
Abstract
Platelets can serve as “first responders” in cancer and metastasis. This is partly due to bioactive lipid metabolism that drives both platelet and cancer biology. The two primary eicosanoid metabolites that maintain platelet rapid response homeostasis are prostacyclin made by endothelial cells that inhibits platelet function, which is counterbalanced by thromboxane produced by platelets during activation, aggregation, and platelet recruitment. Both of these arachidonic acid metabolites are inherently unstable due to their chemical structure. Tumor cells by contrast predominantly make more chemically stable prostaglandin E2, which is the primary bioactive lipid associated with inflammation and oncogenesis. Pharmacological, clinical, and epidemiologic studies demonstrate that non-steroidal anti-inflammatory drugs (NSAIDs), which target cyclooxygenases, can help prevent cancer. Much of the molecular and biological impact of these drugs is generally accepted in the field. Cyclooxygenases catalyze the rate-limiting production of substrate used by all synthase molecules, including those that produce prostaglandins along with prostacyclin and thromboxane. Additional eicosanoid metabolites include lipoxygenases, leukotrienes, and resolvins that can also influence platelets, inflammation, and carcinogenesis. Our knowledge base and technology are now progressing toward identifying newer molecular and cellular interactions that are leading to revealing additional targets. This review endeavors to summarize new developments in the field.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Platelets as “first responders”
Platelets can be thought of as first responders in many hematogenous and inflammatory diseases [1,2,3,4]. Data continue to emerge supporting the notion of platelet bioactive lipid metabolism that is likely central to driving many of these first responder characteristics. The term first responder describes platelet metabolism and biology involved in the hemostasis, wounding, immune, and cancer metastatic processes [1,2,3,4]. Typically, platelets may remain overlooked during in vivo experimental studies or pathologic observations. This can occur due to their small spherical plate-like morphology and size that is 2.6 to 2.9 μm in diameter. The difficulty in establishing a standardized standard interval range for platelet indices across the different clinical methodologies used has also had an impact on the reported platelet distribution. Despite being visible under high magnification light microscopy, electron microscopic ultrastructural analysis is typically used to identify subcellular structural changes although newer methods can potentially be effective [5,6,7,8]. Aggregates and individual activated platelets can be detected by immunohistochemical staining in some cases.
Resting platelets exist as disks that maximize biophysical surface interactions when the plate-face dimension is in contact with vessel walls [9,10,11]. Their small discoid physical characteristics promote platelet segregation near the outer fluid shear fields of flowing blood [12,13,14,15,16,17,18,19]. These shear characteristics cause platelets distribute at 2–3 times greater numbers at the vessel walls than within the core fluid stream dominated by red blood cells. Platelet distribution patterns and excess numbers near vessel walls can increase the probability of detecting vascular wall breaches or wounds. We and others have described the platelet rapid recognition and response properties associated with extracellular matrix exposure and endothelial retraction elsewhere [1,2,3,4]. These rapid response properties are closely linked to the unique stability properties, biochemistry, and metabolism of blood-born bioactive eicosanoids.
2 Keeping rapid platelet responses local: all about epoxide bond instability
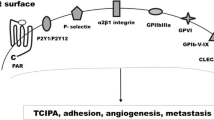
The Honn Laboratory was first to recognize the importance of prostacyclin in metastasis [20]. Prostacyclin is synthesized very rapidly by vascular endothelial cells and is one of the most effective anti-platelet aggregation metabolites of the eicosanoid pathway (Fig. 1). The biological effects of prostacyclin are counterbalanced by thromboxane which is one of the more potent pro-platelet aggregation metabolites of the eicosanoid pathway and is rapidly synthesized by platelets. Both of these molecules contain epoxide groups in their final bioactive form [21,22,23]. These exhibit significant ring strain leading to chemical and metabolic instability [21,22,23,24], which has been experimentally demonstrated in half-life experiments. The half-life of the bioactive (epoxide-containing) forms is around 30 s versus approximately 5 min for the lipid precursor molecule prostaglandin H2 (PGH2) [23, 25]. This intrinsic chemical instability helps to regulate the delicate hematologic balance between pro- and anti-platelet aggregation in the blood stream [26,27,28]. It is nature’s elegant way of maintaining rapid responses of platelets to vascular changes that remain localized near the site of vascular lesion recognition and response (Fig. 2). These platelet rapid response characteristics can be subverted during cancer progression and metastasis. At the First International Conference on Prostaglandins and Cancer, we were the first to report on the inhibition of tumor cell-induced platelet aggregation (TCIPA) by prostacyclin, thromboxane A2, and phosphodiesterase inhibitors [29] and subsequently on the efficacy of prostacyclin on TCIPA and as a deterrent for metastasis [30, 31]. These studies were followed by our report that examined prostacyclin and its synthetic analog carbacyclin and their abilities to inhibit tumor cell-platelet interactions [32]. The extent of cellular interactions during TCIPA was examined ultrastructurally. These studies revealed that tumor cell-platelet interactions began with individual platelets and initiated platelet chain formation in focal association with tumor cell surfaces. By mid-phase aggregation, large homotypic platelet aggregates grew with tumor cells externally positioned at the periphery of emboli. Tumor cell-platelet surface and cytoplasmic interactions became progressively more extensive within growing platelet aggregates. Prostacyclin and carbacyclin showed dose-dependent inhibition of tumor cell platelet interactions. Carbacyclin inhibition of TCIPA lasted longer, but was tenfold less effective than was prostacyclin [32]. Prostacyclin and its analogs significantly decrease blood pressure [33] including some of the more recently described PGI2 mimetics [34]. Some of these effects may also involve endothelium-derived hyperpolarizing factor [35] which may be partly distinct from the direct effects of nitric oxide and prostacyclin [36].

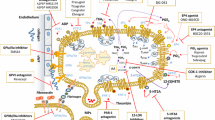
Bioactive eicosanoid and lipid metabolism. Bioactive lipids are derived from metabolic sources that arise from essential dietary fatty acids. These fatty acids include arachidonic acid (AA), docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA) that are transported into cells. Acyl-CoA is coupled to fatty acids by acyl-coenzyme A synthetases (ACSLs). Fatty acids like AA are then inserted as a storage source into membrane phospholipids by fatty acyltransferases (FACTs). After platelet agonist stimulation, the cytoplasmic form of phospholipase A2 (cPLA2) catalyzes the release of AA from membrane phospholipids. Once membrane free, AA is enzymatically converted by cyclooxygenases (COXs) to prostaglandin G2 (PGG2) followed by prostaglandin H2 (PGH2). PGH2 then serves as a substrate multiple PG synthases. In contrast, DHA and EPA are less effective substrates for COX. PG synthases occur in multiple forms, specifically: PGD2 synthases (PGDS), PGE2 synthases (PGES), PGF2α synthase (PGFS), PGI2 synthase (PGIS), or TxA2 synthase (TXS). Both PGI2 and TxA2 contain epoxide bonds (red arrows) that contain significant chemical bond strain that leads to their rapid hydrolysis and short half-life approaching 30 s. Bioactive lipids are exported outside the cell by multidrug resistance-associated protein 4 (MRP4) and other transport molecules. As PGs accumulate in the extracellular microenvironment, they bind to subtype-specific G-protein-coupled receptors. These receptors include DP1, DP2, EP1–4, FP, IP, and TP. Depending on their function, various receptors interact with subtype-specific G-stimulatory (Gs) or G-inhibitory (Gi) proteins. Downstream signaling molecules stimulate cAMP, Ca2+, inositol phosphates, or IP3/Ca2+, and Rho among others. Metabolic breakdown relies on PG transporter (PGT) followed by inactivation involving NAD + -dependent 15-hydroxyprostaglandin dehydrogenase (15-PGDH). Either free AA, DHA, or EPA can be utilized by lipoxygenase (LOX)-5,-12, and -15-1, or -15-2. LOX enzymes generate hydroxyeicosatetraenoic acids (HETEs) or leukotrienes such as LTB4. LOX-15 also uses EPA to make lipoxins or DHA when coupled with 5-LOX to make resolvins. Additional DHA metabolites are protectins and maresins

Platelet first responder bioactive lipid signaling and biology. Platelets circulate near the endothelial cell surface based on their biophysical properties under fluid shear stress. Platelet plasma membranes contain multiple surface receptors that are activated by various agonists or antagonists. These surface receptors can interact with matrix proteins, collagen, other platelets, endothelial cells, immune cells, and tumor cells. Key bioactive receptors include G-protein-coupled receptors. To form eicosanoid metabolites, cyclooxygenase 1 and 2 (COX) couple two oxygens to arachidonic acid to produce PGG2 and then PGH2. In turn, PGH2 is metabolized to various prostaglandins by synthase enzymes. Platelet prostaglandins thromboxane (TX)A2 synthesized by TXA2 synthase (TXAS) and PGE2 synthesized by PGE2 synthase are the potent pro-aggregatory agents. TXA2 and PGE2 stimulate platelet responses through various isoforms of G-protein-coupled TP or EP receptors. TP stimulates G12/13 and Rho-GEF followed by Rho-associated kinase (ROCK), LIM domain kinase (LIMK), and cofilin and that interact with actin to initiate shape change, activation, cytoplasmic process generation contraction and alpha- or dense-granule release. Other interactions include binding to myosin light-chain kinase followed by myosin. Also, EP3 receptors stimulate initiate signal transduction pathways though Gaq-calcium release-linked receptors. Another important Gas-protein-coupled IP receptor prevents platelet aggregation by binding prostacyclin (PGI2) followed by stimulating cyclic adenosine monophosphate (cAMP) synthesis via adenylate cyclase (AC). Another abundant eicosanoid is 12(S)-hydroxyeicosatetraenoic acid [12(S)-HETE] through platelet-type lipoxygenase (p12-LOX) enzymatic activity. Once released, 12-(S)HETE is thought to activate orphan receptor GPR31
3 Inhibition of cyclooxygenase (COX)-1/2
The use of cyclooxygenase-2 (COX-2) inhibitors (COXIBs) to prevent cancer development has strong experimental support. COX-2 inhibition in the tumor and surrounding microenvironment [37, 38] is thought to occur specifically through acetylation of the active site serine side chain (Ser516) and inhibition of prostaglandin biosynthesis at the level of prostaglandin H2 (PGH2) substrate generation [39]. Since PGH2 is the rate-limiting substrate required for all prostaglandin production, its inhibition impacts all PG bioactivities whether they are pro-inflammatory, immunomodulatory, prooncogenic, or otherwise bioactive. Similar chemical acetylation of cyclooxygenase-1 (COX-1) active site serines to limit PGH2 substrate availability for thromboxane production by thromboxane synthase in platelets [40]. This has a more narrow scope of activity that remains a largely unexplored mechanism of limiting carcinogenesis and metastasis [40]. There are also non-enzymatic transacetylation reactions with the N-terminal amino groups of proteins as well as side-chain amino, hydroxyl, and sulfhydryl groups [41], suggesting that other cyclooxygenase-independent effects of aspirin also exist [41, 42].
Prostaglandin synthesis from membrane-derived AA by cyclooxygenase (COX)-1 and COX-2 enzymatic activity can decrease systemic arterial pressure and increase pulmonary arterial pressure in mouse models, and this observation was further supported in KO mouse studies [43]. Mouse models have also been particularly useful in revealing the role of prostaglandin genesis in cancer progression and metastasis. In one mouse model, direct evidence was provided in mice with a truncation mutation in adenomatous polyposis coli at amino acid 716 (ApcΔ716) which predisposes them to adenoma formation in the small intestine [44]. In another mouse model strain, genetic knock-out of COX-2 or pharmaceutical blockage approaches both led to polyp reduction. In this case, both COX-1 null/ApcMin/+ and COX-2 null/ApcMin/+ mice had decreased numbers of intestinal polyps [45]. Genetically engineered and carcinogen-induced animal models consistently show the importance of the COX1/2 pathways in a variety of organ systems [46]. These findings prompted a celecoxib clinical trial in patients with familial adenomatous polyposis (FAP) that has resulted in significantly reduced adenomas [47]. These COX-2-specific inhibitor-related outcomes led the FDA to approve celecoxib for use in FAP patients as an adjunct to surgery. Subsequently, individuals who were previously diagnosed with adenomas also showed reductions in adenoma recurrence in similar COXIB trials, particularly in those patients with advanced adenomas, the recurrence was reduced following treatment with celecoxib. One proposed mechanism of action suggests that this effect results from decreased inflammation and lower levels of pro-inflammatory cytokines downstream of prostaglandin E2 (PGE2)-mediated signal transduction. Although this may help to explain why most COX-1/2 inhibitors prevent cancer in a number of organ sites, the role of platelets in these processes remains to be fully explored [48,49,50].
As metabolically produced bioactive lipids, PGs are derived from arachidonic acid (AA), which is mobilized from membrane phospholipids. Any given prostaglandin pathway subtype activation varies depending on the cell type, tissue involved, and the expression of surface receptors present on target cells. Pro-inflammatory stimuli mobilize bioactive lipid genesis by catalyzing the release of AA from membrane phospholipids via phospholipase A2 [51,52,53,54]. AA released from the phospholipid layer is converted into bioactive lipids by a series of enzymatic reactions. AA is first converted to prostaglandin H2(PGH2) with the incorporation of two oxygen molecules by the COX enzymes. This substrate generated by COX enzymes is used by a number of enzymes downstream of COXs that generate a variety of PGs, each with specific mode of action and a different biological function. COX enzymes catalyze the rate-limiting reactions within this pathway and thereby serve as targets in limiting all PG production.
By targeting the rate-limiting COX activity, both PGE2 pro-inflammatory effects, and those of other critical PGs that influence hemostasis would be attenuated. One key PG that regulates homeostasis is prostacyclin (PGI2). Prostacyclin is continuously produced in nucleated vascular endothelial cells by PGI2 synthase (PGIS; PTGIS) downstream of COX-2. Vascular endothelial cell PGIS synthesizes PGI2, which is transported to the bloodstream. PGI2 has a very short half-life (seconds in solution) and acts locally on blood vessels by inducing vasodilation and inhibiting platelet aggregation.
Because endothelial cells are nucleated and contain all of the gene expression machinery, PGIS is constantly turned over and replaced. NSAIDs and COXIBs are typically competitive inhibitors that occupy the catalytic site of COX enzymes. Pro-inflammatory and pro-oncogenic stimuli stimulate COX-2 synthesis and enzymatic activation.
COX-1 by contrast is a constitutively synthesized housekeeping gene that is elevated primarily in smooth muscle cells and platelets. Antiplatelet activity is counterbalanced by COX-1 that is linked to thromboxane (TxA2) production by TBXAS1 (TXS) in circulation. In circulating platelets, COX-1 synthesizes PGH2 which is then converted to TxA2 by TXS [55]. Aspirin irreversibly acetylates COX-1 at Ser 530 [55] eliminating PGH2 biosynthesis and inhibiting platelet function [56]. This inhibition can have an impact on both inflammatory and carcinogenic processes.
New platelets must be produced by the megakaryocytes in bone marrow to reconstitute platelet function in the circulation. Once produced and exported into circulation, TxA2 activates platelet functions, including aggregation, adhesion, additional platelet recruitment, and vessel contraction. As a whole, COXIBs and NSAIDs can effectively inhibit inflammation due to PGE2 inhibition. In the case of platelet-endothelial cell homeostasis, balance between COX-1/TxA2 production and COX-2/PGI2 is important. Using COX-2-selective COXIBs would inhibit endothelial cell PGI2 synthesis and shift the balance toward platelet TxA2 production leading to cardiovascular thrombosis in certain high-risk individuals. However, acetylation of COX-1 by low-dose aspirin eliminates both downstream TxA2 production by platelets, along with PGI2 and PGE2 while reducing the risk of cardiovascular thrombosis. As an unwanted side effect, aspirin cause severe gastropathy in susceptible individuals, which some studies show can be limited by combining with phosphatidylcholine [57]. The use of these drugs depends on their application in the appropriate clinical context.
4 Thromboxane synthase inhibition
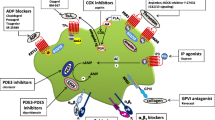
As mentioned above, TXA2 is synthesized by TXAS as a prominent pathway in platelet biology and lies downstream of platelet COX-1. Moreover, thromboxane biosynthesis in activated human platelets does not involve COX-2 [58]. More selective TXAS inhibitors and platelet TXAS have been studied since the 1970s [25, 59, 60]. In particular, Upjohn Company chemists produced a synthetic prostaglandin analog 9,11-azoprosta-5,13-dienoic acid inhibitor. This nitrogen-substituted azo analog structurally resembled PGH2 and was shown to be particularly potent at inhibiting oxygen-based endoperoxide containing PGH2 as well as ADP, epinephrine, and collagen-induced platelet aggregation [25] and as previously mentioned TCIPA [29]. In a follow-up study, platelet cyclooxygenase (TXA2 production) 12-lipoxygenase (12-HETE production) enzyme inhibitors each alone were unable to inhibit TCIPA but when combined inhibited TCIPA even at higher concentrations of tumor cells [61]. In other studies, a novel thromboxane modulator BM-567 (II/II) inhibited platelet function [62] along with TCIPA and TXA2 release [63]. Similarly, 1-alkyl (N-alkyl)-imidazole derivatives such as OKY-046 (Ozagrel) are TXAS inhibitors, especially in human platelets that have been studied since the early 1980s [64, 65] and have been shown to inhibit platelet function, TCIPA [66, 67] and hepatic metastasis [68, 69]. Likewise, R-68070 (Ridogrel) is a combination of TXAS inhibitor-TxA2 receptor antagonist [70, 71] that prevents platelet aggregation. Also, various natural compounds can inhibit platelet TXAS function and aggregation [72].
Finally, platelet surface glycoprotein alpha IIb beta 3 (GPIIb/IIIa) plays an important role in platelet aggregation and surface expression. This platelet integrin serves in the adhesion of tumor cells to platelets and may promote tumor metastasis. Inhibiting this platelet GPIIb/IIIa-mediated interaction with heparin (modified heparins), peptides, or blocking antibodies could prevent TCIPA [73, 74].
5 Prostaglandin E2-related mechanisms
PGE2, the most common PG, is present at high levels in a variety of cancers [75] and serves as a primary driver of carcinogenesis. Activated platelets also release PGE2 contributing to vascular modulation and weakened immune responses [76]. PGE2 regulates tumor cell’s pro-survival and anti-apoptotic pathways by acting on the four E Prostanoid receptors highlighted in later sections. Decreasing PGE2 levels in microenvironment of tumors by various mechanisms can reduce its pro-tumorigenic effects. Anti-inflammatory drugs such as steroids and NSAIDS have cancer-preventive functions as they inhibit PGE levels. The production of PGE2 is dependent on PGE2 synthases that are now gaining importance as more viable target enzymes downstream of COXs without triggering major side effects.
6 Synthesis, transport, and catabolism of prostaglandins
A more targeted approach to reduce PGE2 levels is to directly inhibit PGE2 synthase. This has the added benefit of reducing the toxicity associated with COX inhibition since it does not eliminate PGs that control platelet hemostasis [50, 77, 78]. The first PGE2 synthase identified was microsomal PGE2 synthase-1 (mPGES-1, PTGES-1). Subsequently, two additional isoforms of PGE2 synthases were discovered, cytosolic PGES (cPGES) and mPGES-2 (PTGES-2). The expression of mPGES-1 generally remains low in most normal tissues while cPGES and mPGES-2, by comparison, remain at steady-state expression levels. Stimuli that can be pro-inflammatory or oncogenic in nature can induce the expression of mPGES-1 [79]. The protein structure of these enzymes contains three transmembrane subunits and a glutathione active center [80]. Both COX-2 and mPGES-1 in particular are overexpressed in cancers compared to normal tissue. One of the mechanisms by which mPGES-1 expression could be upregulated is via tumor necrosis factor alpha, a cytokine involved in cancer progression [81]. Several ongoing efforts are focused on developing inhibitors that can specifically target mPGES-1 isoform without targeting the COX enzymes and avoiding the cardiovascular and gastrointestinal side effects of NSAIDs and COXIBs [82, 83]. PGs could also be modulated by eliminating their transport or increasing their catabolism. PGE2 transport and accumulation out of the cell in the tumor microenvironment correlates with oncogenic activity. ATP-binding cassette (ABC) transporter ABCC/multidrug resistance protein-4 (MRP4) is responsible for energy-dependent transport of PGs like PGE2. This transport can also be inhibited by using drugs such as indomethacin and celecoxib [84,85,86]. Similarly, increased expression of organic anion transporter proteins (OATP2A1, OATP3A1, and OATP4A1) can transport PGs and PGE2 into the extracellular microenvironment to influence oncogenesis [87] and these proteins can be inhibited by NSAIDs to improve cancer outcomes. The PGs that are transported into the cell by the PG transporter (PGT) are then inactivated by NAD+-dependent 15-hydroxyprostaglandin dehydrogenase (15-PGDH). This converts PGs into 13,14-dihydro-15-keto-PGs, a stable metabolite excreted in the urine, resulting from the dehydrogenation of PGs at carbon 15. In the case of PGE2, this forms prostaglandin E metabolite (PGEM). It has been shown that aspirin intake reduced incidence of colorectal cancers that were associated with high 15-PGDH expression, but had no influence on low 15-PGDH levels in normal colon mucosa [88] thereby serving as a biomarker of benefit from aspirin chemopreventive use.
7 Prostaglandin receptor antagonists
Eicosanoid receptors are typically G-protein-coupled receptors designated by their PG ligand molecular subclass. This is typically a letter-based identification-based nomenclature. In the case of platelets, surface receptors also facilitate TC-platelet cross talk [89, 90]. Identifying critical receptor-ligand interactions that mediate TC-platelet activation could serve as effective therapeutic targets.
Thromboxane A2 (TP) receptors play a key role in the early and fast platelet activation events. As previously mentioned, TxA2 is produced locally during platelet aggregate formation [23, 25] but is rapidly hydrolyzed in solution to inactive TxB2 in ~ 30 s by epoxide ring opening. TxA2 interactions with TP receptors are among the most rapid proaggregatory platelet agonists [31, 32, 91,92,93]. TxA2 pathway activation is a major stimulus and amplification trigger of heterotypic aggregate formation with tumor cells [31, 32, 91,92,93]. This supports the role of the TxA2 pathway in the rapid responses that mediate TC-platelet heterotypic interactions. TPs can signal through different G protein families of receptors, however, TP mediated platelet activation signals through G12/13 and Rho-GEF followed by its downstream Rho-associated kinase (ROCK), leading to the activation of LIM domain kinase (LIMK) regulating actin reorganization [94]. Additional downstream signaling from the TP-G13 interactions include those with myosin light chain kinase leading to platelet cytoskeletal changes. TP receptor antagonists include terutroban, daltroban, picotamide, sulotroban, CAY10535, Ifetroban, SQ 29,548, BM 567, or pinane.
E prostanoid (EP) receptors mediate the pro-tumorigenic effects of PGE2, which can include direct effects on precancerous and cancer cells. There are four different EP1–4 receptors that respond through G-stimulatory (Gs) or G-inhibitory (Gi) protein coupling and activating the signaling cascade involving second messengers such as cAMP, Ca2+, or inositol phosphates. EP1 receptor signals by regulating Ca2+ flux, EP2 and EP4 receptors increase cAMP levels by coupling to Gs, whereas EP3 receptor has three known isoforms generated by alternative splicing that regulate cAMP levels by being bound to Gi or Gs. EP3 isoforms have been reported to function differently from each other and can also increase IP3/Ca2+ and activate Rho [95]. Platelets also express EP2–4 receptors with the exception of EP1 [76]. EP receptors (1–4) show variable sensitivities and upon PGE2 binding activate different downstream signaling pathways, thereby PGE2-mediated responses could be activating or inhibiting and vary according to the cell type, state, and stage of maturation [96].
8 Prostaglandin influences on immune response modulation
NSAIDs and COXIBs impact on cancer prevention and reducing mortality result not from a single enzyme inhibition but from a combination of pathway interference leading to a complex interaction within the tumor microenvironment. For example, PGE2 helps to influence the local immunosuppressive tumor microenvironment along with systemic responses. When the influence of PGs on the immune system is considered along with the direct effects on tumor cells and platelets, this sheds more light on the potential depth of bioactive lipid-driven mechanisms. In the case of PGE2, it recruits myeloid-derived suppressor cells (MDSCs) into the tumors by CXCR2 signaling [97, 98]. MDSC infiltration plays a critical role in cancer progression as these cells once within the tumor can inhibit CD8+ T-cell mediated cytotoxicity [97, 99, 100]. PGE2, as a mediator of both inflammation and cancer, also suppresses dendritic cell differentiation, and this, combined with inducing MDSC function, promotes tumorigenesis. In other mouse models, the use of COX-2 inhibitors modulated MDSC functions and blocked tumor growth [101], inhibited PGE2 synthesis, and delayed tumor progression [102]. More selective EP2 receptor antagonists were shown to prevent the differentiation of MDSCs and tumor progression [103, 104]. PGE2 can also directly affect CD8 + T cells by suppressing their proliferation, cytotoxicity, and interferon (INF)-γ release. Furthermore, PGE2 causes the acquisition of DNMT3A-dependent tolerogenic functions in human MDSC as an immunological hallmark of cancer [105].
The effects of PGE2 are based on the developmental state of the cell. PGE2 is inhibitory toward immature B cells, induces apoptosis in immature thymocytes, and promotes regulatory T cell development by enriching selective immune cell populations [96, 106]. Elevated circulatory PGE2 levels can also affect T cell signaling responses [107]. Activation of EP2 and EP4 signaling pathways by PGE2 was shown to result in increase in PD-1-mediated immune tolerance in tumor microenvironment. Similarly, PGE2 produced by COX2/mPGES1 pathway was shown to increase PD-L1 in tumor-infiltrating myeloid cells mediating tumor immune evasion [108, 109].
In the case of natural killer (NK) cells, PGE2 can impact their crosstalk with cytotoxic CD8 + T cells and other immune suppressive cells. PGE2 can also directly suppress NK cell function [110, 111]. This suppression of NK cell function was shown to be mediated by EP2 and EP4 receptors, suggesting that NK cell activity can be re-established by specific receptor antagonists. PGE2 can also modulate the activities of regulatory T cells (Treg) that play a key role in immunosuppression [112, 113]. Furthermore, the infiltration of immunosuppressive Treg cells could be inhibited by using COX-2 inhibitors or EP1, EP2, and EP4 receptor antagonists.
NSAIDs can also modulate dendritic cell (DC) activity and regulate their recruitment to the sites of tissue inflammation. In addition, tumor necrosis factor α (TNFα) alteration was also shown in multiple studies [114, 115]. PGE2 modulates DC functions along with their differentiation, maturation, and ability to secrete cytokines which could be reduced with aspirin [116]. In the case of macrophages, plasticity is altered by PGE2 [117]. The conversion of M1 (inflammatory macrophages) to M2 (immunosuppressive macrophages) has been observed in various tumor types [118,119,120].
9 Lipoxygenases and Monooxygenase activity
Monooxygenases such as cytochrome P450,or lipoxygenase (LOX) can act on AA that has been released from the phospholipid bilayer [121, 122]. Unlike COX which inserts two oxygen molecules into AA, monooxygenases insert a single oxygen into a given lipid. Platelet 12-LOX is a key AA metabolizing monooxygenase pathway utilized by platelets to generate 12-hydroxyeicosatetraenoic acid (12-HETE) [123, 124]. The platelet 12-LOX pathway was also shown to promote metastasis [123, 124] and can stimulate vascular endothelial cell growth factor expression [125]. 12-LOX activity in tumor cells stimulates a wide variety of signaling mechanisms and cellular responses along with the production of autocrine motility factor that elevates invasion [126,127,128,129,130,131]. In contrast, 15-LOX-1 is an inducible enzyme that synthesizes 15-hydroxyeicosatetraenoic acid (15-HETE), mediates in reducing or resolving inflammation, and its loss has been observed in various cancers.
As another monooxygenase-based effect, aspirin-acetylation of COX converts the cyclooxygenase into a lipoxygenase that catalyzes the formation of 15-HETE and 11-HETE from AA [132]. In contrast, acetylation of COX-1 renders it inactive. A shunting mechanism also exists following the inhibition of COX which shifts AA pathway to the 5-LOX pathway and leukotriene B4 (LTB4) production. Additionally, in several cancers including colon, lung, breast, and head and neck squamous cell carcinoma, 5-LOX overexpression is also reported [133,134,135]. Thus, monooxygenase activities can play an important role in cancer progression and metastasis.
10 Resolution of inflammation via resolvins and maresins
Inflammation is optimized as a host-defense mechanism to protect against infections and harmful stimuli. By definition, acute inflammation generally resolves quickly. In some cases, inability to resolve the acute phase can lead to chronic inflammation. Precancerous lesions are sometimes influenced by strong chronic inflammatory conditions such as hepatitis, gastritis, colitis, and so on that can result in cancer formation. In some instances, timely resolution of the inflammatory process may help to lower the risk of developing cancer.
Evidence suggests the resolution of inflammation is not a passive process but is activated by turning off or blunting pro-inflammatory stimulatory signals. Inflammation is triggered by Toll-like receptor (TLR) signaling and activation of polymorphonuclear cells (PMNs) and macrophages. These activated immune cells migrate to inflammatory sites and release cytokines, including interleukin (IL)-8, IL-1, and IL-6 [136].
During the inflammatory response period, platelets as “first responders” once activated can recruit PMNs or macrophages to the sites of inflammation [56, 137]. This may occur after platelet infiltration and migration into the tissues through cytokine release or leaky blood vessels or neoangiogenesis [1]. Anti-inflammatory cytokines IL-4, IL-5, IL-10, IL-13, TNFα, and transforming growth factor β (TGF-β) take part in the resolution of inflammation [138, 139]. Bioactive lipids derived from AA take part in both initiation and resolution processes of inflammation. When platelets interact with leukocytes, this leads to lipoxin A4 and B4 formation that promotes resolution. Bioactive lipid functions are governed by tissue-specific isomerases and the receptor profiles on the cells within the microenvironment. LOX activation, which could be enhanced with aspirin, results in leukotriene and lipoxin synthesis. Collectively, these eicosanoids trigger autocrine and paracrine mechanisms to dynamically modulate inflammation, immunity, vascular permeability, smooth muscle contraction, cognition, and synaptic plasticity. Acetylated COX-2 can also act on eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) resulting in the synthesis of resolvins, maresins, and protectins [138, 140,141,142,143,144]. As the name reflects, resolvins initiate the resolution phase of inflammation and can suppress cytokine generation. Together, these resolvins, protectins, and maresins can clear inflammatory signatures, promote homeostasis, and return tissue to normalcy.
Concisely, the exact mechanisms of action for NSAIDs’ and aspirin’s cancer-preventive efficacy are likely to involve a variety of eicosanoid metabolites. These will likely include not only TxA2, PGI2, and PGE2 but other bioactive lipids along with a variety of altered proteins. As new molecular mechanisms emerge, the overall benefit of these drugs may involve multiple targets and microenvironmental influences not the least of which will include platelets and other immune factors during multiple stages of carcinogenesis.
11 Effect on glucose metabolism
The adaptive metabolic shift in cancer cells with a higher glycolytic rate is known as the Warburg effect. Upregulation in the expression of glycolytic enzymes and glucose transporters in cancer cells impart the ability to uptake glucose at a higher rate [145]. Targeting the glycolytic enzymes has been considered as a viable option for targeting cancer cells as their metabolism is much higher than that of the normal cells. Aspirin was shown to inhibit purified 6-phosphofructo-1-kinase (PFK), which regulates the glycolytic pathway [146], in a dose-dependent fashion. Aspirin also acetylates and selectively inhibits glucose-6-phosphate dehydrogenase, a member of the pentose phosphate pathway involved in ribonucleotide biosynthesis, potentially leading to reduced nucleic acid biosynthesis and cell proliferation [147]. Aspirin can also acetylate many other glucose metabolic pathway proteins but this acetylation did not inhibit their enzyme activities [147, 148]. Finally, aspirin has also been reported to affect the subcellular localization of hexokinases which control the first committed step in glycolysis.
The mitochondrial membrane potential (Δψm) dictates the conformations of voltage-dependent anion channel (VDAC) resulting in the exchange of ions and metabolites across the mitochondrial membrane. Due to the high energy demand in cancer cells, HK is translocated into mitochondria in association with VDAC. Aspirin-induced closure of VDAC correlates with the elevation of mitochondrial Ca2+, a strong apoptotic signal. Additionally, aspirin dissociated HK-II from mitochondria that cumulatively decreased cell viability. Treatment with aspirin reduced HK (II) amount in the mitochondria due to VDAC inhibition leading to the alteration of the membrane potential and cell death [149].
Platelets in diabetic (type 2) patients are hypersensitive with elevated surface CD41, Cd42b, CD62, and CD63 receptors and more active compared to those of normal individuals [150]. With highly activated platelets, these patients could benefit more from aspirin treatment. Low-dose aspirin in Japanese diabetic patients was shown to reduce cancer incidence in individuals aged < 65 years of age but not ≥ 65 years [151]. However, high-dose aspirin treatment in type 2 diabetic patients enhanced insulin sensitivity, reduced insulin clearance, and improved their blood glucose levels [152]. Data on aspirin use and dosage recommendations by diabetic patients in reducing their cancer risk and platelet inhibition is limited.
12 Aspirin and NSAIDs for cancer prevention
Aspirin was shown to reduce self-renewal potential and stem cell signaling in pancreatic ductal adenocarcinoma suggesting NSAID use can be effective in targeting cancer stem cell-like properties [153]. Aspirin’s function in acting as a cancer preventive could also be attributed to its ability to acetylate and activate the tumor suppressor protein p53, in mutant or wild-type form in colon cancer cell line [154]. In lineage tracing experiments involving β-galactosidase and fluorescent protein expression in stem cells, sulindac was shown to prevent the progression of aberrant crypts in the colon in becoming cancerous lesions.
Activation of AMPK and mTOR inhibition by aspirin adds to the benefits in the use of aspirin as a chemopreventive in CRC. Aspirin’s inhibition on mTOR signaling was mediated by targeting the phosphorylation of mTOR pathway effectors S6K1, 4E-BP1, and S6. Additionally, the mechanism of mTOR inhibition was shown to be both dependent and independent of AMPK [155]. Aspirin use can suppress cancer cell migration by inhibiting epithelial to mesenchymal transition and induce antiangiogenic activity by inhibiting vascular endothelial growth factor [156, 157]. Aspirin was also shown to induce the production of TGF-β1 by CRC cell lines which in turn mediated the reduction in the viability and entry into apoptosis phase. Caspase 8 levels were also increased in these cells [109].
13 Cancer prevention by inhibiting eicosanoid metabolism
Meta-analyses of randomized clinical trials provide clear support for NSAID-based reduction of gastrointestinal and other cancer incidence, mortality, and metastasis [158,159,160,161,162,163,164,165,166,167,168,169]. The preventive effects of aspirin involve chemical acetylation of cyclooxygenase enzymes 1 or 2 that provide the rate limiting substrate for all prostaglandins. Additional mechanisms are gaining appreciation but there may be some that still needs to be uncovered since aspirin is known to covalently acetylate a variety of different molecules [41, 170,171,172]. With recent advances in technology, there could be a possibility of gaining new insights into aspirin’s mode of action [41, 148, 173, 174]. This article endeavors to describe the mechanisms and supportive evidence that highlights the efficacy of aspirin and other NSAIDs as cancer-preventive drugs.
Aspirin still prevails as one of the most successful drugs ever made, along with serving as an anti-inflammatory agent that can also render effective cancer preventive effects. It is used to prevent cardiovascular disease (CVD) in those who are not at risk for serious bleeding [175, 176]. Several clinical trials and reports show strong evidence that aspirin reduced cancer-associated mortality and prevented CVD with daily use and the beneficial effect improved after 5 years [177]. Remarkably, fatalities from bleeding with aspirin was significantly lower than the control groups. Decreases in cancer incidence [178], particularly for gastrointestinal cancers or cancer-related [179,180,181] or all-cause mortality, were also shown with aspirin.
Aspirin preventive effects on cancer are based on short-term use that inhibited colorectal adenoma formation. For example, hereditary lynch syndrome patients, or those at high risk of developing CRC [182] or adenomas [183, 184], exhibited a reduction in their adenomas with aspirin.
Celecoxib selectively competes for the active site of COX-2 due stearic constraints compared to COX-1 and was shown to suppress adenoma formation in high-risk patients with familial adenomatous polyposis (FAP) [47] as well as post-polypectomy patients [49, 185]. Unfortunate toxicities that occurred during the trails have eliminated the use of some COX-2-selective inhibitors [50, 77, 78]. Despite these issues, in a phase III trial, NSAID sulindac given in combination with difluoromethylornithine (DFMO) was efficacious in preventing cancer. Collectively, these studies support the notion of aspirin and other NSAID use in preventing cancer.
Even after decades of research, the full explanation of NSAID bioactivity in lowering cancer incidence, progression, and metastasis, mortality, remain incomplete. Furthermore, the biologic complexities and signaling pathways that platelet and cancer-derived bioactive lipids impact they are likely to play different roles depending on when they occur during the cancer continuum.
14 Summary
The rapid response of platelets is an essential part of the first responder behavior. This is primarily due to the eicosanoid metabolites TxA2 and PGI2 that maintain hemostatic balance but have very short half-lives in circulation. This is around 30 s due to epoxide ring opening. Both TxA2 and PGI2 are mobilized through membrane lipid sources of arachidonate. Arachidonate is converted by COX enzymes to PGH2, which serves as the substrate for all PG production. PGH2 is converted via COX-1 through TXAS to TxA2 and exported to then bind platelet TP receptors. TP is a very strong G-protein-coupled receptor that triggers platelet activation and aggregation. This is counterbalanced by the conversion of AA to PGI2 through COX-2 and then PGIS that binds to IP G-protein-coupled receptor agonist that prevents platelet activation and aggregation. The TxA2-PGI2 balance is shifted toward TxA2 following interactions with tumor cells that activate platelets by a variety of surface receptor interactions and circulating factors. There are a variety of inhibitors that target all of the molecules within the TxA2 and PGI2 pathways. These serve as more selective targets than COX molecules and may avoid the cardiovascular and GI toxicity associated with targeting COX and eliminating all PG production. Additional eicosanoid metabolites include lipoxygenases, leukotrienes, and resolvins that can also influence platelets, inflammation, and carcinogenesis which can serve as additive targets along with newer molecular and cellular interactions. Nonetheless, continued study of NSAIDs on cancer strengthens the notion that COX-dependent mechanisms can drive the overall process. The fundamental concept that COX-1/2 play key roles in carcinogenesis provides encouragement that other downstream targets may be equally important, but how can we prioritize these? Although COX-2 inhibitor-targeted therapies seem to work, are the other molecular targets of equal or more value? Will these additional targets provide equal or additive overall benefit, be even more selective and eliminate the toxicity associated with NSAIDs? The risk: benefit associated with targeting platelets in effort to impact cancer progression and metastasis presents challenges considering that platelet count and bleeding risk must be considered. The timing and duration for any platelet-based interventions are also critical to consider. Aspirin seems to be an inexpensive option for now but more targeted approaches may ultimately provide less toxic solutions. In the final analysis, a better understanding of platelets and bioactive lipid biology as a key first responder element in cancer and metastasis will add critical knowledge to our prevention and treatment of cancer.
Abbreviations
- 15-PGDH:
-
15-Hydroxyprostaglandin Dehydrogenase
- AP:
-
Activating Protein
- Apc:
-
Adenomatous polyposis coli
- AA:
-
Arachidonic Acid
- ABC:
-
ATP-Binding Cassette
- CCL2:
-
C-C motif Ligand 2
- CVD:
-
Cardiovascular disease
- CRC:
-
Colorectal cancer
- CI:
-
Confidence interval (95%)
- COXIBs:
-
Cyclooxygenase inhibitors
- COX-1:
-
Cyclooxygenase-1
- COX-2:
-
Cyclooxygenase-2
- DC:
-
Dendritic Cells
- DNMT1:
-
DNA methyl transferase
- DHA:
-
Docosahexaenoic acid
- EPA:
-
Eicosapentaenoic acid
- ERK:
-
Extracellular signal regulated kinase
- FAP:
-
Familial adenomatous polyposis
- Gs:
-
G-stimulatory
- HK:
-
Hexokinase
- HR:
-
Hazard Ratio
- HETE:
-
Hydroxyeicosatetraenoic acid
- IκB:
-
Inhibitor of nuclear factor of kappa light chain in B-cells
- LTB4:
-
Leukotriene B4
- LOX:
-
Lipoxygenase
- MSI:
-
Microsatellite instability
- MDSCs:
-
Myeloid-derived suppressor cells
- NK:
-
Natural killer
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- OR:
-
Odds ratio
- OATP:
-
Organic ion transporter protein expression
- PGT:
-
PG transporter
- cPGES/ mPGES:
-
PGE2 synthases, cytosolic/microsomal
- PGIS:
-
PGI2 synthase
- PI3K:
-
Phosphatidylinositol-3-kinase
- PDK1:
-
Phosphoinositide-dependent kinase-1
- PLA2:
-
Phospholipase A2
- PMNs:
-
Polymorphonuclear cells
- PGI2:
-
Prostacyclin
- EP:
-
Prostaglandin (prostanoid) E receptors
- PGEM:
-
Prostaglandin E metabolite
- PGE2:
-
Prostaglandin E2
- PGH2:
-
Prostaglandin H2
- RCT:
-
Randomized controlled trial
- RR:
-
Relative risk
- TCR:
-
T-cell receptor
- TXS/TBXAS1:
-
Thromboxane synthase
- TxA2:
-
Thromboxane
- TLR:
-
Toll-like receptors
- TNF:
-
Tumor necrosis factor
- VDAC:
-
Voltage-dependent anion channels
References
Menter, D. G., Kopetz, S., Hawk, E., Sood, A. K., Loree, J. M., Gresele, P., & Honn, K. V. (2017). Platelet “first responders” in wound response, cancer, and metastasis. Cancer Metastasis Reviews, 36(2), 199–213.
Lam, M., Roszik, J., Kanikarla-Marie, P., Davis, J. S., Morris, J., Kopetz, S., & Menter, D. G. (2017). The potential role of platelets in the consensus molecular subtypes of colorectal cancer. Cancer Metastasis Reviews, 36(2), 273–288.
Kanikarla-Marie, P., Lam, M., Sorokin, A. V., Overman, M. J., Kopetz, S., & Menter, D. G. (2018). Platelet metabolism and other targeted drugs; potential impact on immunotherapy. Frontiers in Oncology, 8, 107.
Haemmerle, M., Stone, R. L., Menter, D. G., Afshar-Kharghan, V., & Sood, A. K. (2018). The platelet lifeline to cancer: challenges and opportunities. Cancer Cell, 33, 965–983.
Leunissen, T. C., Wisman, P. P., van Holten, T. C., de Groot, P. G., Korporaal, S. J., Koekman, A. C., et al. (2016). The effect of P2Y12 inhibition on platelet activation assessed with aggregation- and flow cytometry-based assays. Platelets, 1–9.
Liu, X., Li, Y., Zhu, H., Zhao, Z., Zhou, Y., Zaske, A. M., Liu, L., Li, M., Lu, H., Liu, W., Dong, J. F., Zhang, J., & Zhang, Y. (2015). Use of non-contact hopping probe ion conductance microscopy to investigate dynamic morphology of live platelets. Platelets, 26(5), 480–485.
Lof, A., Muller, J. P., Benoit, M., & Brehm, M. A. (2017). Biophysical approaches promote advances in the understanding of von Willebrand factor processing and function. Advances in Biological Regulation, 63, 81–91.
Heijnen, H., & Korporaal, S. (2017). Platelet morphology and ultrastructure. In P. Gresele, N. Kleiman, J. Lopez, & C. Page (Eds.), Platelets in thrombotic and non-thrombotic disorders (Vol. 2, pp. 21–37). Basel: Springer International Publishing.
O'Brien, S., Kent, N. J., Lucitt, M., Ricco, A. J., McAtamney, C., Kenny, D., & Meade, G. (2012). Effective hydrodynamic shaping of sample streams in a microfluidic parallel-plate flow-assay device: matching whole blood dynamic viscosity. IEEE Transactions on Biomedical Engineering, 59(2), 374–382.
Jen, C. J., & Tai, Y. W. (1992). Morphological study of platelet adhesion dynamics under whole blood flow conditions. Platelets, 3(3), 145–153.
Folie, B. J., & McIntire, L. V. (1989). Mathematical analysis of mural thrombogenesis. Concentration profiles of platelet-activating agents and effects of viscous shear flow. Biophysical Journal, 56(6), 1121–1141.
Fedosov, D. A., Noguchi, H., & Gompper, G. (2014). Multiscale modeling of blood flow: from single cells to blood rheology. Biomechanics and Modeling in Mechanobiology, 13(2), 239–258.
Kumar, A., & Graham, M. D. (2012). Mechanism of margination in confined flows of blood and other multicomponent suspensions. Physical Review Letters, 109(10), 108102.
Tokarev, A. A., Butylin, A. A., & Ataullakhanov, F. I. (2011). Platelet adhesion from shear blood flow is controlled by near-wall rebounding collisions with erythrocytes. Biophysical Journal, 100(4), 799–808.
Tokarev, A. A., Butylin, A. A., Ermakova, E. A., Shnol, E. E., Panasenko, G. P., & Ataullakhanov, F. I. (2011). Finite platelet size could be responsible for platelet margination effect. Biophysical Journal, 101(8), 1835–1843.
Lee, S. Y., Ferrari, M., & Decuzzi, P. (2009). Design of bio-mimetic particles with enhanced vascular interaction. Journal of Biomechanics, 42(12), 1885–1890.
Stukelj, R., Schara, K., Bedina-Zavec, A., Sustar, V., Pajnic, M., Paden, L., et al. (2017). Effect of shear stress in the flow through the sampling needle on concentration of nanovesicles isolated from blood. European Journal of Pharmaceutical Sciences, 98, 17–29.
De Gruttola, S., Boomsma, K., & Poulikakos, D. (2005). Computational simulation of a non-newtonian model of the blood separation process. Artificial Organs, 29(12), 949–959.
Nesbitt, W. S., Westein, E., Tovar-Lopez, F. J., Tolouei, E., Mitchell, A., Fu, J., Carberry, J., Fouras, A., & Jackson, S. P. (2009). A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nature Medicine, 15(6), 665–673.
Honn, K. V., Cicone, B., & Skoff, A. (1981). Prostacyclin: a potent antimetastatic agent. Science, 212(4500), 1270–1272.
Lopp, M. I., Miuraus, A. I., Parve, O. V., Vialimiae, T. K., & Lopp, A. (1988). Synthesis and antiaggregation activity of prostacyclin analogs. 1. Bicyclo [3.2.0] heptane analogs. Bioorganicheskaia Khimiia, 14(2), 222–231.
Dix, T. A., Buck, J. R., & Marnett, L. J. (1986). Hydroperoxide-dependent epoxidation of 3,4-dihydroxy-3,4-dihydrobenzo[a]anthracene by ram seminal vesicle microsomes and by hematin. Biochemical and Biophysical Research Communications, 140(1), 181–187.
Hamberg, M., Svensson, J., & Samuelsson, B. (1975). Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proceedings of the National Academy of Sciences of the United States of America, 72(8), 2994–2998.
Meninno, S., & Lattanzi, A. (2016). Organocatalytic asymmetric reactions of epoxides: recent progress. Chemistry, 22(11), 3632–3642.
Gorman, R. R., Bundy, G. L., Peterson, D. C., Sun, F. F., Miller, O. V., & Fitzpatrick, F. A. (1977). Inhibition of human platelet thromboxane synthetase by 9,11-azoprosta-5,13-dienoic acid. Proceedings of the National Academy of Sciences of the United States of America, 74(9), 4007–4011.
Schror, K. (1985). Prostaglandins and endothelial cells. Z Kardiol, 74(Suppl 7), 93–97.
El Tahir, K. E., Williams, K. I., & Betteridge, D. J. (1983). Differences in the stability of prostacyclin in human, rabbit and rat plasma. Prostaglandins, Leukotrienes, and Medicine, 10(2), 109–114.
Lawson, J. A., Patrono, C., Ciabattoni, G., & Fitzgerald, G. A. (1986). Long-lived enzymatic metabolites of thromboxane B2 in the human circulation. Analytical Biochemistry, 155(1), 198–205.
Menter, D. G., Neagos, G., Dunn, J., Palazzo, R., Tchen, T. T., Taylor, J. D., et al. (1982). Tumor cell induced platelet aggregation: inhibition by prostacyclin, thromboxane A2 and phosphodiesterase inhibitors. In T. J. Powles, R. S. Bockman, K. V. Honn, & P. Ramwell (Eds.), Prostaglandins and cancer: first international conference (pp. 809–813). New York: Alan R. Liss.
Honn, K. V., Menter, D. G., Onoda, J. M., Taylor, J. D., & Sloane, B. F. (1983). Role of prostacyclin as a natural deterrent to hematogenous tumor metastasis. Symposium on Fundamental Cancer Research, 36, 361–388.
Menter, D. G., Onoda, J. M., Taylor, J. D., & Honn, K. V. (1984). Effects of prostacyclin on tumor cell-induced platelet aggregation. Cancer Research, 44(2), 450–456.
Menter, D. G., Harkins, C., Onoda, J., Riorden, W., Sloane, B. F., Taylor, J. D., & Honn, K. V. (1987). Inhibition of tumor cell induced platelet aggregation by prostacyclin and carbacyclin: an ultrastructural study. Invasion & Metastasis, 7(2), 109–128.
Murata, T., Murai, T., Kanai, T., Ogaki, Y., Sanai, K., Kanda, H., Sato, S., Kajikawa, N., Umetsu, T., & Matsuura, H. (1989). General pharmacology of beraprost sodium. 2nd communication: effect on the autonomic, cardiovascular and gastrointestinal systems, and other effects. Arzneimittelforschung, 39(8), 867–876.
Kashiwagi, H., Yuhki, K., Kojima, F., Kumei, S., Takahata, O., Sakai, Y., Narumiya, S., & Ushikubi, F. (2015). The novel prostaglandin I2 mimetic ONO-1301 escapes desensitization in an antiplatelet effect due to its inhibitory action on thromboxane A2 synthesis in mice. The Journal of Pharmacology and Experimental Therapeutics, 353(2), 269–278.
Takaki, A., Morikawa, K., Murayama, Y., Yamagishi, H., Hosoya, M., Ohashi, J., & Shimokawa, H. (2008). Roles of endothelial oxidases in endothelium-derived hyperpolarizing factor responses in mice. Journal of Cardiovascular Pharmacology, 52(6), 510–517.
Brandes, R. P., Schmitz-Winnenthal, F. H., Feletou, M., Godecke, A., Huang, P. L., Vanhoutte, P. M., Fleming, I., & Busse, R. (2000). An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proceedings of the National Academy of Sciences of the United States of America, 97(17), 9747–9752.
Eberhart, C. E., Coffey, R. J., Radhika, A., Giardiello, F. M., Ferrenbach, S., & DuBois, R. N. (1994). Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology, 107(4), 1183–1188.
Wang, D., & Dubois, R. N. (2010). Eicosanoids and cancer. Nature Reviews. Cancer, 10(3), 181–193.
Blobaum, A. L., & Marnett, L. J. (2007). Structural and functional basis of cyclooxygenase inhibition. Journal of Medicinal Chemistry, 50(7), 1425–1441.
Dovizio, M., Bruno, A., Tacconelli, S., & Patrignani, P. (2013). Mode of action of aspirin as a chemopreventive agent. Recent Results in Cancer Research, 191, 39–65.
Ornelas, A., Zacharias-Millward, N., Menter, D. G., Davis, J. S., Lichtenberger, L., Hawke, D., Hawk, E., Vilar, E., Bhattacharya, P., & Millward, S. (2017). Beyond COX-1: the effects of aspirin on platelet biology and potential mechanisms of chemoprevention. Cancer Metastasis Reviews, 36(2), 289–303.
Alfonso, L. F., Srivenugopal, K. S., & Bhat, G. J. (2009). Does aspirin acetylate multiple cellular proteins? (review). Molecular Medicine Reports, 2(4), 533–537.
Baber, S. R., Deng, W., Rodriguez, J., Master, R. G., Bivalacqua, T. J., Hyman, A. L., & Kadowitz, P. J. (2005). Vasoactive prostanoids are generated from arachidonic acid by COX-1 and COX-2 in the mouse. American Journal of Physiology. Heart and Circulatory Physiology, 289(4), H1476–H1487.
Oshima, M., Dinchuk, J. E., Kargman, S. L., Oshima, H., Hancock, B., Kwong, E., Trzaskos, J. M., Evans, J. F., & Taketo, M. M. (1996). Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell, 87(5), 803–809.
Williams, C. S., Luongo, C., Radhika, A., Zhang, T., Lamps, L. W., Nanney, L. B., Beauchamp, R. D., & DuBois, R. (1996). Elevated cyclooxygenase-2 levels in min mouse adenomas. Gastroenterology, 111(4), 1134–1140.
Fischer, S. M., Hawk, E. T., & Lubet, R. A. (2011). Coxibs and other nonsteroidal anti-inflammatory drugs in animal models of cancer chemoprevention. Cancer Prevention Research (Philadelphia, Pa.), 4(11), 1728–1735.
Phillips, R. K., Wallace, M. H., Lynch, P. M., Hawk, E., Gordon, G. B., Saunders, B. P., Wakabayashi, N., Shen, Y., Zimmerman, S., Godio, L., Rodrigues-Bigas, M., Su, L. K., Sherman, J., Kelloff, G., Levin, B., Steinbach, G., & FAP Study Group. (2002). A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut, 50(6), 857–860.
Bertagnolli, M. M. (2003). Colorectal cancer prevention studies: the importance of defining disease risk. Annals of Surgical Oncology, 10(8), 829–830.
Bertagnolli, M. M., Eagle, C. J., Zauber, A. G., Redston, M., Solomon, S. D., Kim, K., Tang, J., Rosenstein, R. B., Wittes, J., Corle, D., Hess, T. M., Woloj, G. M., Boisserie, F., Anderson, W. F., Viner, J. L., Bagheri, D., Burn, J., Chung, D. C., Dewar, T., Foley, T. R., Hoffman, N., Macrae, F., Pruitt, R. E., Saltzman, J. R., Salzberg, B., Sylwestrowicz, T., Gordon, G. B., Hawk, E. T., & APC Study Investigators. (2006). Celecoxib for the prevention of sporadic colorectal adenomas. The New England Journal of Medicine, 355(9), 873–884.
Bresalier, R. S., Sandler, R. S., Quan, H., Bolognese, J. A., Oxenius, B., Horgan, K., Lines, C., Riddell, R., Morton, D., Lanas, A., Konstam, M. A., Baron, J. A., & Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators. (2005). Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. The New England Journal of Medicine, 352(11), 1092–1102.
Milde, M., Werthmann, R. C., von Hayn, K., & Bunemann, M. (2014). Dynamics of adenylate cyclase regulation via heterotrimeric G-proteins. Biochemical Society Transactions, 42(2), 239–243.
Brummett, A. M., Navratil, A. R., Bryan, J. D., & Woolard, M. D. (2014). Janus kinase 3 activity is necessary for phosphorylation of cytosolic phospholipase A2 and prostaglandin E2 synthesis by macrophages infected with Francisella tularensis live vaccine strain. Infection and Immunity, 82(3), 970–982.
Elimam, H., Papillon, J., Takano, T., & Cybulsky, A. V. (2013). Complement-mediated activation of calcium-independent phospholipase A2gamma: role of protein kinases and phosphorylation. The Journal of Biological Chemistry, 288(6), 3871–3885.
Flower, R. J., & Blackwell, G. J. (1976). The importance of phospholipase-A2 in prostaglandin biosynthesis. Biochemical Pharmacology, 25(3), 285–291.
Loll, P. J., Picot, D., & Garavito, R. M. (1995). The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nature Structural Biology, 2(8), 637–643.
Menter, D. G., Tucker, S. C., Kopetz, S., Sood, A. K., Crissman, J. D., & Honn, K. V. (2014). Platelets and cancer: a casual or causal relationship: revisited. Cancer Metastasis Reviews, 33(1), 231–269.
Lichtenberger, L. M., Zhou, Y., Jayaraman, V., Doyen, J. R., O'Neil, R. G., Dial, E. J., Volk, D. E., Gorenstein, D. G., Boggara, M. B., & Krishnamoorti, R. (2012). Insight into NSAID-induced membrane alterations, pathogenesis and therapeutics: characterization of interaction of NSAIDs with phosphatidylcholine. Biochimica et Biophysica Acta, 1821(7), 994–1002.
Patrignani, P., Sciulli, M. G., Manarini, S., Santini, G., Cerletti, C., & Evangelista, V. (1999). COX-2 is not involved in thromboxane biosynthesis by activated human platelets. Journal of Physiology and Pharmacology, 50(4), 661–667.
Fitzpatrick, F. A., & Gorman, R. R. (1977). Platelet rich plasma transforms exogenous prostaglandin endoperoxide H2 into thromboxane A2. Prostaglandins, 14(5), 881–889.
Alusy, U. D., & Hammarstrom, S. (1977). Inhibitors of thromboxane synthase in human platelets. FEBS Letters, 82(1), 107–110.
Steinert, B. W., Tang, D. G., Grossi, I. M., Umbarger, L. A., & Honn, K. V. (1993). Studies on the role of platelet eicosanoid metabolism and integrin alpha IIb beta 3 in tumor-cell-induced platelet aggregation. International Journal of Cancer, 54(1), 92–101.
Dogne, J. M., de Leval, X., Kolh, P., Sanna, V., Rolin, S., Michaux, C., et al. (2003). Pharmacological evaluation of the novel thromboxane modulator BM-567 (I/II). Effects of BM-567 on platelet function. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 68(1), 49–54.
de Leval, X., Benoit, V., Delarge, J., Julemont, F., Masereel, B., Pirotte, B., et al. (2003). Pharmacological evaluation of the novel thromboxane modulator BM-567 (II/II). Effects of BM-567 on osteogenic sarcoma-cell-induced platelet aggregation. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 68(1), 55–59.
Iizuka, K., Akahane, K., Momose, D., Nakazawa, M., Tanouchi, T., Kawamura, M., Ohyama, I., Kajiwara, I., Iguchi, Y., Okada, T., Taniguchi, K., Miyamoto, T., & Hayashi, M. (1981). Highly selective inhibitors of thromboxane synthetase. 1. Imidazole derivatives. Journal of Medicinal Chemistry, 24(10), 1139–1148.
Uyama, O., Nagatsuka, K., Nakabayashi, S., Isaka, Y., Yoneda, S., Kimura, K., & Abe, H. (1985). The effect of a thromboxane synthetase inhibitor, OKY-046, on urinary excretion of immunoreactive thromboxane B2 and 6-keto-prostaglandin F1 alpha in patients with ischemic cerebrovascular disease. Stroke, 16(2), 241–244.
Kosugi, T., Nakamura, M., Saitoh, S., Noguchi, S., Shimoji, T., & Minei, S. (1991). Changes in parameters of the coagulation-fibrinolysis system and platelet function after OKY-046 administration to patients with ruptured aneurysm of the cerebral artery. International Journal of Tissue Reactions, 13(1), 51–57.
Mehta, P., Lawson, D., Ward, M. B., Lee-Ambrose, L., & Kimura, A. (1986). Effects of thromboxane A2 inhibition on osteogenic sarcoma cell-induced platelet aggregation. Cancer Research, 46(10), 5061–5063.
Hoshino, M., Koyama, Y., Igarashi, W., Ono, T., Sato, N., Hatakeyama, Y., & Abe, R. (1993). An experimental study evaluating the efficacy of platelet aggregation inhibitory (OKY-046) for hepatic metastasis of VX2 carcinoma. Gan to Kagaku Ryoho, 20(11), 1578–1581.
Koyama, Y., Hoshino, M., Yamaki, T., Igarashi, W., Ono, T., Sato, N., Hatakeyama, Y., & Abe, R. (1994). An experimental study of the efficacy of platelet aggregating inhibitor on hepatic metastasis. Gan to Kagaku Ryoho, 21(13), 2124–2127.
Aoki, N., Johnson 3rd, G., Siegfried, M. R., & Lefer, A. M. (1989). Protective effects of a combination thromboxane synthesis inhibitor-receptor antagonist, R-68070, during murine traumatic shock. Eicosanoids, 2(3), 169–174.
Janssens, W. J., Cools, F. J., Hoskens, L. A., & Van Nueten, J. M. (1990). Effect of ridogrel on vascular contractions caused by vasoactive substances released during platelet activation. Thrombosis and Haemostasis, 64(1), 91–96.
Applova, L., Karlickova, J., Riha, M., Filipsky, T., Macakova, K., Spilkova, J., et al. (2017). The isoflavonoid tectorigenin has better antiplatelet potential than acetylsalicylic acid. Phytomedicine, 35, 11–17.
Lonsdorf, A. S., Kramer, B. F., Fahrleitner, M., Schonberger, T., Gnerlich, S., Ring, S., et al. (2012). Engagement of alphaIIbbeta3 (GPIIb/IIIa) with alphanubeta3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. The Journal of Biological Chemistry, 287(3), 2168–2178.
Zhang, C., Liu, Y., Gao, Y., Shen, J., Zheng, S., Wei, M., & Zeng, X. L. (2009). Modified heparins inhibit integrin alpha(IIb)beta(3) mediated adhesion of melanoma cells to platelets in vitro and in vivo. International Journal of Cancer, 125(9), 2058–2065.
Cha, Y. I., Kim, S. H., Sepich, D., Buchanan, F. G., Solnica-Krezel, L., & DuBois, R. N. (2006). Cyclooxygenase-1-derived PGE2 promotes cell motility via the G-protein-coupled EP4 receptor during vertebrate gastrulation. Genes & Development, 20(1), 77–86.
Friedman, E. A., Ogletree, M. L., Haddad, E. V., & Boutaud, O. (2015). Understanding the role of prostaglandin E2 in regulating human platelet activity in health and disease. Thrombosis Research, 136(3), 493–503.
Kerr, D. J., Dunn, J. A., Langman, M. J., Smith, J. L., Midgley, R. S., Stanley, A., Stokes, J. C., Julier, P., Iveson, C., Duvvuri, R., McConkey, C., & VICTOR Trial Group. (2007). Rofecoxib and cardiovascular adverse events in adjuvant treatment of colorectal cancer. The New England Journal of Medicine, 357(4), 360–369.
Solomon, S. D., Pfeffer, M. A., McMurray, J. J., Fowler, R., Finn, P., Levin, B., et al. (2006). Effect of celecoxib on cardiovascular events and blood pressure in two trials for the prevention of colorectal adenomas. Circulation, 114(10), 1028–1035.
Jakobsson, P. J., Thoren, S., Morgenstern, R., & Samuelsson, B. (1999). Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proceedings of the National Academy of Sciences of the United States of America, 96(13), 7220–7225.
Prage, E. B., Pawelzik, S. C., Busenlehner, L. S., Kim, K., Morgenstern, R., Jakobsson, P. J., & Armstrong, R. N. (2011). Location of inhibitor binding sites in the human inducible prostaglandin E synthase, MPGES1. Biochemistry, 50(35), 7684–7693.
Fournier, T., Fadok, V., & Henson, P. M. (1997). Tumor necrosis factor-alpha inversely regulates prostaglandin D2 and prostaglandin E2 production in murine macrophages. Synergistic action of cyclic AMP on cyclooxygenase-2 expression and prostaglandin E2 synthesis. The Journal of Biological Chemistry, 272(49), 31065–31072.
Chell, S., Kadi, A., Caroline Williams, A., & Paraskeva, C. (2006). Mediators of PGE2 synthesis and signalling downstream of COX-2 represent potential targets for the prevention/treatment of colorectal cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, 1766(1), 104–119.
Psarra, A., Nikolaou, A., Kokotou, M. G., Limnios, D., & Kokotos, G. (2017). Microsomal prostaglandin E2 synthase-1 inhibitors: a patent review. Expert Opinion on Therapeutic Patents, 27(9), 1047–1059.
El-Sheikh, A. A., van den Heuvel, J. J., Koenderink, J. B., & Russel, F. G. (2007). Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. The Journal of Pharmacology and Experimental Therapeutics, 320(1), 229–235.
Russel, F. G., Koenderink, J. B., & Masereeuw, R. (2008). Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends in Pharmacological Sciences, 29(4), 200–207.
Reid, G., Wielinga, P., Zelcer, N., van der Heijden, I., Kuil, A., de Haas, M., Wijnholds, J., & Borst, P. (2003). The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proceedings of the National Academy of Sciences of the United States of America, 100(16), 9244–9249.
Kleberg, K., Jensen, G. M., Christensen, D. P., Lundh, M., Grunnet, L. G., Knuhtsen, S., Poulsen, S. S., Hansen, M. B., & Bindslev, N. (2012). Transporter function and cyclic AMP turnover in normal colonic mucosa from patients with and without colorectal neoplasia. BMC Gastroenterology, 12, 78.
Fink, S. P., Yamauchi, M., Nishihara, R., Jung, S., Kuchiba, A., Wu, K., et al. (2014). Aspirin and the risk of colorectal cancer in relation to the expression of 15-hydroxyprostaglandin dehydrogenase (HPGD). Sci Transl Med, 6(233), 233re232.
Grossi, I. M., Fitzgerald, L. A., Kendall, A., Taylor, J. D., Sloane, B. F., & Honn, K. V. (1987). Inhibition of human tumor cell induced platelet aggregation by antibodies to platelet glycoproteins Ib and IIb/IIIa. Proceedings of the Society for Experimental Biology and Medicine, 186(3), 378–383.
Bendas, G., & Borsig, L. (2012). Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. International Journal of Cell Biology, 2012, 676731.
Menter, D. G., Onoda, J. M., Moilanen, D., Sloane, B. F., Taylor, J. D., & Honn, K. V. (1987). Inhibition by prostacyclin of the tumor cell-induced platelet release reaction and platelet aggregation. Journal of the National Cancer Institute, 78(5), 961–969.
Menter, D. G., Schilsky, R. L., & DuBois, R. N. (2010). Cyclooxygenase-2 and cancer treatment: understanding the risk should be worth the reward. Clinical Cancer Research, 16(5), 1384–1390.
Umar, A., Steele, V. E., Menter, D. G., & Hawk, E. T. (2016). Mechanisms of nonsteroidal anti-inflammatory drugs in cancer prevention. Seminars in Oncology, 43(1), 65–77.
Huang, J. S., Ramamurthy, S. K., Lin, X., & Le Breton, G. C. (2004). Cell signalling through thromboxane A2 receptors. Cellular Signalling, 16(5), 521–533.
Sugimoto, Y., & Narumiya, S. (2007). Prostaglandin E receptors. The Journal of Biological Chemistry, 282(16), 11613–11617.
Kalinski, P. (2012). Regulation of immune responses by prostaglandin E2. Journal of Immunology, 188(1), 21–28.
Katoh, H., Wang, D., Daikoku, T., Sun, H., Dey, S. K., & Dubois, R. N. (2013). CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell, 24(5), 631–644.
Wang, D., Wang, H., Brown, J., Daikoku, T., Ning, W., Shi, Q., Richmond, A., Strieter, R., Dey, S. K., & DuBois, R. N. (2006). CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. The Journal of Experimental Medicine, 203(4), 941–951.
Obermajer, N., & Kalinski, P. (2012). Key role of the positive feedback between PGE(2) and COX2 in the biology of myeloid-derived suppressor cells. Oncoimmunology, 1(5), 762–764.
Obermajer, N., Wong, J. L., Edwards, R. P., Odunsi, K., Moysich, K., & Kalinski, P. (2012). PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunological Investigations, 41(6–7), 635–657.
Veltman, J. D., Lambers, M. E., van Nimwegen, M., Hendriks, R. W., Hoogsteden, H. C., Aerts, J. G., et al. (2010). COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer, 10, 464.
Fujita, M., Kohanbash, G., Fellows-Mayle, W., Hamilton, R. L., Komohara, Y., Decker, S. A., Ohlfest, J. R., & Okada, H. (2011). COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Research, 71(7), 2664–2674.
Sinha, P., Clements, V. K., Fulton, A. M., & Ostrand-Rosenberg, S. (2007). Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Research, 67(9), 4507–4513.
Ostrand-Rosenberg, S., & Sinha, P. (2009). Myeloid-derived suppressor cells: linking inflammation and cancer. Journal of Immunology, 182(8), 4499–4506.
Rodriguez-Ubreva, J., Catala-Moll, F., Obermajer, N., Alvarez-Errico, D., Ramirez, R. N., Company, C., et al. (2017). Prostaglandin E2 leads to the acquisition of DNMT3A-dependent tolerogenic functions in human myeloid-derived suppressor cells. Cell Reports, 21(1), 154–167.
Harris, S. G., Padilla, J., Koumas, L., Ray, D., & Phipps, R. P. (2002). Prostaglandins as modulators of immunity. Trends in Immunology, 23(3), 144–150.
Moltu, K., Henjum, K., Oberprieler, N. G., Bjornbeth, B. A., & Tasken, K. (2017). Proximal signaling responses in peripheral T cells from colorectal cancer patients are affected by high concentrations of circulating prostaglandin E2. Human Immunology, 78(2), 129–137.
Prima, V., Kaliberova, L. N., Kaliberov, S., Curiel, D. T., & Kusmartsev, S. (2017). COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proceedings of the National Academy of Sciences of the United States of America, 114(5), 1117–1122.
Wang, J., Zhang, L., Kang, D., Yang, D., & Tang, Y. (2018). Activation of PGE2/EP2 and PGE2/EP4 signaling pathways positively regulate the level of PD-1 in infiltrating CD8(+) T cells in patients with lung cancer. Oncology Letters, 15(1), 552–558.
Goto, T., Herberman, R. B., Maluish, A., & Strong, D. M. (1983). Cyclic AMP as a mediator of prostaglandin E-induced suppression of human natural killer cell activity. Journal of Immunology, 130(3), 1350–1355.
Targan, S. R. (1981). The dual interaction of prostaglandin E2 (PGE2) and interferon (IFN) on NK lytic activation: enhanced capacity of effector-target lytic interactions (recycling) and blockage of pre-NK cell recruitment. Journal of Immunology, 127(4), 1424–1428.
Baratelli, F., Lin, Y., Zhu, L., Yang, S. C., Heuze-Vourc'h, N., Zeng, G., Reckamp, K., Dohadwala, M., Sharma, S., & Dubinett, S. M. (2005). Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. Journal of Immunology, 175(3), 1483–1490.
Sharma, S., Yang, S. C., Zhu, L., Reckamp, K., Gardner, B., Baratelli, F., Huang, M., Batra, R. K., & Dubinett, S. M. (2005). Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Research, 65(12), 5211–5220.
Harizi, H., & Gualde, N. (2002). Dendritic cells produce eicosanoids, which modulate generation and functions of antigen-presenting cells. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 66(5–6), 459–466.
Slattery, M. L., Lundgreen, A., Bondurant, K. L., & Wolff, R. K. (2011). Tumor necrosis factor-related genes and colon and rectal cancer. International Journal of Molecular Epidemiology and Genetics, 2(4), 328–338.
Hackstein, H., Morelli, A. E., Larregina, A. T., Ganster, R. W., Papworth, G. D., Logar, A. J., Watkins, S. C., Falo, L. D., & Thomson, A. W. (2001). Aspirin inhibits in vitro maturation and in vivo immunostimulatory function of murine myeloid dendritic cells. Journal of Immunology, 166(12), 7053–7062.
Vasandan, A. B., Jahnavi, S., Shashank, C., Prasad, P., Kumar, A., & Prasanna, S. J. (2016). Human mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Scientific Reports, 6, 38308.
Liu, L., Ge, D., Ma, L., Mei, J., Liu, S., Zhang, Q., Ren, F., Liao, H., Pu, Q., Wang, T., & You, Z. (2012). Interleukin-17 and prostaglandin E2 are involved in formation of an M2 macrophage-dominant microenvironment in lung cancer. Journal of Thoracic Oncology, 7(7), 1091–1100.
Heusinkveld, M., de Vos van Steenwijk, P. J., Goedemans, R., Ramwadhdoebe, T. H., Gorter, A., Welters, M. J., et al. (2011). M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. Journal of Immunology, 187(3), 1157–1165.
Torroella-Kouri, M., Silvera, R., Rodriguez, D., Caso, R., Shatry, A., Opiela, S., Ilkovitch, D., Schwendener, R. A., Iragavarapu-Charyulu, V., Cardentey, Y., Strbo, N., & Lopez, D. M. (2009). Identification of a subpopulation of macrophages in mammary tumor-bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Research, 69(11), 4800–4809.
Yoshida, N., & Aoki, N. (1978). Release of arachidonic acid from human platelets. A key role for the potentiation of platelet aggregability in normal subjects as well as in those with nephrotic syndrome. Blood, 52(5), 969–977.
Derksen, A., & Cohen, P. (1975). Patterns of fatty acid release from endogenous substrates by human platelet homogenates and membranes. The Journal of Biological Chemistry, 250(24), 9342–9347.
Honn, K. V., Grossi, I. M., Diglio, C. A., Wojtukiewicz, M., & Taylor, J. D. (1989). Enhanced tumor cell adhesion to the subendothelial matrix resulting from 12(S)-HETE-induced endothelial cell retraction. The FASEB Journal, 3(11), 2285–2293.
Honn, K. V., Tang, D. G., Gao, X., Butovich, I. A., Liu, B., Timar, J., & Hagmann, W. (1994). 12-lipoxygenases and 12(S)-HETE: role in cancer metastasis. Cancer Metastasis Reviews, 13(3–4), 365–396.
McCabe, N. P., Selman, S. H., & Jankun, J. (2006). Vascular endothelial growth factor production in human prostate cancer cells is stimulated by overexpression of platelet 12-lipoxygenase. Prostate, 66(7), 779–787.
Honn, K. V., Timar, J., Rozhin, J., Bazaz, R., Sameni, M., Ziegler, G., et al. (1994). A lipoxygenase metabolite, 12-(S)-HETE, stimulates protein kinase C-mediated release of cathepsin B from malignant cells. Experimental Cell Research, 214(1), 120–130.
Raso, E., Tovari, J., Toth, K., Paku, S., Trikha, M., Honn, K. V., et al. (2001). Ectopic alphaIIbbeta3 integrin signaling involves 12-lipoxygenase- and PKC-mediated serine phosphorylation events in melanoma cells. Thrombosis and Haemostasis, 85(6), 1037–1042.
Silletti, S., Timar, J., Honn, K. V., & Raz, A. (1994). Autocrine motility factor induces differential 12-lipoxygenase expression and activity in high- and low-metastatic K1735 melanoma cell variants. Cancer Research, 54(22), 5752–5756.
Tang, D. G., Tarrien, M., Dobrzynski, P., & Honn, K. V. (1995). Melanoma cell spreading on fibronectin induced by 12(S)-HETE involves both protein kinase C- and protein tyrosine kinase-dependent focal adhesion formation and tyrosine phosphorylation of focal adhesion kinase (pp125FAK). Journal of Cellular Physiology, 165(2), 291–306.
Timar, J., Raso, E., Fazakas, Z. S., Silletti, S., Raz, A., & Honn, K. V. (1996). Multiple use of a signal transduction pathway in tumor cell invasion. Anticancer Research, 16(6A), 3299–3306.
Timar, J., Trikha, M., Szekeres, K., Bazaz, R., Tovari, J., Silletti, S., Raz, A., & Honn, K. V. (1996). Autocrine motility factor signals integrin-mediated metastatic melanoma cell adhesion and invasion. Cancer Research, 56(8), 1902–1908.
Rowlinson, S. W., Crews, B. C., Goodwin, D. C., Schneider, C., Gierse, J. K., & Marnett, L. J. (2000). Spatial requirements for 15-(R)-hydroxy-5Z,8Z,11Z, 13E-eicosatetraenoic acid synthesis within the cyclooxygenase active site of murine COX-2. Why acetylated COX-1 does not synthesize 15-(R)-hete. The Journal of Biological Chemistry, 275(9), 6586–6591.
Park, S. W., Heo, D. S., & Sung, M. W. (2012). The shunting of arachidonic acid metabolism to 5-lipoxygenase and cytochrome p450 epoxygenase antagonizes the anti-cancer effect of cyclooxygenase-2 inhibition in head and neck cancer cells. Cellular Oncology (Dordrecht), 35(1), 1–8.
Ganesh, R., Marks, D. J., Sales, K., Winslet, M. C., & Seifalian, A. M. (2012). Cyclooxygenase/lipoxygenase shunting lowers the anti-cancer effect of cyclooxygenase-2 inhibition in colorectal cancer cells. World Journal of Surgical Oncology, 10, 200.
Melstrom, L. G., Bentrem, D. J., Salabat, M. R., Kennedy, T. J., Ding, X. Z., Strouch, M., Rao, S. M., Witt, R. C., Ternent, C. A., Talamonti, M. S., Bell, R. H., & Adrian, T. A. (2008). Overexpression of 5-lipoxygenase in colon polyps and cancer and the effect of 5-LOX inhibitors in vitro and in a murine model. Clinical Cancer Research, 14(20), 6525–6530.
Tam, V. C. (2013). Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Seminars in Immunology, 25(3), 240–248.
Evangelista, V., Celardo, A., Dell'Elba, G., Manarini, S., Mironov, A., de Gaetano, G., & Cerletti, C. (1999). Platelet contribution to leukotriene production in inflammation: in vivo evidence in the rabbit. Thrombosis and Haemostasis, 81(3), 442–448.
Spite, M., Claria, J., & Serhan, C. N. (2014). Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metabolism, 19(1), 21–36.
Umar, A. (2012). Is 15-LOX-1 a tumor suppressor? Journal of the National Cancer Institute, 104(9), 645–647.
Serhan, C. N., Yang, R., Martinod, K., Kasuga, K., Pillai, P. S., Porter, T. F., Oh, S. F., & Spite, M. (2009). Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. The Journal of Experimental Medicine, 206(1), 15–23.
Spite, M., & Serhan, C. N. (2010). Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circulation Research, 107(10), 1170–1184.
Krishnamoorthy, N., Burkett, P. R., Dalli, J., Abdulnour, R. E., Colas, R., Ramon, S., et al. (2015). Cutting edge: maresin-1 engages regulatory T cells to limit type 2 innate lymphoid cell activation and promote resolution of lung inflammation. Journal of Immunology, 194(3), 863–867.
Serhan, C. N. (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature, 510(7503), 92–101.
Serhan, C. N., Gotlinger, K., Hong, S., Lu, Y., Siegelman, J., Baer, T., Yang, R., Colgan, S. P., & Petasis, N. A. (2006). Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. Journal of Immunology, 176(3), 1848–1859.
Cerella, C., Michiels, C., Dashwood, R. H., Surh, Y. J., & Diederich, M. (2013). Metabolism and cancer: old and new players. International Journal of Cell and Biology, 2013, 293201.
Spitz, G. A., Furtado, C. M., Sola-Penna, M., & Zancan, P. (2009). Acetylsalicylic acid and salicylic acid decrease tumor cell viability and glucose metabolism modulating 6-phosphofructo-1-kinase structure and activity. Biochemical Pharmacology, 77(1), 46–53.
Marimuthu, S., Chivukula, R. S., Alfonso, L. F., Moridani, M., Hagen, F. K., & Bhat, G. J. (2011). Aspirin acetylates multiple cellular proteins in HCT-116 colon cancer cells: Identification of novel targets. International Journal of Oncology, 39(5), 1273–1283.
Wang, J., Zhang, C. J., Zhang, J., He, Y., Lee, Y. M., Chen, S., Lim, T. K., Ng, S., Shen, H. M., & Lin, Q. (2015). Mapping sites of aspirin-induced acetylations in live cells by quantitative acid-cleavable activity-based protein profiling (QA-ABPP). Scientific Reports, 5, 7896.
Tewari, D., Majumdar, D., Vallabhaneni, S., & Bera, A. K. (2017). Aspirin induces cell death by directly modulating mitochondrial voltage-dependent anion channel (VDAC). Scientific Reports, 7, 45184.
Soma, P., Swanepoel, A. C., du Plooy, J. N., Mqoco, T., & Pretorius, E. (2016). Flow cytometric analysis of platelets type 2 diabetes mellitus reveals ‘angry’ platelets. Cardiovascular Diabetology, 15, 52.
Okada, S., Morimoto, T., Ogawa, H., Sakuma, M., Matsumoto, C., Soejima, H., Nakayama, M., Doi, N., Jinnouchi, H., Waki, M., Masuda, I., Saito, Y., & JPAD Trial Investigators. (2018). Effect of aspirin on cancer chemoprevention in Japanese patients with type 2 diabetes: 10-year observational follow-up of a randomized controlled trial. Diabetes Care, 41(8), 1757–1764.
Hundal, R. S., Petersen, K. F., Mayerson, A. B., Randhawa, P. S., Inzucchi, S., Shoelson, S. E., & Shulman, G. I. (2002). Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. The Journal of Clinical Investigation, 109(10), 1321–1326.
Zhang, Y., Liu, L., Fan, P., Bauer, N., Gladkich, J., Ryschich, E., Bazhin, A. V., Giese, N. A., Strobel, O., Hackert, T., Hinz, U., Gross, W., Fortunato, F., & Herr, I. (2015). Aspirin counteracts cancer stem cell features, desmoplasia and gemcitabine resistance in pancreatic cancer. Oncotarget, 6(12), 9999–10015.
Ai, G., Dachineni, R., Kumar, D. R., Marimuthu, S., Alfonso, L. F., & Bhat, G. J. (2016). Aspirin acetylates wild type and mutant p53 in colon cancer cells: identification of aspirin acetylated sites on recombinant p53. Tumour Biology, 37(5), 6007–6016.
Din, F. V., Valanciute, A., Houde, V. P., Zibrova, D., Green, K. A., Sakamoto, K., et al. (2012). Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology, 142(7), 1504–1515 e1503.
Khan, K. M., Kothari, P., Du, B., Dannenberg, A. J., & Falcone, D. J. (2012). Matrix metalloproteinase-dependent microsomal prostaglandin E synthase-1 expression in macrophages: role of TNF-alpha and the EP4 prostanoid receptor. Journal of Immunology, 188(4), 1970–1980.
Pathi, S., Jutooru, I., Chadalapaka, G., Nair, V., Lee, S. O., & Safe, S. (2012). Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (Sp) transcription factors. PLoS One, 7(10), e48208.
Bousser, M. G., Amarenco, P., Chamorro, A., Fisher, M., Ford, I., Fox, K. M., Hennerici, M. G., Mattle, H. P., Rothwell, P. M., de Cordoüe, A., & Fratacci, M. D. (2011). Terutroban versus aspirin in patients with cerebral ischaemic events (PERFORM): a randomised, double-blind, parallel-group trial. Lancet, 377(9782), 2013–2022.
Chan, A. T., Arber, N., Burn, J., Chia, W. K., Elwood, P., Hull, M. A., Logan, R. F., Rothwell, P. M., Schror, K., & Baron, J. A. (2012). Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prevention Research (Philadelphia, Pa.), 5(2), 164–178.
Chan, A. T., & Cook, N. R. (2012). Are we ready to recommend aspirin for cancer prevention? Lancet, 379(9826), 1569–1571.
Cuzick, J., Thorat, M. A., Bosetti, C., Brown, P. H., Burn, J., Cook, N. R., Ford, L. G., Jacobs, E. J., Jankowski, J. A., la Vecchia, C., Law, M., Meyskens, F., Rothwell, P. M., Senn, H. J., & Umar, A. (2015). Estimates of benefits and harms of prophylactic use of aspirin in the general population. Annals of Oncology, 26(1), 47–57.
Goldstein, L. B., & Rothwell, P. M. (2012). Advances in prevention and health services delivery 2010-2011. Stroke, 43(2), 298–299.
Langley, R. E., & Rothwell, P. M. (2013). Potential biomarker for aspirin use in colorectal cancer therapy. Nature Reviews. Clinical Oncology, 10(1), 8–10.
Langley, R. E., & Rothwell, P. M. (2014). Aspirin in gastrointestinal oncology: new data on an old friend. Current Opinion in Oncology, 26(4), 441–447.
Rothwell, P. M. (2013). Alternate-day, low-dose aspirin and cancer risk. Annals of Internal Medicine, 159(2), 148–150.
Walker, J., Hutchison, P., Ge, J., Zhao, D., Wang, Y., Rothwell, P. M., et al. (2018). Aspirin: 120 years of innovation. A report from the 2017 Scientific Conference of the International Aspirin Foundation, 14 September 2017, Charite, Berlin. Ecancermedicalscience, 12, p. 813.
Chan, A. T., & Moayyedi, P. (2018). Signature celebration of gastroenterology, colorectal cancer. Gastroenterology, 154(4), 767–770.
Drew, D. A., Cao, Y., & Chan, A. T. (2016). Aspirin and colorectal cancer: the promise of precision chemoprevention. Nature Reviews. Cancer, 16(3), 173–186.
Cao, Y., Nishihara, R., Wu, K., Wang, M., Ogino, S., Willett, W. C., Spiegelman, D., Fuchs, C. S., Giovannucci, E. L., & Chan, A. T. (2016). Population-wide impact of long-term use of aspirin and the risk for cancer. JAMA Oncology, 2(6), 762–769.
Pinckard, R. N., Hawkins, D., & Farr, R. S. (1968). In vitro acetylation of plasma proteins, enzymes and DNA by aspirin. Nature, 219(5149), 68–69.
Macdonald, J. M., LeBlanc, D. A., Haas, A. L., & London, R. E. (1999). An NMR analysis of the reaction of ubiquitin with [acetyl-1-13C]aspirin. Biochemical Pharmacology, 57(11), 1233–1244.
Liyasova, M. S., Schopfer, L. M., & Lockridge, O. (2010). Reaction of human albumin with aspirin in vitro: mass spectrometric identification of acetylated lysines 199, 402, 519, and 545. Biochemical Pharmacology, 79(5), 784–791.
Lei, J., Zhou, Y., Xie, D., & Zhang, Y. (2015). Mechanistic insights into a classic wonder drug-aspirin. Journal of the American Chemical Society, 137(1), 70–73.
Kovacs, E. G., Katona, E., Bereczky, Z., Homorodi, N., Balogh, L., Toth, E., et al. (2013). New direct and indirect methods for the detection of cyclooxygenase 1 acetylation by aspirin; the lack of aspirin resistance among healthy individuals. Thrombosis Research, 131(4), 320–324.
Antithrombotic Trialists, C., Baigent, C., Blackwell, L., Collins, R., Emberson, J., Godwin, J., et al. (2009). Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet, 373(9678), 1849–1860.
Force, U. S. P. S. T. (2009). Aspirin for the prevention of cardiovascular disease: U.S. preventive services task force recommendation statement. Annals of Internal Medicine, 150(6), 396–404.
Rothwell, P. M., Price, J. F., Fowkes, F. G., Zanchetti, A., Roncaglioni, M. C., Tognoni, G., et al. (2012). Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet, 379(9826), 1602–1612.
Bosetti, C., Rosato, V., Gallus, S., Cuzick, J., & La Vecchia, C. (2012). Aspirin and cancer risk: a quantitative review to 2011. Annals of Oncology, 23(6), 1403–1415.
Bardia, A., Ebbert, J. O., Vierkant, R. A., Limburg, P. J., Anderson, K., Wang, A. H., Olson, J. E., Vachon, C. M., & Cerhan, J. R. (2007). Association of aspirin and nonaspirin nonsteroidal anti-inflammatory drugs with cancer incidence and mortality. Journal of the National Cancer Institute, 99(11), 881–889.
Chan, A. T., Manson, J. E., Feskanich, D., Stampfer, M. J., Colditz, G. A., & Fuchs, C. S. (2007). Long-term aspirin use and mortality in women. Archives of Internal Medicine, 167(6), 562–572.
Jacobs, E. J., Newton, C. C., Gapstur, S. M., & Thun, M. J. (2012). Daily aspirin use and cancer mortality in a large US cohort. Journal of the National Cancer Institute, 104(16), 1208–1217.
Sandler, R. S., Halabi, S., Baron, J. A., Budinger, S., Paskett, E., Keresztes, R., Petrelli, N., Pipas, J. M., Karp, D. D., Loprinzi, C. L., Steinbach, G., & Schilsky, R. (2003). A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. The New England Journal of Medicine, 348(10), 883–890.
Baron, J. A., Cole, B. F., Sandler, R. S., Haile, R. W., Ahnen, D., Bresalier, R., McKeown-Eyssen, G., Summers, R. W., Rothstein, R., Burke, C. A., Snover, D. C., Church, T. R., Allen, J. I., Beach, M., Beck, G. J., Bond, J. H., Byers, T., Greenberg, E. R., Mandel, J. S., Marcon, N., Mott, L. A., Pearson, L., Saibil, F., & van Stolk, R. U. (2003). A randomized trial of aspirin to prevent colorectal adenomas. The New England Journal of Medicine, 348(10), 891–899.
Benamouzig, R., Deyra, J., Martin, A., Girard, B., Jullian, E., Piednoir, B., Couturier, D., Coste, T., Little, J., & Chaussade, S. (2003). Daily soluble aspirin and prevention of colorectal adenoma recurrence: one-year results of the APACC trial. Gastroenterology, 125(2), 328–336.
Arber, N., Eagle, C. J., Spicak, J., Racz, I., Dite, P., Hajer, J., et al. (2006). Celecoxib for the prevention of colorectal adenomatous polyps. The New England Journal of Medicine, 355(9), 885–895.
Acknowledgements
Grant and Other Support: Boone Pickens Distinguished Chair for Early Prevention of Cancer, Duncan Family Institute, Colorectal Cancer Moon Shot, 1R01CA187238-01, 5R01CA172670-03 and 1R01CA184843-01A1, CA177909, and the American Cancer Society Research Professor Award, Cancer Center Support Grant (P30 CA016672).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kanikarla-Marie, P., Kopetz, S., Hawk, E.T. et al. Bioactive lipid metabolism in platelet “first responder” and cancer biology. Cancer Metastasis Rev 37, 439–454 (2018). https://doi.org/10.1007/s10555-018-9755-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-018-9755-8