald, C., Homoe, P., & Hallas, J. (2019). Angiotensin II receptor blockers are safe in patients with prior angioedema related to angiotensin-converting enzyme inhibitors - a nationwide registry-based cohort study. Journal of Internal Medicine, 285(5), 553–561. https://doi.org/10.1111/joim.12867.
Barreras, A., & Gurk-Turner, C. (2003). Angiotensin II receptor blockers. Proc (Bayl Univ Med Cent), 16(1), 123–126. https://doi.org/10.1080/08998280.2003.11927893.
Ino, K., Shibata, K., Kajiyama, H., Yamamoto, E., Nagasaka, T., Nawa, A., et al. (2006). Angiotensin II type 1 receptor expression in ovarian cancer and its correlation with tumour angiogenesis and patient survival. British Journal of Cancer, 94(4), 552–560. https://doi.org/10.1038/sj.bjc.6602961.
Arrieta, O., Villarreal-Garza, C., Vizcaino, G., Pineda, B., Hernandez-Pedro, N., Guevara-Salazar, P., et al. (2015). Association between AT1 and AT2 angiotensin II receptor expression with cell proliferation and angiogenesis in operable breast cancer. Tumour Biology, 36(7), 5627–5634. https://doi.org/10.1007/s13277-015-3235-3.
Rocken, C., Rohl, F. W., Diebler, E., Lendeckel, U., Pross, M., Carl-McGrath, S., et al. (2007). The angiotensin II/angiotensin II receptor system correlates with nodal spread in intestinal type gastric cancer. Cancer Epidemiology, Biomarkers & Prevention, 16(6), 1206–1212. https://doi.org/10.1158/1055-9965.epi-05-0934.
Feng, E., Sui, C., Wang, T., & Sun, G. (2017). Temozolomide with or without radiotherapy in patients with newly diagnosed glioblastoma Multiforme: a meta-analysis. European Neurology, 77(3–4), 201–210. https://doi.org/10.1159/000455842.
Ghaffari, S. (2011). Cancer, stem cells and cancer stem cells: old ideas, new developments. F1000 Med Rep, 3, 23. https://doi.org/10.3410/M3-23.
Nakada M., M. T, Pyko I., Hayashi Y. and Hamada J. (2011). The pivotal roles of GSK3β in glioma biology In M. Garami (Ed.), Molecular Targets of CNS Tumors IntechOpen.
Vashishtha, V., Jinghan, N., & A, K. Y. (2018). Antagonistic role of GSK3 isoforms in glioma survival. Journal of Cancer, 9(10), 1846–1855. https://doi.org/10.7150/jca.21248.
Llorens-Martin, M., Jurado, J., Hernandez, F., & Avila, J. (2014). GSK-3beta, a pivotal kinase in Alzheimer disease. Frontiers in Molecular Neuroscience, 7, 46. https://doi.org/10.3389/fnmol.2014.00046.
del Ser, T., Steinwachs, K. C., Gertz, H. J., Andres, M. V., Gomez-Carrillo, B., Medina, M., et al. (2013). Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. Journal of Alzheimer's Disease, 33(1), 205–215. https://doi.org/10.3233/JAD-2012-120805.
Tolosa, E., Litvan, I., Hoglinger, G. U., Burn, D., Lees, A., Andres, M. V., et al. (2014). A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Movement Disorders, 29(4), 470–478. https://doi.org/10.1002/mds.25824.
Lovestone, S., Boada, M., Dubois, B., Hull, M., Rinne, J. O., Huppertz, H. J., et al. (2015). A phase II trial of tideglusib in Alzheimer’s disease. Journal of Alzheimer's Disease, 45(1), 75–88. https://doi.org/10.3233/jad-141959.
Mathuram, T. L., Ravikumar, V., Reece, L. M., Karthik, S., Sasikumar, C. S., & Cherian, K. M. (2016). Tideglusib induces apoptosis in human neuroblastoma IMR32 cells, provoking sub-G0/G1 accumulation and ROS generation. Environmental Toxicology and Pharmacology, 46, 194–205. https://doi.org/10.1016/j.etap.2016.07.013.
Zhou, A., Lin, K., Zhang, S., Chen, Y., Zhang, N., Xue, J., et al. (2016). Nuclear GSK3beta promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nature Cell Biology, 18(9), 954–966. https://doi.org/10.1038/ncb3396.
Chalhoub, R. M., Bahmad, H. F., Harati, H., Assi, S., Araji, T., Bou-Gharios, J., et al. (2019). Specific inhibition of GSK-3β by Tideglusib: potential therapeutic target for neuroblastoma cancer stem cells. Submitted.
Hata, A. N., & Breyer, R. M. (2004). Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacology & Therapeutics, 103(2), 147–166. https://doi.org/10.1016/j.pharmthera.2004.06.003.
Amano, H., Hayashi, I., Endo, H., Kitasato, H., Yamashina, S., Maruyama, T., et al. (2003). Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. The Journal of Experimental Medicine, 197(2), 221–232. https://doi.org/10.1084/jem.20021408.
Seliger, C., & Hau, P. (2018). Drug repurposing of metabolic agents in malignant glioma. International Journal of Molecular Sciences, 19(9). https://doi.org/10.3390/ijms19092768.
Li, J., Kim, S. G., & Blenis, J. (2014). Rapamycin: One drug, many effects. Cell Metabolism, 19(3), 373–379. https://doi.org/10.1016/j.cmet.2014.01.001.
Yang, Z., Hackshaw, A., Feng, Q., Fu, X., Zhang, Y., Mao, C., et al. (2017). Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: a meta-analysis. International Journal of Cancer, 140(12), 2805–2819. https://doi.org/10.1002/ijc.30691.
Yalon, M., Rood, B., MacDonald, T. J., McCowage, G., Kane, R., Constantini, S., et al. (2013). A feasibility and efficacy study of rapamycin and erlotinib for recurrent pediatric low-grade glioma (LGG). Pediatric Blood & Cancer, 60(1), 71–76. https://doi.org/10.1002/pbc.24142.
Mollashahi, B., Aghamaleki, F. S., & Movafagh, A. (2019). The roles of miRNAs in medulloblastoma: a systematic review. J Cancer Prev, 24(2), 79–90. https://doi.org/10.15430/jcp.2019.24.2.79.
Zulch, K. J. (1980). Principles of the new World Health Organization (WHO) classification of brain tumors. Neuroradiology, 19(2), 59–66. https://doi.org/10.1007/bf00342596.
DeAngelis, L. M. (2001). Brain tumors. The New England Journal of Medicine, 344(2), 114–123. https://doi.org/10.1056/nejm200101113440207.
Pomeroy, S. L., Tamayo, P., Gaasenbeek, M., Sturla, L. M., Angelo, M., McLaughlin, M. E., et al. (2002). Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature, 415(6870), 436–442. https://doi.org/10.1038/415436a.
Thompson, M. C., Fuller, C., Hogg, T. L., Dalton, J., Finkelstein, D., Lau, C. C., et al. (2006). Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. Journal of Clinical Oncology, 24(12), 1924–1931. https://doi.org/10.1200/jco.2005.04.4974.
Kool, M., Koster, J., Bunt, J., Hasselt, N. E., Lakeman, A., van Sluis, P., et al. (2008). Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One, 3(8), e3088. https://doi.org/10.1371/journal.pone.0003088.
Cho, Y. J., Tsherniak, A., Tamayo, P., Santagata, S., Ligon, A., Greulich, H., et al. (2011). Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. Journal of Clinical Oncology, 29(11), 1424–1430. https://doi.org/10.1200/jco.2010.28.5148.
Taylor, M. D., Northcott, P. A., Korshunov, A., Remke, M., Cho, Y. J., Clifford, S. C., et al. (2012). Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathologica, 123(4), 465–472. https://doi.org/10.1007/s00401-011-0922-z.
Thomas, A., & Noel, G. (2019). Medulloblastoma: optimizing care with a multidisciplinary approach. Journal of Multidisciplinary Healthcare, 12, 335–347. https://doi.org/10.2147/jmdh.s167808.
Bao, S., Wu, Q., McLendon, R. E., Hao, Y., Shi, Q., Hjelmeland, A. B., et al. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 444(7120), 756–760. https://doi.org/10.1038/nature05236.
Singh, S. K., Hawkins, C., Clarke, I. D., Squire, J. A., Bayani, J., Hide, T., et al. (2004). Identification of human brain tumour initiating cells. Nature, 432(7015), 396–401. https://doi.org/10.1038/nature03128.
Abouantoun, T. J., Castellino, R. C., & MacDonald, T. J. (2011). Sunitinib induces PTEN expression and inhibits PDGFR signaling and migration of medulloblastoma cells. Journal of Neuro-Oncology, 101(2), 215–226. https://doi.org/10.1007/s11060-010-0259-9.
Abouantoun, T. J., & MacDonald, T. J. (2009). Imatinib blocks migration and invasion of medulloblastoma cells by concurrently inhibiting activation of platelet-derived growth factor receptor and transactivation of epidermal growth factor receptor. Molecular Cancer Therapeutics, 8(5), 1137–1147. https://doi.org/10.1158/1535-7163.mct-08-0889.
Wolle, D., Lee, S. J., Li, Z., Litan, A., Barwe, S. P., & Langhans, S. A. (2014). Inhibition of epidermal growth factor signaling by the cardiac glycoside ouabain in medulloblastoma. Cancer Medicine, 3(5), 1146–1158. https://doi.org/10.1002/cam4.314.
Huang, L., Garrett Injac, S., Cui, K., Braun, F., Lin, Q., Du, Y., et al. (2018). Systems biology-based drug repositioning identifies digoxin as a potential therapy for groups 3 and 4 medulloblastoma. Science Translational Medicine, 10(464). https://doi.org/10.1126/scitranslmed.aat0150.
Takwi, A. A., Li, Y., Becker Buscaglia, L. E., Zhang, J., Choudhury, S., Park, A. K., et al. (2012). A statin-regulated microRNA represses human c-Myc expression and function. EMBO Molecular Medicine, 4(9), 896–909. https://doi.org/10.1002/emmm.201101045.
Bar, E. E., Chaudhry, A., Farah, M. H., & Eberhart, C. G. (2007). Hedgehog signaling promotes medulloblastoma survival via Bc/II. The American Journal of Pathology, 170(1), 347–355. https://doi.org/10.2353/ajpath.2007.060066.
Bar, E. E., & Stearns, D. (2008). New developments in medulloblastoma treatment: the potential of a cyclopamine-lovastatin combination. Expert Opinion on Investigational Drugs, 17(2), 185–195. https://doi.org/10.1517/13543784.17.2.185.
Sheikholeslami, K., Ali Sher, A., Lockman, S., Kroft, D., Ganjibakhsh, M., Nejati-Koshki, K., et al. (2019). Simvastatin induces apoptosis in medulloblastoma brain tumor cells via mevalonate cascade prenylation substrates. Cancers (Basel), 11(7). https://doi.org/10.3390/cancers11070994.
Bai, R. Y., Staedtke, V., Rudin, C. M., Bunz, F., & Riggins, G. J. (2015). Effective treatment of diverse medulloblastoma models with mebendazole and its impact on tumor angiogenesis. Neuro-Oncology, 17(4), 545–554. https://doi.org/10.1093/neuonc/nou234.
Larsen, A. R., Bai, R. Y., Chung, J. H., Borodovsky, A., Rudin, C. M., Riggins, G. J., et al. (2015). Repurposing the antihelmintic mebendazole as a hedgehog inhibitor. Molecular Cancer Therapeutics, 14(1), 3–13. https://doi.org/10.1158/1535-7163.mct-14-0755-t.
Bell, J. B., Rink, J. S., Eckerdt, F., Clymer, J., Goldman, S., Thaxton, C. S., et al. (2018). HDL nanoparticles targeting sonic hedgehog subtype medulloblastoma. Scientific Reports, 8(1), 1211. https://doi.org/10.1038/s41598-017-18100-8.
Rossi, A., Russo, G., Puca, A., La Montagna, R., Caputo, M., Mattioli, E., et al. (2009). The antiretroviral nucleoside analogue Abacavir reduces cell growth and promotes differentiation of human medulloblastoma cells. International Journal of Cancer, 125(1), 235–243. https://doi.org/10.1002/ijc.24331.
Baryawno, N., Sveinbjornsson, B., Eksborg, S., Orrego, A., Segerstrom, L., Oqvist, C. O., et al. (2008). Tumor-growth-promoting cyclooxygenase-2 prostaglandin E2 pathway provides medulloblastoma therapeutic targets. Neuro-Oncology, 10(5), 661–674. https://doi.org/10.1215/15228517-2008-035.
Yang, M. Y., Lee, H. T., Chen, C. M., Shen, C. C., & Ma, H. I. (2014). Celecoxib suppresses the phosphorylation of STAT3 protein and can enhance the radiosensitivity of medulloblastoma-derived cancer stem-like cells. International Journal of Molecular Sciences, 15(6), 11013–11029. https://doi.org/10.3390/ijms150611013.
Eslin, D., Lee, C., Sankpal, U. T., Maliakal, P., Sutphin, R. M., Abraham, L., et al. (2013). Anticancer activity of tolfenamic acid in medulloblastoma: a preclinical study. Tumour Biology, 34(5), 2781–2789. https://doi.org/10.1007/s13277-013-0836-6.
Kaplan, J. H. (2002). Biochemistry of Na,K-ATPase. Annual Review of Biochemistry, 71, 511–535. https://doi.org/10.1146/annurev.biochem.71.102201.141218.
Lingrel, J. B. (2010). The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annual Review of Physiology, 72, 395–412. https://doi.org/10.1146/annurev-physiol-021909-135725.
Mijatovic, T., Van Quaquebeke, E., Delest, B., Debeir, O., Darro, F., & Kiss, R. (2007). Cardiotonic steroids on the road to anti-cancer therapy. Biochimica et Biophysica Acta, 1776(1), 32–57. https://doi.org/10.1016/j.bbcan.2007.06.002.
Yamada, M., Ikeuchi, T., & Hatanaka, H. (1997). The neurotrophic action and signalling of epidermal growth factor. Progress in Neurobiology, 51(1), 19–37.
Wong, R. W., & Guillaud, L. (2004). The role of epidermal growth factor and its receptors in mammalian CNS. Cytokine & Growth Factor Reviews, 15(2–3), 147–156. https://doi.org/10.1016/j.cytogfr.2004.01.004.
Gilbertson, R. J., Perry, R. H., Kelly, P. J., Pearson, A. D., & Lunec, J. (1997). Prognostic significance of HER2 and HER4 coexpression in childhood medulloblastoma. Cancer Research, 57(15), 3272–3280.
Waage, I. S., Vreim, I., & Torp, S. H. (2013). C-erbB2/HER2 in human gliomas, medulloblastomas, and meningiomas: a minireview. International Journal of Surgical Pathology, 21(6), 573–582. https://doi.org/10.1177/1066896913492196.
Bal, M. M., Das Radotra, B., Srinivasan, R., & Sharma, S. C. (2006). Does c-erbB-2 expression have a role in medulloblastoma prognosis? Indian Journal of Pathology & Microbiology, 49(4), 535–539.
Ivanov, D. P., Coyle, B., Walker, D. A., & Grabowska, A. M. (2016). In vitro models of medulloblastoma: choosing the right tool for the job. Journal of Biotechnology, 236, 10–25. https://doi.org/10.1016/j.jbiotec.2016.07.028.
Zeki, A. A., Yeganeh, B., Kenyon, N. J., & Ghavami, S. (2017). Editorial: new insights into a classical pathway: key roles of the mevalonate cascade in different diseases (part II). Current Molecular Pharmacology, 10(2), 74–76. https://doi.org/10.2174/187446721002170301204357.
Matusewicz, L., Meissner, J., Toporkiewicz, M., & Sikorski, A. F. (2015). The effect of statins on cancer cells—review. Tumour Biology, 36(7), 4889–4904. https://doi.org/10.1007/s13277-015-3551-7.
Chan, K. K., Oza, A. M., & Siu, L. L. (2003). The statins as anticancer agents. Clinical Cancer Research, 9(1), 10–19.
Bjarnadottir, O., Kimbung, S., Johansson, I., Veerla, S., Jonsson, M., Bendahl, P. O., et al. (2015). Global transcriptional changes following statin treatment in breast cancer. Clinical Cancer Research, 21(15), 3402–3411. https://doi.org/10.1158/1078-0432.ccr-14-1403.
Wang, T., Seah, S., Loh, X., Chan, C. W., Hartman, M., Goh, B. C., et al. (2016). Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget, 7(3), 2532–2544. https://doi.org/10.18632/oncotarget.6304.
de Bont, J. M., Packer, R. J., Michiels, E. M., den Boer, M. L., & Pieters, R. (2008). Biological background of pediatric medulloblastoma and ependymoma: a review from a translational research perspective. Neuro-Oncology, 10(6), 1040–1060. https://doi.org/10.1215/15228517-2008-059.
Cochrane, C. R., Szczepny, A., Watkins, D. N., & Cain, J. E. (2015). Hedgehog signaling in the maintenance of cancer stem cells. Cancers (Basel), 7(3), 1554–1585. https://doi.org/10.3390/cancers7030851.
Northcott, P. A., Dubuc, A. M., Pfister, S., & Taylor, M. D. (2012). Molecular subgroups of medulloblastoma. Expert Review of Neurotherapeutics, 12(7), 871–884. https://doi.org/10.1586/ern.12.66.
Guerini, A. E., Triggiani, L., Maddalo, M., Bonu, M. L., Frassine, F., Baiguini, A., et al. (2019). Mebendazole as a candidate for drug repurposing in oncology: an extensive review of current literature. Cancers (Basel), 11(9). https://doi.org/10.3390/cancers11091284.
Kohler, P. (2001). The biochemical basis of anthelmintic action and resistance. International Journal for Parasitology, 31(4), 336–345. https://doi.org/10.1016/s0020-7519(01)00131-x.
Goel, H. L., & Mercurio, A. M. (2013). VEGF targets the tumour cell. Nature Reviews. Cancer, 13(12), 871–882. https://doi.org/10.1038/nrc3627.
Bai, R. Y., Staedtke, V., Wanjiku, T., Rudek, M. A., Joshi, A., Gallia, G. L., et al. (2015). Brain penetration and efficacy of different mebendazole polymorphs in a mouse brain tumor model. Clinical Cancer Research, 21(15), 3462–3470. https://doi.org/10.1158/1078-0432.ccr-14-2681.
Rohatgi, R., Milenkovic, L., & Scott, M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science, 317(5836), 372–376. https://doi.org/10.1126/science.1139740.
Yuen, G. J., Weller, S., & Pakes, G. E. (2008). A review of the pharmacokinetics of abacavir. Clinical Pharmacokinetics, 47(6), 351–371. https://doi.org/10.2165/00003088-200847060-00001.
Phatak, P., & Burger, A. M. (2007). Telomerase and its potential for therapeutic intervention. British Journal of Pharmacology, 152(7), 1003–1011. https://doi.org/10.1038/sj.bjp.0707374.
Tendian, S. W., & Parker, W. B. (2000). Interaction of deoxyguanosine nucleotide analogs with human telomerase. Molecular Pharmacology, 57(4), 695–699. https://doi.org/10.1124/mol.57.4.695.
Shay, J. W., & Keith, W. N. (2008). Targeting telomerase for cancer therapeutics. British Journal of Cancer, 98(4), 677–683. https://doi.org/10.1038/sj.bjc.6604209.
Chang, Q., Pang, J. C., Li, J., Hu, L., Kong, X., & Ng, H. K. (2004). Molecular analysis of PinX1 in medulloblastomas. International Journal of Cancer, 109(2), 309–314. https://doi.org/10.1002/ijc.11689.
Witzig, T. E., Timm, M., Stenson, M., Svingen, P. A., & Kaufmann, S. H. (2000). Induction of apoptosis in malignant B cells by phenylbutyrate or phenylacetate in combination with chemotherapeutic agents. Clinical Cancer Research, 6(2), 681–692.
Shay, J. W., & Wright, W. E. (2005). Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis, 26(5), 867–874. https://doi.org/10.1093/carcin/bgh296.
Epling-Burnette, P. K., Liu, J. H., Catlett-Falcone, R., Turkson, J., Oshiro, M., Kothapalli, R., et al. (2001). Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. The Journal of Clinical Investigation, 107(3), 351–362. https://doi.org/10.1172/jci9940.
Kim, K. W., Mutter, R. W., Cao, C., Albert, J. M., Shinohara, E. T., Sekhar, K. R., et al. (2006). Inhibition of signal transducer and activator of transcription 3 activity results in down-regulation of Survivin following irradiation. Molecular Cancer Therapeutics, 5(11), 2659–2665. https://doi.org/10.1158/1535-7163.mct-06-0261.
Chen, K. H., Hsu, C. C., Song, W. S., Huang, C. S., Tsai, C. C., Kuo, C. D., et al. (2010). Celecoxib enhances radiosensitivity in medulloblastoma-derived CD133-positive cells. Child's Nervous System, 26(11), 1605–1612. https://doi.org/10.1007/s00381-010-1190-2.
Abdelrahim, M., Baker, C. H., Abbruzzese, J. L., & Safe, S. (2006). Tolfenamic acid and pancreatic cancer growth, angiogenesis, and Sp protein degradation. Journal of the National Cancer Institute, 98(12), 855–868. https://doi.org/10.1093/jnci/djj232.
Shelake, S., Sankpal, U. T., Paul Bowman, W., Wise, M., Ray, A., & Basha, R. (2017). Targeting specificity protein 1 transcription factor and survivin using tolfenamic acid for inhibiting Ewing sarcoma cell growth. Investigational New Drugs, 35(2), 158–165. https://doi.org/10.1007/s10637-016-0417-9.
Yao, J. C., Wang, L., Wei, D., Gong, W., Hassan, M., Wu, T. T., et al. (2004). Association between expression of transcription factor Sp1 and increased vascular endothelial growth factor expression, advanced stage, and poor survival in patients with resected gastric cancer. Clinical Cancer Research, 10(12 Pt 1), 4109–4117. https://doi.org/10.1158/1078-0432.ccr-03-0628.
Patil, S., Sankpal, U. T., Hurtado, M., Bowman, W. P., Murray, J., Borgmann, K., et al. (2019). Combination of clotam and vincristine enhances anti-proliferative effect in medulloblastoma cells. Gene, 705, 67–76. https://doi.org/10.1016/j.gene.2019.04.037.
Colon, N. C., & Chung, D. H. (2011). Neuroblastoma. Advances in Pediatrics, 58(1), 297–311. https://doi.org/10.1016/j.yapd.2011.03.011.
Zaatiti, H., Abdallah, J., Nasr, Z., Khazen, G., Sandler, A., & Abou-Antoun, T. J. (2018). Tumorigenic proteins upregulated in the MYCN-amplified IMR-32 human neuroblastoma cells promote proliferation and migration. International Journal of Oncology, 52(3), 787–803. https://doi.org/10.3892/ijo.2018.4236.
Abou-Antoun, T. J., Nazarian, J., Ghanem, A., Vukmanovic, S., & Sandler, A. D. (2018). Molecular and functional analysis of anchorage independent, treatment-evasive neuroblastoma tumorspheres with enhanced malignant properties: a possible explanation for radio-therapy resistance. PLoS One, 13(1), e0189711. https://doi.org/10.1371/journal.pone.0189711.
Lopez-Barcons, L., Maurer, B. J., Kang, M. H., & Reynolds, C. P. (2017). P450 inhibitor ketoconazole increased the intratumor drug levels and antitumor activity of fenretinide in human neuroblastoma xenograft models. International Journal of Cancer, 141(2), 405–413. https://doi.org/10.1002/ijc.30706.
Michaelis, M., Agha, B., Rothweiler, F., Löschmann, N., Voges, Y., Mittelbronn, M., et al. (2015). Identification of flubendazole as potential anti-neuroblastoma compound in a large cell line screen. Scientific Reports, 5, 8202–8202. https://doi.org/10.1038/srep08202.
Zhong, X., Zhao, E., Tang, C., Zhang, W., Tan, J., Dong, Z., et al. (2016). Antibiotic drug tigecycline reduces neuroblastoma cells proliferation by inhibiting Akt activation in vitro and in vivo. Tumor Biology, 37(6), 7615–7623. https://doi.org/10.1007/s13277-015-4613-6.
Di Zanni, E., Bianchi, G., Ravazzolo, R., Raffaghello, L., Ceccherini, I., & Bachetti, T. (2017). Targeting of PHOX2B expression allows the identification of drugs effective in counteracting neuroblastoma cell growth. Oncotarget, 8(42), 72133–72146. https://doi.org/10.18632/oncotarget.19922.
Aveic, S., Pantile, M., Polo, P., Sidarovich, V., De Mariano, M., Quattrone, A., et al. (2018). Autophagy inhibition improves the cytotoxic effects of receptor tyrosine kinase inhibitors. Cancer Cell International, 18, 63–63. https://doi.org/10.1186/s12935-018-0557-4.
Saulnier Sholler, G. L., Kalkunte, S., Greenlaw, C., McCarten, K., & Forman, E. (2006). Antitumor activity of Nifurtimox observed in a patient with neuroblastoma. Journal of Pediatric Hematology/Oncology, 28(10).
Saulnier Sholler, G. L., Brard, L., Straub, J. A., Dorf, L., Illeyne, S., Koto, K., et al. (2009). Nifurtimox induces apoptosis of neuroblastoma cells in vitro and in vivo. Journal of Pediatric Hematology/Oncology, 31(3), 187–193. https://doi.org/10.1097/MPH.0b013e3181984d91.
Cabanillas Stanchi, K. M., Bruchelt, G., Handgretinger, R., & Holzer, U. (2015). Nifurtimox reduces N-Myc expression and aerobic glycolysis in neuroblastoma. Cancer Biology & Therapy, 16(9), 1353–1363. https://doi.org/10.1080/15384047.2015.1070987.
Kong, E., Zhu, J., Wu, W., Ren, H., Jiao, X., Wang, H., et al. (2019). Nifurtimox inhibits the progression of neuroblastoma in vivo. Journal of Cancer, 10(10), 2194–2204. https://doi.org/10.7150/jca.27851.
Bassiri, H., Benavides, A., Haber, M., Gilmour, S. K., Norris, M. D., & Hogarty, M. D. (2015). Translational development of difluoromethylornithine (DFMO) for the treatment of neuroblastoma. Translational pediatrics, 4(3), 226–238. https://doi.org/10.3978/j.issn.2224-4336.2015.04.06.
Lozier, A. M., Rich, M. E., Grawe, A. P., Peck, A. S., Zhao, P., Chang, A. T.-T., et al. (2015). Targeting ornithine decarboxylase reverses the LIN28/Let-7 axis and inhibits glycolytic metabolism in neuroblastoma. Oncotarget, 6(1), 196–206. https://doi.org/10.18632/oncotarget.2768.
Larsson, K., Kock, A., Idborg, H., Arsenian Henriksson, M., Martinsson, T., Johnsen, J. I., et al. (2015). COX/mPGES-1/PGE2 pathway depicts an inflammatory-dependent high-risk neuroblastoma subset. Proceedings of the National Academy of Sciences of the United States of America, 112(26), 8070–8075. https://doi.org/10.1073/pnas.1424355112.
Mooney, M. R., Geerts, D., Kort, E. J., & Bachmann, A. S. (2019). Anti-tumor effect of sulfasalazine in neuroblastoma. Biochemical Pharmacology, 162, 237–249. https://doi.org/10.1016/j.bcp.2019.01.007.
Komar-Stossel, C., Gross, E., Dery, E., Corchia, N., Meir, K., Fried, I., et al. (2014). TL-118 and gemcitabine drug combination display therapeutic efficacy in a MYCN amplified orthotopic neuroblastoma murine model—evaluation by MRI. PLoS One, 9(3), e90224–e90224. https://doi.org/10.1371/journal.pone.0090224.
Campos-Arroyo, D., Maldonado, V., Bahena, I., Quintanar, V., Patiño, N., Carlos Martinez-Lazcano, J., et al. (2016). Probenecid sensitizes neuroblastoma cancer stem cells to cisplatin. Cancer Investigation, 34(3), 155–166. https://doi.org/10.3109/07357907.2016.1139717.
Rodríguez-Hernández, C. J., Mateo-Lozano, S., García, M., Casalà, C., Briansó, F., Castrejón, N., et al. (2016). Cinacalcet inhibits neuroblastoma tumor growth and upregulates cancer-testis antigens. Oncotarget, 7(13), 16112–16129. https://doi.org/10.18632/oncotarget.7448.
Nishio, N., Fujita, M., Tanaka, Y., Maki, H., Zhang, R., Hirosawa, T., et al. (2012). Zoledronate sensitizes neuroblastoma-derived tumor-initiating cells to cytolysis mediated by human γδ T cells. Journal of Immunotherapy, 35(8).
Alizadeh, J., Zeki, A. A., Mirzaei, N., Tewary, S., Rezaei Moghadam, A., Glogowska, A., et al. (2017). Mevalonate Cascade inhibition by simvastatin induces the intrinsic apoptosis pathway via depletion of isoprenoids in tumor cells. Scientific Reports, 7, 44841–44841. https://doi.org/10.1038/srep44841.
Su, C., Shi, A., Cao, G., Tao, T., Chen, R., Hu, Z., et al. (2015). Fenofibrate suppressed proliferation and migration of human neuroblastoma cells via oxidative stress dependent of TXNIP upregulation. Biochemical and Biophysical Research Communications, 460(4), 983–988. https://doi.org/10.1016/j.bbrc.2015.03.138.
Costa, D., Gigoni, A., Würth, R., Cancedda, R., Florio, T., & Pagano, A. (2014). Metformin inhibition of neuroblastoma cell proliferation is differently modulated by cell differentiation induced by retinoic acid or overexpression of NDM29 non-coding RNA. Cancer Cell International, 14, 59–59. https://doi.org/10.1186/1475-2867-14-59.
Kumar, A., Al-Sammarraie, N., DiPette, D. J., & Singh, U. S. (2014). Metformin impairs Rho GTPase signaling to induce apoptosis in neuroblastoma cells and inhibits growth of tumors in the xenograft mouse model of neuroblastoma. Oncotarget, 5(22), 11709–11722. https://doi.org/10.18632/oncotarget.2606.
Vujic, I., Sanlorenzo, M., Posch, C., Esteve-Puig, R., Yen, A. J., Kwong, A., et al. (2015). Metformin and trametinib have synergistic effects on cell viability and tumor growth in NRAS mutant cancer. Oncotarget, 6(2), 969–978. https://doi.org/10.18632/oncotarget.2824.
Mouhieddine, T. H., Nokkari, A., Itani, M. M., Chamaa, F., Bahmad, H., Monzer, A., et al. (2015). Metformin and Ara-a effectively suppress brain cancer by targeting cancer stem/progenitor cells. Frontiers in Neuroscience, 9, 442–442. https://doi.org/10.3389/fnins.2015.00442.
Binlateh, T., Tanasawet, S., Rattanaporn, O., Sukketsiri, W., & Hutamekalin, P. (2019). Metformin promotes neuronal differentiation via crosstalk between Cdk5 and Sox6 in neuroblastoma cells. Evidence-based complementary and alternative medicine : eCAM, 2019, 1765182–1765182. https://doi.org/10.1155/2019/1765182.
Vella, S., Conaldi, P. G., Florio, T., & Pagano, A. (2016). PPAR gamma in neuroblastoma: the translational perspectives of hypoglycemic drugs. PPAR Research, 2016, 3038164–3038164. https://doi.org/10.1155/2016/3038164.
Wolter, J. K., Wolter, N. E., Blanch, A., Partridge, T., Cheng, L., Morgenstern, D. A., et al. (2014). Anti-tumor activity of the beta-adrenergic receptor antagonist propranolol in neuroblastoma. Oncotarget, 5(1), 161–172. https://doi.org/10.18632/oncotarget.1083.
Vella, S., Penna, I., Longo, L., Pioggia, G., Garbati, P., Florio, T., et al. (2015). Perhexiline maleate enhances antitumor efficacy of cisplatin in neuroblastoma by inducing over-expression of NDM29 ncRNA. Scientific Reports, 5, 18144–18144. https://doi.org/10.1038/srep18144.
Gu, S., Tian, Y., Chlenski, A., Salwen, H. R., Lu, Z., Raj, J. U., et al. (2012). Valproic acid shows a potent antitumor effect with alteration of DNA methylation in neuroblastoma. Anti-Cancer Drugs, 23(10), 1054–1066. https://doi.org/10.1097/CAD.0b013e32835739dd.
Shah, R. D., Jagtap, J. C., Mruthyunjaya, S., Shelke, G. V., Pujari, R., Das, G., et al. (2013). Sodium valproate potentiates staurosporine-induced apoptosis in neuroblastoma cells via Akt/survivin independently of HDAC inhibition. Journal of Cellular Biochemistry, 114(4), 854–863. https://doi.org/10.1002/jcb.24422.
Groh, T., Hrabeta, J., Ashraf Khalil, M., Doktorova, H., Eckschlager, T., & Stiborova, M. (2015). The synergistic effects of DNA-damaging drugs cisplatin and etoposide with a histone deacetylase inhibitor valproate in high-risk neuroblastoma cells. International Journal of Oncology, 47(1), 343–352.
Fang, E., Wang, J., Hong, M., Zheng, L., & Tong, Q. (2019). Valproic acid suppresses Warburg effect and tumor progression in neuroblastoma. Biochemical and Biophysical Research Communications, 508(1), 9–16. https://doi.org/10.1016/j.bbrc.2018.11.103.
Bayat Mokhtari, R., Baluch, N., Ka Hon Tsui, M., Kumar, S., S Homayouni, T., Aitken, K., et al. (2017). Acetazolamide potentiates the anti-tumor potential of HDACi, MS-275, in neuroblastoma. BMC Cancer, 17(1), 156–156, https://doi.org/10.1186/s12885-017-3126-7.
Bilir, A., Erguven, M., Yazihan, N., Aktas, E., Oktem, G., & Sabanci, A. (2010). Enhancement of vinorelbine-induced cytotoxicity and apoptosis by clomipramine and lithium chloride in human neuroblastoma cancer cell line SH-SY5Y. Journal of Neuro-Oncology, 100(3), 385–395. https://doi.org/10.1007/s11060-010-0209-6.
Zheng, X., Naiditch, J., Czurylo, M., Jie, C., Lautz, T., Clark, S., et al. (2013). Differential effect of long-term drug selection with doxorubicin and vorinostat on neuroblastoma cells with cancer stem cell characteristics. Cell Death & Disease, 4(7), e740–e740. https://doi.org/10.1038/cddis.2013.264.
Sidarovich, V., De Mariano, M., Aveic, S., Pancher, M., Adami, V., Gatto, P., et al. (2018). A high-content screening of anticancer compounds suggests the multiple tyrosine kinase inhibitor Ponatinib for repurposing in neuroblastoma therapy. Molecular Cancer Therapeutics, 17(7), 1405–1415. https://doi.org/10.1158/1535-7163.mct-17-0841.
Bahmad, H. F., Mouhieddine, T. H., Chalhoub, R. M., Assi, S., Araji, T., Chamaa, F., et al. (2018). The Akt/mTOR pathway in cancer stem/progenitor cells is a potential therapeutic target for glioblastoma and neuroblastoma. Oncotarget, 9(71), 33549-33561, https://doi.org/10.18632/oncotarget.26088.
Cerna, T., Hrabeta, J., Eckschlager, T., Frei, E., Schmeiser, H. H., Arlt, V. M., et al. (2018). The histone deacetylase inhibitor valproic acid exerts a synergistic cytotoxicity with the DNA-damaging drug Ellipticine in neuroblastoma cells. International Journal of Molecular Sciences, 19(1), 164. https://doi.org/10.3390/ijms19010164.
Chen, Y. U. N., Tsai, Y.-H., & Tseng, S.-H. (2011). Combined Valproic acid and celecoxib treatment induced synergistic cytotoxicity and apoptosis in neuroblastoma cells. Anticancer Research, 31(6), 2231–2239.
He, W., Wu, Y., Tang, X., Xia, Y., He, G., Min, Z., et al. (2016). HDAC inhibitors suppress c-Jun/Fra-1-mediated proliferation through transcriptionally downregulating MKK7 and Raf1 in neuroblastoma cells. Oncotarget, 7(6), 6727–6747. https://doi.org/10.18632/oncotarget.6797.
Dedoni, S., Marras, L., Olianas, M. C., Ingianni, A., & Onali, P. (2019). Downregulation of TrkB expression and signaling by Valproic acid and other histone deacetylase inhibitors. Journal of Pharmacology and Experimental Therapeutics, 370(3), 490. https://doi.org/10.1124/jpet.119.258129.
Khalil, M. A., Hraběta, J., Groh, T., Procházka, P., Doktorová, H., & Eckschlager, T. (2016). Valproic acid increases CD133 positive cells that show low sensitivity to cytostatics in neuroblastoma. PLoS One, 11(9), e0162916–e0162916. https://doi.org/10.1371/journal.pone.0162916.
Lange, I., Espinoza-Fuenzalida, I., Ali, M. W., Serrano, L. E., & Koomoa, D.-L. T. (2017). FTY-720 induces apoptosis in neuroblastoma via multiple signaling pathways. Oncotarget, 8(66), 109985–109999. https://doi.org/10.18632/oncotarget.22452.
Kanno, H., Nishihara, H., Oikawa, M., Ozaki, Y., Murata, J., Sawamura, Y., et al. (2012). Expression of O6-methylguanine DNA methyltransferase (MGMT) and immunohistochemical analysis of 12 pineal parenchymal tumors. Neuropathology, 32(6), 647–653. https://doi.org/10.1111/j.1440-1789.2012.01315.x.
DeBoer, R., Batjer, H., Marymont, M., Goldman, S., Walker, M., Gottardi-Littell, N., et al. (2009). Response of an adult patient with pineoblastoma to vorinostat and retinoic acid. Journal of Neuro-Oncology, 95(2), 289–292. https://doi.org/10.1007/s11060-009-9921-5.
Mohankumar, K. M., Currle, D. S., White, E., Boulos, N., Dapper, J., Eden, C., et al. (2015). An in vivo screen identifies ependymoma oncogenes and tumor-suppressor genes. Nature Genetics, 47(8), 878–887. https://doi.org/10.1038/ng.3323.
Nimmervoll, B. V., Boulos, N., Bianski, B., Dapper, J., DeCuypere, M., Shelat, A., et al. (2018). Establishing a preclinical multidisciplinary board for brain tumors. Clinical Cancer Research, 24(7), 1654. https://doi.org/10.1158/1078-0432.CCR-17-2168.
Acknowledgments
We would like to thank all members in Dr. Abou-Kheir’s Laboratory (The WAK Lab) and Dr. Abou-Antoun’s Laboratory for their help on this work.
Author information
Authors and Affiliations
Contributions
WAK and TAA conceived the concept and idea of the present review. HFB, TAA, and WAK worked on the study design strategy and selected the topics to be discussed. HFB and MKE did literature searches and screened titles and abstracts for relevance. HFB, MKE, TEZ, and JB abstracted the data from the eligible full text articles, analyzed and interpreted the data, and drafted the manuscript. TAA and WAK critically revised the manuscript with input from the entire team. All authors have read and approved the final draft.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bahmad, H.F., Elajami, M.K., El Zarif, T. et al. Drug repurposing towards targeting cancer stem cells in pediatric brain tumors. Cancer Metastasis Rev 39, 127–148 (2020). https://doi.org/10.1007/s10555-019-09840-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-019-09840-2