
Abstract
Apoptotic caspases have long been studied for their roles in programmed cell death and tumor suppression. With recent discoveries, however, it is becoming apparent these cell death executioners are involved in additional biological pathways beyond killing cells. In some cases, apoptotic cells secrete growth signals to stimulate proliferation of neighboring cells. This pathway functions to regenerate tissues in multiple organisms, but it also poses problems in tumor resistance to chemo- and radiotherapy. Additionally, it was found that activation of caspases does not irreversibly lead to cell death, contrary to the established paradigm. Sub-lethal activation of caspases is evident in cell differentiation and epigenetic reprogramming. Furthermore, evidence indicates spontaneous, unprovoked activation of caspases in many cancer cells, which plays pivotal roles in maintaining their tumorigenicity and metastasis. These unexpected findings challenge current cancer therapy approaches aimed at activation of the apoptotic pathway. At the same time, the newly discovered functions of caspases suggest new treatment approaches for cancer and other pathological conditions in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Apoptotic caspases, the established paradigm
Apoptosis is a process for multicellular organisms to get rid of damaged or unwanted cells. Apoptotic cells die in an orderly fashion with features of pyknosis, karyorrhexis, and karyolysis; no inflammation follows, which is a main characteristic that sets it apart from necrosis. There is no disturbance of neighboring cells, as apoptotic bodies are taken up by macrophages.
Caspases, a family of proteases that are pivotal executioners of apoptosis, comprise of the initiators and the effectors [1]. The initiator caspases include caspase-2, -8, -9, and -10, and the effector caspases include caspase-3, -6, and -7. The apoptotic process in mammalian cells is mediated through either the intrinsic or the extrinsic pathway. Briefly, the intrinsic pathway is usually activated as a result of pro-apoptotic signals that activate several proteins inside the mitochondria to be released into the cytoplasm. A key mitochondrial protein released being cytochrome c, which binds to APAF1 that causes APAF1 to further bind to ATP/dATP, forming the apoptosome [2]. The apoptosis then recruits and activates caspase 9. Of note, additional proteins that were released from the mitochondria in addition to cytochrome c include apoptosis-inducing factor (AIF), endonuclease G, high temperature requirement protein A2, second mitochondria-derived activator of caspase, and direct inhibitor of apoptosis binding protein with low pH [3]. Activation of caspase-9 leads to caspase-3 activation, which is the executioner caspase that kills the host cell.
On the other hand, the extrinsic pathway is initiated by extracellular signals. Extracellular molecules, such as the well-known Fas Ligand binds to the Fas receptor on cellular surface [4]. Upon binding, cytosolic factors caspase-8 and FADD associate with the activated homotrimeric receptor to form the death-inducing signaling complex or DISC [5]. Subsequently, DISC cleaves caspase 8 and allows it to activate the effector caspase 3, which leads to dismantling of critical cellular infrastructure and eventual cell death.
In addition to eliminating damaged cells, programmed cell death is essential in morphogenesis and organ sculpting during development. Studies have used model organisms, including Caenorhabditis elegans [6], Drosophila [7], zebrafish [8], and mouse [9], to demonstrate that this role of apoptosis is highly conserved in evolution [10]. During gametogenesis, apoptosis operates to eliminate defective cells and limit the number of oocytes that are permitted to grow [11,12,13,14,15]. During organogenesis, apoptosis is pivotal in orchestrating the formation of the organs [16]. This programmed cell death also plays essential roles in development in humans, in regard to sex differentiation [17, 18], hearing acquisition [19], limb development [20], and immune system maturation [21].
Given the crucial roles of apoptosis, its disruption may be deeply problematic. Indeed, evading apoptosis is deemed a hallmark of cancer [22], and caspases are thought to be tumor-suppressive because of their roles for facilitating apoptosis. This makes sense intuitively, as cancer forms most often when p53 or BAX (both pro-apoptotic genes) are mutated [23]. In support of this view, the disruption of the apoptotic pathway has been implicated in many diseases, including neurodegenerative diseases [24,25,26], autoimmune diseases [27,28,29], and many types of cancer [30,31,32,33].
Based on the assumption that activating or enhancing the apoptotic pathway is beneficial for cancer treatment, much work has gone into developing therapeutic approaches to activate or re-enable the apoptosis pathway within cancer cells. One well-studied target is p53; the high percentage of mutated p53 in cancer and subsequent loss of its ability to induce apoptosis has led to much effort in finding a way to restore its normal function. Multiple approaches have been tested, including drug therapy, gene therapy, and p53-based immunotherapy, but as of yet, no clinical trial has yielded a noteworthy drug, and the FDA has yet to approve any p53-based therapeutic agent [34]. Another seemingly promising target is the IAP, with the reasoning that inhibition of the inhibitors of apoptosis may lead to greater induction of apoptosis via stressors. But despite promising results in xenografts and pre-clinical trials, no therapeutic agents targeting IAPs have yet progressed to the clinic either [35].
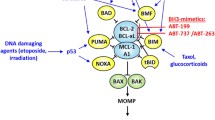
However, one avenue of investigation into activating the apoptosis pathway has led to a viable and approved therapeutic. BCL-2 is a protein that plays a critical role in the regulation of apoptosis as it prevents apoptosis by inhibiting pro-apoptotic molecules such as Bax and Bak [36]. Based on an understanding of the apoptotic mechanism and players, many pharmaceutical companies are looking to inhibit anti-apoptotic BCL-2 members and to use BH3 mimetics. Venetoclax, a small molecule that blocks Bcl-2 function, has been granted Breakthrough Therapy Designation by the FDA and approved for the treatment of chronic lymphocytic leukemia in patients with the 17p genetic mutation [37]. Letai et al. discussed the need to tailor the use of BH3 mimetics to specific cancers and genotypes. The authors developed “BH3 profiling” that allows for the determination of the cancer cell’s dependence on specific anti-apoptotic proteins and predict sensitivity to BCL-2 antagonists [38, 39].
2 Novel roles of apoptotic caspases: promoting neighboring cellular proliferation and tissue regeneration
Despite the well-entrenched views on apoptotic caspases above, a series of recent unanticipated findings have shifted the views of them as purely executioners of cell death. Indeed, more and more they are viewed as mediators of a diverse array of biological functions. In one study, Li et al. proposed the “Phoenix Rising” pathway, in which the dying cells promote wound healing and tissue regeneration in a paracrine manner in mice [40]. Caspase-3 and -7 hold keys in triggering the release of the growth signal PGE2 to promote stem or progenitor cell proliferation and tissue generation (Fig. 1). In the absence of either of these caspases, mice become deficient in wound healing and liver regeneration.

A schematic representation of the “Phoenix Rising” pathway of cell death-induced tissue regeneration. In injured tissues, apoptotic cells activate caspase-3 and -7 through either the intrinsic or the extrinsic pathways. Activated caspase-3 and -7 subsequently cleave and activate iPLA2, which generates arachidonic acid. Arachidonic acid is then converted into PGH2 by cyclooxygenases 1 and 2 (Cox1&2). PGE2 synthase converts PGH2 into PGE2, which stimulates stem cell proliferation and tissue regeneration. Adapted from ref. 40
The role of apoptotic caspases in tissue regeneration is not unique in mice. It was previously discovered that damaged imaginal discs in Drosophila induce the proliferation of the neighboring viable cells via Dronc-dependent p53 pathway [41]. Xenopus laevis requires the activation of caspase-3 to regenerate its tail [42]. Apoptotic caspases were also found to control the tissue regeneration and remodeling process in planarian [43]. Decapitated Hydra was found to regenerate its head from growth signals induced by apoptotic cells via caspase-dependent activation of the Wnt3 pathway [44]. These discoveries in diverse organisms provide strong support for the counterintuitive hypothesis that tissue regeneration induced by apoptotic cell death may be an evolutionally conserved pathway in multi-cellular organisms.
3 Novel roles of apoptotic caspases: stimulating tumor cell repopulation
The pathway in which the apoptotic cells release growth factors to promote tissue regeneration is clearly beneficial in normal tissue regeneration and wound healing. However, the “Phoenix Rising” pathway was also found to be hijacked by cancer cells to repopulate tumors during radiotherapy [45]. Huang et al. found that apoptotic tumor cells generate potent growth signals to stimulate repopulation of cancer after radiotherapy, through the same caspase-activated iPLA2-AA-PGE2 axis that is observed in normal tissues (Fig. 1). Indeed, inhibiting caspase-3 activities caused significant tumor sensitivity to radiotherapy in xenograft tumors. Consistent with the animal studies, authors found that breast and head and neck cancer patients with higher levels of activated caspase-3 in tumor tissues demonstrated significantly increased rate of recurrence and deaths.
Caspase-3 has since been found to promote growth of surviving tumor cells in many types of cancer. Feng et al. uncovered that dying glioma cells promote post-irradiation angiogenesis in a caspase-3-dependent manner, and inhibition of caspase-3 ablated pro-angiogenic effects of dying glioma cells in vivo and in vitro. The authors identified the NF-κB-COX2-PGE2 axis as the key pathway mediating tumor response after irradiation [46]. In another study, Li et al. demonstrated that caspase-3 in dying tumor cells drove a pro-angiogenic response via VEGF-A and Akt signaling after irradiation [47]. Kurtova et al. revealed a mechanism in which bladder cancer stem cells actively contribute to chemoresistance via a proliferative response to repopulate killed tumor cells. This repopulation pathway was also found to be mediated by PGE2 signaling [48]. Taken together, there is solid evidence from multiple groups to support the counterintuitive observation that apoptotic caspases promote neighboring tumor cell repopulation and confer therapeutic resistance in radiotherapy and chemotherapy.
Direct secretion of growth signals is not the only way that apoptotic cells use to communicate; they also secrete exosomes to maintain the viability of the neighboring cells. Exosomes are nano-sized lipid vesicles containing proteins, nucleic acids, tumor suppressor genes, micron RNAs, etc. These substances, particularly the nucleic acids, which are secreted both by tumor cells and the stromal cells can promote tumorigenesis and metastasis of cancers. Yu et al. found increased level of exosomal survivin, a protein that inhibits caspase activation, in breast cancer cells [49]. In a recent review paper, Lynch et al. discuss the specific role of apoptotic cell-derived EVs (Apo-EVs), which they define as heterogeneous vesicles ranging from 50 nm to several microns that are apoptosis dependent, which provide cells undergoing apoptosis with the capability of sending signals over significant distances. It has been shown that apoptotic bodies from tumor cells can induce p53 deficiency in fibroblasts, which promotes tumor growth, thus highlighting the possibility that cells that receive the Apo-EVs can transiently express the same genes from dying tumor cells and this may lead to sustained transformation [50, 51]. More research is needed, but it is clear that Apo-EVs are capable of producing important activating molecules that modulate the tumor’s microenvironment.
4 Novel roles of apoptotic caspases: promoting metastasis
There is increasing evidence that apoptotic caspases are closely associated with cancer progression. A recent meta-analysis revealed that increased caspase-3 expression was statistically correlated with worse prognosis of breast cancer [52]. Zhou et al. provided direct experimental evidence in support of a pro-metastatic role of caspases by demonstrating that colon cancer cells with CASP3 knockout were markedly less invasive and more sensitive to radiotherapy in vitro and in vivo. More interestingly, cells deficient in caspase-3 were less prone to generate pulmonary metastasis when inoculated subcutaneously or intravenously. Authors also found significantly increased E-cadherin expression, reduced N-cadherin, Snail, Slug, and ZEB1 expression in caspase-3-deficient cancer cells, suggesting that the reduced EMT phenotype was implicated in the mechanism in which caspase-3 promoted metastasis [53]. Another recent study by Rudraptna et al. showed effector caspase activity drives cell invasion without initiating apoptosis in a Drosophila model. The study linked effector caspases to matrix metalloproteinase Mmp1 and Jnk pathway [54]. In the same vein of thinking, studies have also identified caspase-8 as a key regulator in integrin internationalization, cell motility, and metastasis [55]. Caspase-8 interacts with multiprotein complex to enhance cleavage of focal adhesion substrates and cell migration. Furthermore, caspase-8 knockdown disrupts metastasis in neuroblastoma in vivo [56]. Senft et al. reported that caspase-8 contributes to cell migration via interaction with p85, a subunit of phosphatidylinositol 3-kinase and an established cell migration component [57, 58]. In glioblastomas with poor response to radiochemotherapy, caspases were found to be constitutively active in vivo and in vitro in the absence of external stress or pro-apoptotic stimuli. Gydnia et al. reported that inhibition of caspase-3 and -8 decreased glioblastoma cell migration and invasion. This caspase-dependent motility was mediated by a constant cleavage of the motility-associated gelsolin protein [59]. In non-small cell lung cancers, elevated caspase-8 predicted early metastasis to the brain [60]. Such findings provide a rationale for elevated expression of apoptotic caspases in many malignant tumors in the absence of treatment. They also suggest inhibition of apoptotic caspases as a plausible approach to reduce cancer metastasis.
5 Activation of apoptotic caspase without cell death
Programmed cell death was previously believed to be irreversible after mitochondrial permeabilization and caspase activation [61,62,63,64,65]. Recent discoveries challenge the notion of the irreversibility of apoptosis and create a more complicated picture of our understanding of the conventional “apoptotic” caspases.
Tang et al. demonstrated the surprising results that even after cells underwent late-stage apoptosis, marked by mitochondrial permeabilization, caspase-3 activation, and DNA damage, the vast majority of cells actually recover when the inducers were washed away. This phenomenon was observed for many types of cells and inducers. The authors coined this unanticipated mechanism “Anastasia” [61]. Notably, some surviving cells acquired permanent genetic alterations and/or underwent oncogenic transformation. Independently, many other groups also investigated the roles of sub-lethal activation of effector caspases in various aspects including pluripotency maintenance, response to irradiation, genetic instability, and carcinogenesis. To more directly track the activation of caspase-3 in vivo, Ding et al. designed CasExpress, which drives fluorescent protein expression in cells that survive caspase-3 activation in Drosophila. Authors provided direct evidence of widespread sub-lethal caspase-3 activation in most tissues of every animal [65].
Sub-lethal activation of caspase activation is not without its consequences. Lovric et al. investigated the effects of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) on surviving glioma cells. TRAIL induces extrinsic apoptosis pathway by ligating death receptors that recruit FADD, caspase-8, and/or caspase-10 to form the “death-inducing signaling complex” [66]. Authors observed increased DNA damage and mutations in surviving tumor cells with activated caspase-8 after sub-lethal exposure to TRAIL or FasL [67]. Orth et al. demonstrated that prolonged mitotic arrest using antimitotic drugs partially actives the apoptotic pathway. This sub-lethal activation of caspases increased DNA damage by partly activating CAD, a downstream factor of caspase-3 [68].
6 Roles of non-lethal caspase activation in cellular differentiation
In Drosophila, it was found that caspases and cytochrome c aid the final stage of spermatid terminal differentiation, in which the bulk cytoplasm is removed. In human tissues, caspases are involved in the differentiation from human monocytes to macrophages [69]. Furthermore, caspase-8 plays a fundamental role in heart muscle differentiation [70], and caspase-14 was associated with terminal differentiation of keratinocytes [71, 72].
Fujita et al. reported that in order for embryonic stem cells (ESCs) to undergo germ layer-specific differentiation, caspase-3 is needed to induce cleavage of Nanog transcription factor. Stem cells lacking this Casp3 gene showed marked defects in differentiation [73]. This finding is significant because Nanog has been shown in multiple studies to be important in maintaining pluripotency in stem cells. Silva et al. demonstrated that Nanog stimulated pluripotent gene activation from somatic cell genomes and increased Nanog is sufficient to reset neural stem cell epigenome to reset into a state of pluripotency [74]. Another study also found that Nanog expression resulted in increased ES-cell-like gene expression and DNA methylation patterns after inducing mouse fibroblast into iPS cells [75].
Dejosez et al. described another caspase-3-mediated ESC differentiation pathway. Their study showed that caspase-3 can cleave Ronin, a nuclear protein that possesses a zinc-finger DNA-binding motif (THAP domain), binds directly to HCF-1 (a key transcriptional regulator), and plays an essential role in embryogenesis and maintaining ESC pluripotency [76].
In addition to their role in ESC differentiation, caspases are also proven to be important in cell differentiation in other cell types. Kang et al. found that caspase-8 mediated differentiation in myelomonocytic lineage in bone-marrow cells [77]. Furthermore, a different group found that caspase-3 was required for RANKL-induced osteoclast differentiation. RANKL cleaves procaspase-3, and the activated protein is localized to the plasma membrane and cytosol. In procaspase-3 knockout cells or with caspase-3 inhibitors, primary osteoclasts failed to express TRAP or become multinucleated even with RANKL induction [78].
7 Roles of non-lethal activation of caspases in cellular de-differentiation
In addition to promoting cellular differentiation, caspases also facilitate the opposite process: de-differentiation. Li et al. reported that caspase-3 and -8 play critical roles in the induction of induced pluripotent stem (iPS) cells from human fibroblasts [79]. When Oct-4, a key iPSC transcription factor, is transduced in human fibroblasts, a portion of cells die in the reprogramming process. However, a significant number of cells with persistent caspase-3 activation survive and become iPSC. Surprisingly, inhibition of caspases-3 or -8 activation impeded iPSC induction, suggesting caspases-3 and -8 are necessary in the induction of pluripotent cells. The authors identified the tumor suppressor protein Rb as a key downstream target of Casp3/8 whose cleavage and inactivation plays key roles in epigenetic reprogramming. In addition to defining a key role for Casp3/8 in induction of iPSCs, the authors also observed consistently elevated expression of caspase-3 in iPSCs and H9 ESC, a well-known ESC line, consistent with the study of Fujita et al. [73].
At first glance, a role for effector caspases in both ESC differentiation and iPSC induction from human fibroblasts, which is basically a de-differentiation process, is puzzling. However, if one view caspases as an instrument of the cellular epigenetic machinery to go back and forth between differentiation and de-differentiation, it makes some sense. Obviously, many further studies are needed to clearly define the roles of caspases in regulating cellular differentiation and transcriptional reprogramming.
8 Role of non-lethal activation of caspases in maintenance of stemness and tumorigenicity of cancer cells
Cancer stem cells (CSCs) are important in tumorigenesis, metastasis, recurrence, and chemoresistance [80]. Cancer stem cells were shown to possess over-activated signaling pathways, including JAK/STAT, wnt/beta-catenin, Nanog, and Notch [81,82,83,84,85]. Even though they are often a very fraction of the overall cancer cell population, CSCs often play key roles in tumor growth and metastasis [86]. Moreover, CSCs were shown to be resistant to chemotherapy and radiotherapy [87,88,89]. However, the molecular mechanisms that sustain the stemness of cancer cells are now clearly defined.
In a recent study, Liu et al. demonstrated a surprising role of caspases in maintaining the tumorigenicity and stemness of breast cancer and glioma cells [90]. The authors found that many cancer cells have persistent effector caspase activation in the absence of any external stressors, much like those observed in iPSC and ESCs (Li et al., Fujita et al.). Similar to what is observed in ESCs, activated Casp3/7 did not kill the host cells. Instead, limited caspase activation leads to self-inflicted DNA double-strand breaks, which induces persistent ATM activation, resulting in Stat3 activation and in elevated CD133 expression in glioma CSCs [91].
The findings of Liu et al. directly challenges the established paradigm on the roles of apoptotic caspases in tumor growth and treatment. Furthermore, they also revealed the dark side of DNA damage response (DDR). They found that in many tumor cells, low level of cytochrome c leakage from the mitochondria leads to sub-lethal caspases activation, which causes DNA DSBs and activation ATM, a central player in the DDR. This occurs without exposure to any external stressors. When caspase-3, -6, and -7 were knocked out using CRISPR, the authors found significant reduction in DNA damage, reflected by gamma H2AX foci. Furthermore, caspases knockout breast tumor cells, EndoG/CAD (two apoptotic endonucleases downstream of Casp3/7 [92, 93]) knockout cells, and ATM knockout cells all showed significantly reduced tumor growth in vivo. Thus, authors established the functional relevance of caspases and spDSB on the growth and tumorigenic abilities of cancer cells (Fig. 2).

An illustration of spontaneous DNA double-strand break induction and their roles in maintaining the stemness and tumorigenicity of cancer cells. Mitochondrial permeability changes in cancer cells allow spontaneous, sub-lethal activation of the apoptotic cascade that includes the cytoplasmic leakage of cytochrome c, caspase-3 activation, CAD activation, and EndoG nuclear translocation. The presence of spontaneous DNA double-strand breaks activates ATM, a key player in DNA damage repair pathway. Phosphorylated ATM activates NF-κB and Stat3, two factors well known to the maintenance of tumorigenicity and stemness of cancer cells. Adapted from ref. 88
The identification of DNA damage/ATM activation/NF-kB/Stat3 activation/secretion of pro-tumor cytokines is reminiscent of the senescence-associated secretory phenotype (SASP) pathway, where the cell damage activates p53, triggering permanent growth arrest, which leads to the senescent state, and cells secrete signaling factors in the senescent state that leads to various pathological conditions including tumor promotion [94]. Similarly, our studies in tumor cells point to a pathway that the sub-lethal activation of caspase-3, -6, -7, and endonucleases (CAD and EndoG) leads to DNA double-strand breaking. This activates ATM and downstream factors (NF-kB and STAT3) and results in pro-inflammatory signaling factors secretion, which promote tumor growth (Fig. 2).
These unexpected roles of caspases in promoting cancer aggressiveness raise the questions on some of the current strategies of cancer therapeutics development. Thus, the singular approach of caspase activation in cancer treatment may need to be reassessed. In many instances, it might be beneficial to inhibit rather than activate effector caspases.
9 Role of sub-lethal caspase activation in carcinogenesis
Not only can caspases promote cancer aggression and metastasis, they may also be directly involved in promoting cancer development. Studies have found that pro-apoptotic pathways directly promote tumorigenesis in multiple types of malignancies. Biswas et al. demonstrated that loss of pro-apoptotic Bid surprisingly increases the latency of leukemogenic in Atm knockout mice. The authors suggest that a loss of Bid inhibits T cell tumorigenesis by increasing clearance of damaged cells [95]. Labi et al. also found that apoptosis actively drives tumor formation. The investigators demonstrated that mice defective in p53-induced apoptosis resist gamma-irradiation-induced lymphomagenesis, whereas repeated irradiation in wild-type animals leads to lymphoma formation by inducing expansion of hematopoietic stem cells [96]. Another study showed that the loss of Puma, a pro-apoptotic BH3-only protein of the Bcl-2 family, ablated tumorigenesis [97]. These studies strongly support that apoptosis promote cancer through its ability to generate compensatory proliferation of neighboring cancer cells or provide a vacant niche for cancer growth [98].
Mitochondrial outer membrane permeabilization (MOMP) was historically considered the point of no return in the apoptotic mechanism because the release of mitochondrial proteins including cytochrome c activates caspases [99] and leads to rapid cell death. However, Ichim et al. found that instead of killing the cells, a minority MOMP actually induces caspase-dependent DNA damage, promoting genomic instability and tumorigenesis [100]. Consistently, by use of a non-invasive caspase-3 reporter, Liu et al. demonstrated importance of caspase-3 activation in radiation-induced and chemical-induced malignant transformation of mammalian cells [101]. With low-dose radiation, a significant fraction of mammalian cells not only survive despite caspase-3 activation, but the surviving cells also demonstrated persistent DNA damage up to 3 months after exposure. More importantly, authors provided evidence that caspase-3 facilitates oncogenic transformation of human mammary epithelial cells. After exposure to irradiation, MCF10A cells were cultured and plated into soft agar, a well-established method to examine anchorage-independent growth in malignant transformation [102]. These irradiated cells readily formed soft agar colonies, and those with higher caspase-3 activation formed colonies at a significantly higher frequency. Furthermore, this ability to form soft agar colonies was significantly attenuated when caspase-3 was inhibited. In addition to irradiation-induced carcinogenesis, the authors also showed that caspase-3 facilitated two-stage chemically induced skin carcinogenesis in vitro. EndoG was found to be the downstream factor of caspase-3 role in mediating radiation-induced DNA damage and transformation (Fig. 3).

A schematic diagram illustrating how abortive apoptosis facilitate stress-induced genetic instability and oncogenic transformation. Left panel shows the conventional scenario where mitochondrial permeability changes lead to activation of Casp3 and leakage of endonuclease G that kills the host cells. Right panel, on the other hand, shows partial leakage and survival of the cells with secondary genetic damage and oncogenic transformation
The facilitative role of caspase-3 in carcinogenesis was also confirmed in Myc-induced transformation of human mammary epithelial cells [103]. In that study, the authors demonstrated that an overexpression of Myc oncogene induces chromosome aberrations and gammaH2AX foci in non-transformed human mammary epithelial cells in a caspase-3-dependent manner. Furthermore, the authors show that Casp3 activation and activation of its downstream factor endoG is absolutely required for Myc-induced carcinogenesis. This finding thus resolves a long-standing dilemma in Myc-induced oncogenesis: Myc’s role as both a powerful inducer of apoptosis and a powerful oncogenic factor.
10 Implications and future directions
Recent studies have significantly shifted the established paradigm based on the simplistic view caspases are strictly instruments of cell death. It is clear that they play unexpected roles in promoting, rather than suppressing carcinogenesis, epigenetic reprogramming, and genetic instability. One important clinical implication of these discoveries is that current anti-oncogenic therapies aimed at activating caspases to kill cancer cells are at best a flawed strategy. In fact, established cancer treatment such as radiotherapy and chemotherapy may select for cancer cells that could survive the treatments and become stronger by acquiring new mutations or become more stem cell-like based on sub-lethal caspase activation. Indeed, caspase inhibition may be a viable strategy to enhance current cancer therapy. In support of this view, new studies that look into caspase inhibition as an adjunct therapeutic approach have found promising results. Flanagan et al. demonstrated that selective inhibition of caspase-3 or its downstream effectors markedly reduced the expression of proliferation markers in colorectal tumor explant. Additionally, metastatic CRC patients with low level of active caspase-3 had increased disease-free survival, especially in patients who received 5-FU chemotherapy [104]. All these findings point to a new direction of caspase inhibition as an adjunct anticancer option in advanced cancer.
Taken together, apoptotic caspases play a far more complicated role than simply being an instrument of cell death. More studies are clearly needed to achieve a better understanding of their non-lethal biological involvements, see (Fig. 4) for a summary. Only after achieving a better understanding can we begin to target them effectively to achieve therapeutic gains.

A summary of the more established and recently identified non-canonical pathways related to apoptotic caspases
References
Riedl, S. J., & Shi, Y. (2004). Molecular mechanisms of caspase regulation during apoptosis. Nature Reviews. Molecular Cell Biology, 5.11, 897–907.
Zou, H., et al. (1999). An Apaf-1. Cytochrome C multimeric complex is a functional apoptosome that activates procaspase-9. The Journal of Biological Chemistry, 274.17, 11549–11556.
Wang, X. (2001). The expanding role of mitochondria in apoptosis. Genes & Development, 15.22, 2922–2933.
Nagata, S. (1999). Fas ligand-induced apoptosis. Annual Review of Genetics, 33, 29–55.
Peter, M. E., & Krammer, P. H. (2003). The Cd95(Apo-1/Fas) disc and beyond. Cell Death and Differentiation, 10.1, 26–35.
Horvitz, H. R. (2003). Nobel lecture. Worms, life and death. Bioscience Reports, 23.-65, 239–303.
Lee, C. Y., Cooksey, B. A., & Baehrecke, E. H. (2002). Steroid regulation of midgut cell death during Drosophila development. Developmental Biology, 250.1, 101–111.
Pyati, U. J., et al. (2006). Sustained bmp signaling is essential for cloaca development in zebrafish. Development, 133.11, 2275–2284.
Magnus, T., et al. (2002). Astrocytes are less efficient in the removal of apoptotic lymphocytes than microglia cells: implications for the role of glial cells in the inflamed central nervous system. Journal of Neuropathology and Experimental Neurology, 61.9, 760–766.
Shklover, J., Levy-Adam, F., & Kurant, E. (2015). Apoptotic cell clearance in development. Current Topics in Developmental Biology, 114, 297–334.
Arama, E., Agapite, J., & Steller, H. (2003). Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Developmental Cell, 4, 687–697.
Arama, E., Bader, M., Rieckhof, G. E., & Steller, H. (2007). A ubiquitin ligase complex regulates caspase activation during sperm differentiation in Drosophila. PLoS Biology, 5, e251.
Baum, J. S., Arama, E., Steller, H., & McCall, K. (2007). The Drosophila caspases Strica and Dronc function redundantly in programmed cell death during oogenesis. Cell Death and Differentiation, 14, 1508–1517.
Bergmann, A., Agapite, J., & Steller, H. (1998). Mechanisms and control of programmed cell death in invertebrates. Oncogene, 17, 3215–3223.
Mccall, K., & Steller, H. (1998). Requirement for DCP-1 caspase during Drosophila oogenesis. Science, 279, 230–234.
Zakeri, Z., et al. (2015). What cell death does in development. The International Journal of Developmental Biology, 59.1-3, 11–22.
DYCHE, W. J. (1979). A comparative study of the differentiation and involution of the Mullerian duct and Wolffian duct in the male and female fetal mouse. Journal of Morphology, 162, 175–209.
Teixeira, J., Maheswaran, S., & Donahoe, P. K. (2001). Mullerian inhibiting substance: an instructive developmental hormone with diagnostic and possible therapeutic applications. Endocrine Reviews, 22, 657–674.
Mammano, F., & Bortolozzi, M. (2018). Ca (2+) signaling, apoptosis and autophagy in the developing cochlea: milestones to hearing acquisition. Cell Calcium, 70, 117–126.
Zuzarte-Luis, V., & Hurle, J. M. (2002). Programmed cell death in the developing limb. The International Journal of Developmental Biology, 46.7, 871–876.
Iversen, O. H. (1996). Cell death in vivo: terminal maturation, necrosis and apoptosis. East African Medical Journal, 73.5(Suppl), S5–S6.
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144.5, 646–674.
Lengauer, C., Kinzler, K. W., & Vogelstein, B. (1997). Genetic instability in colorectal cancers. Nature, 386, 623–627.
Guenette, S. Y., & Tanzi, R. E. (1999). Progress toward valid transgenic mouse models for Alzheimer’s disease. Neurobiology of Aging, 20.2, 201–211.
Sathasivam, K., et al. (1999). Transgenic models of Huntington’s disease. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 354.1386, 963–969.
Borchelt, D. R., et al. (1998). Transgenic mouse models of Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Pathology, 8.4, 735–757.
Wang, J., et al. (1999). Inherited human caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type Ii. Cell, 98.1, 47–58.
Furlan, R., et al. (1999). Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. Journal of Immunology, 163.5, 2403–2409.
Liadis, N., et al. (2005). Caspase-3-dependent beta-cell apoptosis in the initiation of autoimmune diabetes mellitus. Molecular and Cellular Biology, 25.9, 3620–3629.
Taghiyev, A. F., Rokhlin, O. W., & Glover, R. B. (2011). Caspase-2-based regulation of the androgen receptor and cell cycle in the prostate cancer cell line Lncap. Genes & Cancer, 2.7, 745–752.
Mathiasen, I. S., Lademann, U., & Jaattela, M. (1999). Apoptosis induced by vitamin D compounds in breast cancer cells is inhibited by Bcl-2 but does not involve known caspases or P53. Cancer Research, 59.19, 4848–4856.
King, D., et al. (1998). Processing/activation of caspases, -3 and -7 and -8 but not caspase-2, in the induction of apoptosis in B-chronic lymphocytic leukemia cells. Leukemia, 12.10, 1553–1560.
Ozaki, T., & Nakagawara, A. (2011). Role of P53 in cell death and human cancers. Cancers (Basel), 3.1, 994–1013.
Wong, R. S. (2011). Apoptosis in cancer: from pathogenesis to treatment. Journal of Experimental & Clinical Cancer Research, 30, 87.
de Almagro, M. C., & Vucic, D. (2012). The inhibitor of apoptosis (Iap) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Experimental Oncology, 34.3, 200–211.
Montero, J., & Letai, A. (2018). Why do Bcl-2 inhibitors work and where should we use them in the clinic? Cell Death and Differentiation, 25.1, 56–64.
Deeks, E. D. (2016). Venetoclax: first global approval. Drugs, 76.9, 979–987.
Del Gaizo Moore, V., et al. (2007). Chronic lymphocytic leukemia requires Bcl2 to sequester Prodeath Bim, explaining sensitivity to Bcl2 antagonist Abt-737. The Journal of Clinical Investigation, 117.1, 112–121.
Davids, M. S., et al. (2012). Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood, 120.17, 3501–3509.
Li, F., et al. (2010). Apoptotic cells activate the “Phoenix rising” pathway to promote wound healing and tissue regeneration. Science Signaling, 3.110, ra13.
Wells, B. S., Yoshida, E., & Johnston, L. A. (2006). Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent P53 activity. Current Biology, 16.16, 1606–1615.
Tseng, A. S., et al. (2007). Apoptosis is required during early stages of tail regeneration in Xenopus Laevis. Developmental Biology, 301.1, 62–69.
Hwang, J. S., Kobayashi, C., Agata, K., Ikeo, K., & Gojobori, T. (2004). Detection of apoptosis during planarian regeneration by the expression of apoptosis-related genes and Tunel assay. Gene, 333, 15–25.
Chera, S., et al. (2009). Apoptotic cells provide an unexpected source of Wnt3 signaling to drive Hydra head regeneration. Developmental Cell, 17.2, 279–289.
Huang, Q., et al. (2011). Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nature Medicine, 17.7, 860–866.
Feng, X., Yu, Y., He, S., Cheng, J., Gong, Y., Zhang, Z., Yang, X., Xu, B., Liu, X., Li, C. Y., Tian, L., & Huang, Q. (2017). Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Letters, 385, 12–20.
Feng, X., et al. (2015). Caspase 3 in dying tumor cells mediates post-irradiation angiogenesis. Oncotarget, 6.32, 32353–32367.
Kurtova, A. V., et al. (2015). Blocking Pge2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature, 517.7533, 209–213.
Yu, D. D., et al. (2015). Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Science, 106.8, 959–964.
Bergsmedh, A., et al. (2001). Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proceedings of the National Academy of Sciences of the United States of America, 98.11, 6407–6411.
Lynch, C., Panagopoulou, M., & Gregory, C. D. (2017). Extracellular vesicles arising from apoptotic cells in tumors: roles in cancer pathogenesis and potential clinical applications. Frontiers in Immunology, 8, 1174.
Yang, X., et al. (2018). Caspase-3 over-expression is associated with poor overall survival and clinicopathological parameters in breast cancer: a meta-analysis of 3091 cases. Oncotarget, 9.9, 8629–8641.
Zhou, M., et al. (2018). Caspase-3 regulates the migration, invasion, and metastasis of colon cancer cells. International Journal of Cancer, https://doi.org/10.1002/ijc.31374. [Epub ahead of print].
Rudrapatna, V. A., Bangi, E., & Cagan, R. L. (2013). Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Reports, 14.2, 172–177.
Torres, V. A., et al. (2010). Rab5 mediates caspase-8-promoted cell motility and metastasis. Molecular Biology of the Cell, 21.2, 369–376.
Barbero, S., et al. (2009). Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Research, 69.9, 3755–3763.
Helfer, B., et al. (2006). Caspase-8 promotes cell motility and Calpain activity under nonapoptotic conditions. Cancer Research, 66.8, 4273–4278.
Senft, J., Helfer, B., & Frisch, S. M. (2007). Caspase-8 interacts with the P85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Research, 67.24, 11505–11509.
Gdynia, G., et al. (2007). Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Molecular Cancer Research, 5.12, 1232–1240.
Liao, Y., et al. (2015). The impact of caspase-8 on non-small cell lung cancer brain metastasis in Ii/Iii stage patient. Neoplasma. https://doi.org/10.4149/neo_2125_043.
Tang, H. L., et al. (2012). Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Molecular Biology of the Cell, 23.12, 2240–2252.
Green, D. R., & Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science, 305.5684, 626–629.
Taylor, R. C., Cullen, S. P., & Martin, S. J. (2008). Apoptosis: controlled demolition at the cellular level. Nature Reviews. Molecular Cell Biology, 9.3, 231–241.
Chipuk, J. E., et al. (2010). The Bcl-2 family reunion. Molecular Cell, 37.3, 299–310.
Ding, A. X., et al. (2016). Casexpress reveals widespread and diverse patterns of cell survival of caspase-3 activation during development in vivo. Elife, 5. pii: e10936. https://doi.org/10.7554/elife.10936.
Ashkenazi, A., Holland, P., & Eckhardt, S. G. (2008). Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/tumor necrosis factor-related apoptosis-inducing ligand (Rhapo2l/Trail). Journal of Clinical Oncology, 26.21, 3621–3630.
Lovric, M. M., & Hawkins, C. J. (2010). Trail treatment provokes mutations in surviving cells. Oncogene, 29.36, 5048–5060.
Orth, J. D., et al. (2012). Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and P53 induction. Molecular Biology of the Cell, 23.4, 567–576.
Sordet, O., et al. (2002). Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood, 100.13, 4446–4453.
Varfolomeev, E. E., et al. (1998). Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the Tnf receptors, Fas/Apo1, and Dr3 and is lethal prenatally. Immunity, 9.2, 267–276.
Rendl, M., et al. (2002). Caspase-14 expression by epidermal keratinocytes is regulated by Retinoids in a differentiation-associated manner. The Journal of Investigative Dermatology, 119.5, 1150–1155.
Eckhart, L., et al. (2000). Terminal differentiation of human keratinocytes and stratum corneum formation is associated with caspase-14 activation. The Journal of Investigative Dermatology, 115.6, 1148–1151.
Fujita, J., et al. (2008). Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell, 2.6, 595–601.
Silva, J., et al. (2006). Nanog promotes transfer of pluripotency after cell fusion. Nature, 441.7096, 997–1001.
Okita, K., Ichisaka, T., & Yamanaka, S. (2007). Generation of germline-competent induced pluripotent stem cells. Nature, 4487151, 313–317.
Dejosez, M., et al. (2008). Ronin is essential for embryogenesis and the pluripotency of mouse embryonic stem cells. Cell, 133.7, 1162–1174.
Kang, T. B., et al. (2004). Caspase-8 serves both apoptotic and nonapoptotic roles. Journal of Immunology, 173.5, 2976–2984.
Szymczyk, K. H., et al. (2006). Active caspase-3 is required for osteoclast differentiation. Journal of Cellular Physiology, 209.3, 836–844.
Li, F., et al. (2010). Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell, 7.4, 508–520.
Li, Z. (2013). Cd133: a stem cell biomarker and beyond. Experimental Hematology & Oncology, 2.1, 17.
Kim, J., Jeon, H., & Kim, H. (2015). The molecular mechanisms underlying the therapeutic resistance of cancer stem cells. Archives of Pharmacal Research, 38(3), 389–401.
Karamboulas, C., & Ailles, L. (2013). Developmental signaling pathways in cancer stem cells of solid tumors. Biochimica et Biophysica Acta, 1830(2), 2481–2495.
Hernandez-Vargas, H., Ouzounova, M., Le Calvez-Kelm, F., et al. (2011). Methylome analysis reveals Jak-STAT pathway deregulation in putative breast cancer stem cells. Epigenetics, 6(4), 428–439.
Watabe, T., & Miyazono, K. (2009). Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Research, 19(1), 103–115.
Mo, J., Park, H., & Guan, K. (2014). The hippo signaling pathway in stem cell biology and cancer. EMBO Reports, 15(6), 642–656.
Norton, K. A., & Popel, A. S. (2014). An agent-based model of cancer stem cell initiated avascular tumour growth and metastasis: the effect of seeding frequency and location. Journal of the Royal Society, Interface, 11.100, 20140640.
Ma, R., et al. (2014). Stemness in human thyroid cancers and derived cell lines: the role of asymmetrically dividing cancer stem cells resistant to chemotherapy. The Journal of Clinical Endocrinology and Metabolism, 99.3, E400–E409.
Morrison, R., et al. (2011). Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. Journal of Oncology, (2011), 941876.
Smit, J. K., et al. (2013). Prediction of response to radiotherapy in the treatment of esophageal cancer using stem cell markers. Radiotherapy and Oncology, 107.3, 434–441.
Liu, X., Li, F., Huang, Q., Zhang, Z., Zhou, L., Deng, Y., Zhou, M., Fleenor, D. E., Wang, H., Kastan, M. B., & Li, C. Y. (2017). Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Research, 27, 764–783.
Stagni, V., Manni, I., Oropallo, V., Mottolese, M., di Benedetto, A., Piaggio, G., Falcioni, R., Giaccari, D., di Carlo, S., Sperati, F., Cencioni, M. T., & Barilà, D. (2015). Atm kinase sustains Her2 tumorigenicity in breast cancer. Nature Communications, 6, 6886.
Arnoult, D., et al. (2003). Mitochondrial release of Aif and Endog requires caspase activation downstream of Bax/Bak-mediated permeabilization. The EMBO Journal, 22.17, 4385–4399.
Uegaki, K., et al. (2000). Structure of the Cad domain of caspase-activated Dnase and interaction with the Cad domain of its inhibitor. Journal of Molecular Biology, 297.5, 1121–1128.
Coppe, J. P., et al. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual Review of Pathology, 5, 99–118.
Biswas, S., et al. (2013). The loss of the Bh3-only Bcl-2 family member bid delays T-cell leukemogenesis in Atm−/− mice. Cell Death and Differentiation, 20.7, 869–877.
Labi, V., et al. (2010). Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes & Development, 24.15, 1602–1607.
Michalak, E. M., et al. (2010). Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes & Development, 24.15, 1608–1613.
Ichim, G., & Tait, S. W. (2016). A fate worse than death: apoptosis as an oncogenic process. Nature Reviews. Cancer, 16.8, 539–548.
Tait, S. W., & Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nature Reviews. Molecular Cell Biology, 11.9, 621–632.
Ichim, G., et al. (2015). Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Molecular Cell, 57.5, 860–872.
Liu, X., et al. (2015). Caspase-3 promotes genetic instability and carcinogenesis. Molecular Cell, 58.2, 284–296.
Cifone, M. A., & Fidler, I. J. (1980). Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proceedings of the National Academy of Sciences of the United States of America, 77.2, 1039–1043.
Cartwright, I. M., et al. (2017). Essential roles of caspase-3 in facilitating Myc-induced genetic instability and carcinogenesis. Elife, 6 pii:e26371. https://doi.org/10.7554/eLife.26731.
Flanagan, L., Meyer, M., Fay, J., Curry, S., Bacon, O., Duessmann, H., John, K., Boland, K. C., McNamara, D. A., Kay, E. W., Bantel, H., Schulze-Bergkamen, H., & Prehn, J. H. M. (2016). Low levels of caspase-3 predict favourable response to 5fu-based chemotherapy in advanced colorectal cancer: caspase-3 inhibition as a therapeutic approach. Cell Death & Disease, 7, e2087.
Funding
The study is supported in part by grants ES024015, CA208852, and CA216876 from the US National Institutes of Health (to C-Y. Li).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhao, R., Kaakati, R., Lee, A.K. et al. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev 37, 227–236 (2018). https://doi.org/10.1007/s10555-018-9736-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-018-9736-y