
Abstract
Purpose of Review
To summarize recent findings on novel roles of caspases in stem cell biology, tumor repopulation, and tissue regeneration. Contrary to the long-held notion that apoptotic caspases are exclusively executioners of programmed cell death, an abundance of evidence is emerging that activation of caspases does not inevitably lead to cell death.
Recent Findings
It is now known that sublethal activation of caspases occurs in development, stem cell differentiation, epigenetic reprogramming, and a whole host of other key biological processes. Important for cancer biology, recent studies show that activation of caspases in tumors facilitates carcinogenesis, metastasis, and tumor relapse after cancer treatment. We have found that apoptotic cells secrete prostaglandins to stimulate proliferation of neighboring cells. This pathway functions to regenerate tissues and stem cells in multiple organisms, but it also poses problems in emerging tumor resistance to chemotherapy and radiotherapy.
Summary
Novel findings on caspases are contrary to established paradigms and might explain why cancer therapies aimed at activating apoptotic caspases have not been very successful in the clinic. In this brief review, we summarize some novel findings regarding caspases with the hope of stimulating more interest in this nascent but increasingly important research area. Better understanding of the diverse roles of caspases may one day help us establish novel approaches for treating cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
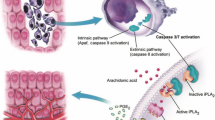
Apoptosis is a tightly regulated physiological process whereby multicellular organisms get rid of unwanted or DNA damaged cells. After apoptosis, no inflammation ensues and neighboring cells are left undisturbed, distinguishing it from necrosis. Caspases are a family of cysteine proteases that execute mid to late stages of apoptosis and are composed of the initiator caspases 2, 8, 9 and the executor caspases 3, 6, and 7 [1]. There are two pathways that mediate apoptosis—the intrinsic and extrinsic pathways. The intrinsic pathway is mediated by the mitochondria as pro-apoptotic signals result in the release of mitochondrial cytochrome c which binds to APAF1, that in turn causes APAF1 to bind ATP/dATP and form the apoptosome. The apoptosome leads to activation of caspase 9, which results in the downstream activation of caspase 3—the primary executioner of apoptosis [2]. In contrast to the intrinsic pathway, the extrinsic pathway is initiated by extracellular signals such as the well-studied Fas ligand that binds to Fas receptor on cells. Upon activation of the receptor, caspase 8 and the Fas-associated death domain (FADD) protein interact with the receptor to form the death-inducing signaling complex (DISC). DISC subsequently cleaves caspase 8, which then activates caspase 3, thus resulting in initiation of apoptosis [3, 4].
In addition to eliminating unwanted or damaged cells, apoptosis is pivotal in morphogenesis during development and in fine-tuning the properties of stem cells. During organogenesis, apoptosis is essential in arranging the formation of organs [5], sex differentiation in humans [6], limb development [7], and immune system maturation [8]. Thus, with the many essential roles of apoptosis, its disruption is typically detrimental to an organism during development. However, the evasion of apoptosis is also a hallmark of cancer [9], and caspases are traditionally thought to be tumor-suppressive as cancer frequently occurs when the Tp53 gene, which encodes a key initiator of the intrinsic apoptosis pathway [10], is mutated. In support of this view, the disruption of the apoptotic pathway has been implicated in the formation of many types of cancers [11, 12]. However, studies in the past decade have shown that the roles apoptosis play during carcinogenesis as well as during/following chemotherapy and radiotherapy treatments are much more complicated, with caspase activation in many scenarios acting to promote chemo- and/or ionizing radiation (IR)-induced genetic instability and carcinogenesis. In this review, we discuss this shifting paradigm of the roles of caspases in DNA damaging agent-induced tumor relapse as well as their roles in stem cell biology.
Caspase Involvement in Stem Cell Differentiation, Epigenetic Reprogramming, and De-differentiation
Stem cells are remarkable cells that have the potential to develop into any cell type during time periods of early embryogenesis, growth, or repair. Stem cells renew themselves through symmetric cell division and can be induced to become tissue-specific cells under certain circumstances through asymmetric cell divisions. There are three major types of stem cells: embryonic stem cells (ESCs), adult/somatic stem cells, and induced pluripotent stem cells (iPSCs) [13, 14]. ESCs are present in early life and are pluripotent, capable of differentiating into any cell type, whereas adult stem cells are capable of self-renewal but are usually restricted to differentiating into the cells of the host tissue in which they are housed [15]. Thus, adult stem cells are “unipotent” or “multipotent” rather than “pluripotent” and are usually in a state of dormancy/quiescence until they receive proliferative signals from their resident “stem cell niche.” The niche is a microenvironment that usually consists of differentiated cells in the tissue, other stem cells, extracellular matrix, cytokines, and other factors that induce these adult stem cells to differentiate and replenish the tissue when needed [16,17,18]. iPSCs are artificially reprogrammed pluripotent stem cells that are derived from differentiated cells by the use of multiple exogenous transcription factors such as Oct4, Sox2, Klf4, c-Myc, Nanog, and Lin28 [19, 20]. Interestingly, these factors contain active caspase cleavage sites that modulate stem cell properties [21, 22]. This begs the question, could caspases, traditionally known as “the irreversible death initiators”, be involved in the regulation of transcription factors related to stem cell proliferation and differentiation? To answer this question, one must first compare the phenotypic similarities between these seemingly opposing processes.
Apoptosis has multiple defining cytological features such as the stereotypical characteristics of pyknosis, karyorrhexis, and karyolysis followed by the eventual consumption of apoptotic bodies by macrophages. Macrophages are designed to recognize phosphatidylserines exposed on the outer cell membrane that have translocated from the inner membrane during apoptosis. Coincidently, many of the same processes also occur in stem cell differentiation [23]. Jeong et al. provides evidence that phosphatidylserine translocation to the outer cell membrane is important in myoblast fusion into myotubes, a key step of muscle differentiation. They also show that myotubes are formed when phosphatidylserine is supplied to in vitro cultures, as well as increases in the number of myonuclei and width of myotubes [24]. In parallel to the formation of apoptotic bodies, Marzesco et al. report that during the early stages of neurogenesis, neural stem cells can secrete membrane vesicles similar to apoptotic bodies that contain the stem cell marker prominin-1, which may play a role in tissue development and maintenance [25]. Lastly, another characteristic shared between apoptosis and differentiation is that both involve the intrinsic induction of significant DNA double-strand breaks (DSBs; which are also the critical genotoxic lesions induced by IR and many chemotherapeutic agents, either directly or during their subsequent repair). Multiple researchers have independently demonstrated that several cellular differentiation pathways rely on enzymatically mediated DNA DSB induction, including those for myoblasts [26], lymphocytes [27], keratinocytes [28], granulocytes [29], and monocytes [30].
In addition to these biochemical similarities, there are other direct links between caspase pathway induction and differentiation. In 1998, Raff et al. provided some of the earliest evidence that caspase 3 activity is pivotal for stem cell differentiation in lens epithelial cells [31]. Many studies followed soon after to support the notion that a transient increase in caspase 3 is necessary for stem cell differentiation across multiple cell lines and species, including skeletal muscle [32], osteoblasts [33], neurons [34], astrocytes [35], hematopoietic stem cells [36], and cardiomyocyte progenitors [37, 38]. Another direct demonstration of the importance of caspase 3 in cellular differentiation was shown by the observation that caspase 3 activation and cleavage of the stem cell factor Nanog were critical for the differentiation of H9 embryonic stem cells [39•]. Fujita et al. further discovered that embryonic stem cells that lacked functional caspases 3 and 9 were unable to differentiate. They also found that mutations in Nanog that prevented caspase 3 cleavage also attenuated embryonic stem cell differentiation. These results also appeared to apply in vivo, as it was found that during development in Drosophila, there was widespread activation of multiple caspases in cells that nonetheless eventually survived [40•].
Therefore, there is an abundance of evidence indicating facilitative roles for caspases in cellular proliferation and differentiation. What about the reverse process, de-differentiation, that occurs e.g., during the induction of iPSCs? While investigating molecular mechanisms of the induction of pluripotent stem cells from human fibroblast cells in our lab, we have shown that in cells transduced with stem cell reprogramming factors, caspases 3 and 8 were significantly activated, but these cells likewise survived. Interestingly, the iPSCs had caspase 3 remaining active at persistently high levels. Most importantly, caspase 3 and 8 activation appeared to be required, as inhibition of either caspase 3 or 8 significantly attenuated iPSC induction from the fibroblasts. It was further discovered that caspases 3 and 8 play critical roles by cleaving and deactivating Rb, the protein encoded by retinoblastoma susceptibility gene Rb1 (itself also a very frequent target of inactivation during carcinogenesis), which is an epigenetic barrier that must be overcome to allow epigenetic reprogramming to occur [21].
Non-lethal Activation of Caspases and Its Role in the Maintenance of Cancer Stem Cells
Cancer stem cells (CSCs) are tumor cells found within cancers that possess stem cell-like characteristics and the potent ability to form tumors, while at the same time preserving the ability to differentiate into various cell types. They are known for being chemo- and radio-resistant and critically involved in tumorigenesis, metastasis, and recurrence [41, 42]. CSCs have been shown to possess over-activated signaling pathways, including JAK/STAT, Wnt/beta-catenin, Nanog, and Notch [43,44,45]. Despite their importance, the molecular mechanisms involved in CSC maintenance are not well understood. Recently, our lab demonstrated an important role of caspases in maintaining the tumorigenicity and stemness of breast cancer and glioma CSCs. Briefly, we found evidence for spontaneous leakage of mitochondrial cytochrome c, leading to sublethal activation of apoptotic caspases in cancer cells and the generation of DSBs. It is very well established that DSBs play an essential role in both tumorigenicity and stemness of cancer through their activation of ATM and various downstream DNA damage response (DDR) pathways [46•]. One study found that radioresistant glioma stem cells had higher levels of DNA repair proteins [47], and we propose that a reason for this could be caspase-mediated DSB induction driving ATM activation, thereby upregulating DDR pathway activation in these cells allowing them to be better equipped to deal with IR-induced DNA damage. Similarly, the stemness of glioma CSCs may be dependent on caspase-mediated DSB-induced ATM activation. Using CRISPR-mediated knockouts of caspase 3 and ATM, we also found that the expression of stem cell markers was markedly reduced. We suggest that higher levels of ATM activation may correlate with higher malignant potential of CSCs, which could partially explain why human breast and colon tumors with higher levels of ATM activation have a poorer prognosis [48, 49]. To summarize, we propose that increased ATM and subsequent activation of DNA damage response pathways, instead of promoting apoptosis progression, act to maintain cancer cell stemness and promote tumorigenicity. The identification of increased ATM activation and DSB levels is reminiscent of the pathway leading to the senescence-associated secretory phenotype (SASP), in which DNA damage activates p53, subsequently causing permanent cell cycle arrest and increased production and secretion of pro-inflammatory cytokines [50]. Our studies show that sublethal activation of caspases leads to increased levels of caspase-mediated DSBs that activate ATM and downstream factors such as STAT-3 that can result in pro-inflammatory SASP that promotes stemness of host cancer cells.
A Counterintuitive Role of Caspases: Promoting Cellular Proliferation and Tissue Regeneration
With the gradual realization that apoptotic caspases can have non-apoptotic roles, an increasing number of studies are showing that caspases can actually promote cellular growth. Our lab has previously proposed the “phoenix rising” pathway, in which dying cells promote wound healing and tissue regeneration in a paracrine-dependent manner in mice. The activation of apoptotic caspases 3 and 7 results in the release of prostaglandin E2 (PGE2), which is a growth signal that promotes stem cell proliferation and tissue regeneration. In the absence of either of these caspases, mice become deficient in wound healing and liver regeneration [51]. In a follow-up study, we also showed that phoenix rising pathway activation occurs in IR-treated breast cancer cells [52••]. Furthermore, Feng et al. discovered that dying glioma cells promote post-irradiation angiogenesis in a caspase 3–dependent manner. Inhibition of caspase 3 in vitro and in vivo decreased the pro-angiogenic effects of dying glioma cells. The authors identified activation of the NFκB-COX2-PGE2 signaling cascade as the primary driver of caspase activation mediating this post-irradiation response [53]. More recently, we utilized B16F10 melanoma cells stably transduced with shRNA-encoding minigenes targeting caspase 3 to examine the roles of caspase 3 in melanoma radiation responses. Irradiation of in vitro cultures induced a significant amount PGE2 in control cells (Fig. 1a), as expected. However, the induction was abolished in B16F10 shCasp3 caspase 3-deficient cells. Furthermore, when we injected B16F10 shCasp3 cells into syngeneic C57BL/6 mice and exposed the injected tumor volume with 8-Gy X-rays (225 KeV), a significantly longer tumor growth delay was observed compared to the shCasp3 controls (Fig. 1b). These results confirm our earlier breast cancer model results [52••]. Similar findings have also been reported using a bladder cancer model, where Kurtova et al. found that bladder cancer stem cells mediate tumor resistance to chemotherapy by proliferating in response to increased PGE2 production [54•].

Effect of Casp3 downregulation on the in vitro and in vivo post-radiation responses of B16F10 melanoma cells. a B16F10 and B16F10 shCaspase3 cells were either untreated or irradiated with 10 Gy X-ray in vitro. Supernatant from the cells was collected 72 h after radiation. PGE2 concentration in the supernatant was measured following the manufacturer protocol from the ELISA kit from MyBioSource Inc. (San Diego, CA), which was then normalized by cell numbers. b About 1 × 105 cells of either B16F10 WT or shCaspase3 were implanted subcutaneously into the hind legs of C57BL/6 mice. Five days after the tumor implantation, the mice were irradiated with 8 Gy X-ray. Once tumors were palpable, tumor volume was regularly measured with a caliper (*P < 0.05, **P < 0.01, and ***P < 0.001)
Further support for the concept of caspase activation promoting cell survival under stress comes from Khalil et al. who demonstrated that in response to cellular stress and organ damage, caspase 3 activates Akt kinase, an anti-apoptotic molecule. The caspase 3–deficient mouse model showed an increased level of cell death and severe colitis, where the protective mechanism was shown to be mediated through the caspase-3 cleavage of p120 RasGAP protein [55]. In addition, Oficjalska et al. independently provided evidence that mice deficient in caspase 11, an inflammatory caspase, have an increased susceptibility to dextran sodium sulfate treatment with reduced rates of intestinal cell proliferation. The authors attributed the pro-survival role of caspase 11 to its ability to modify levels of cytokines IL-18 and IL-1b [56].
In an earlier study from our lab, we investigated the role of angiogenesis in wild-type or caspase 3–knockout mouse embryonic fibroblasts (MEFs) that were lethally irradiated. After 2 weeks, irradiated WT MEFs had induced significant vascular growth in the host, while caspase 3–knockout MEFs induced minimal host vascular growth [51]. Moreover, Bonner et al. found that paracrine signaling by effector caspases was essential for regeneration of beta cells in the pancreas. They provided evidence that beta cell apoptosis stimulated subsequent regeneration through caspase-dependent apoptosis via the induction of regenerative genes [57]. Additionally, as outlined by Zhao et al., many organisms use this counterintuitive function of caspases for tissue regeneration [58], for example, Xenopus laevis required the activation of caspase 3 to regenerate its tail [59], and decapitated Hydra regenerated its head from growth signals induced by apoptotic cells via caspase-dependent activation of the Wnt3 pathway [60]. Thus, all these studies demonstrate how tissues can tolerate cell death by compensating through proliferation and growth [61], but it can come at the cost of cancer resistance to chemotherapy and radiation therapy.
Caspases and Cancer Metastasis
Metastasis is a dreaded word in the oncology community as it typically confers a dismal survival prognosis. Briefly, it is a process where tumor cells derive the ability to leave their tissue of origin to seed other tissues in the body, often relying on neoangiogenesis to survive. Surprisingly, caspases have been shown to aid in the development of metastasis by promoting cell migration and angiogenesis. From a migratory standpoint, Kang et al. demonstrated that mouse embryos deficient in caspase 8 die from circulatory failure, as caspase 8 is needed for the migration of endothelial cells as well as B lymphocyte progenitors [62]. Caspase 9 can also help cell migration. In a Drosophila model, Geisbrecht et al. demonstrated that caspase 9 is crucial in oocyte development, where it is needed for the movement of border cells in the ovaries [63]. In terms of a cancer-promoting standpoint, a recent study by Rudraptna et al. showed effector caspase activity drives cell invasion without initiating apoptosis, linking caspases to the matrix metalloproteinase 1 (MMP1) and Jnk pathways [64]. Another study by Oh et al. provides evidence that inhibition of death receptor 5 (DR5), a key player in the apoptosis-promoting DISC complex, prevented the formation of the metastasis and invasion signaling complex (MISC). These authors provided direct data that FADD and caspase 8 can recruit TRAF2 and other proteins that eventually result in the activation of MMPs that enables tumor metastasis [65]. Caspase 8 also promotes metastasis by regulating integrin internalization and cell motility [66].
Caspase 3 also has been shown to play a pro-metastatic role as well. Using A549 lung cancer cells, Cheng et al. showed that knocking down caspase 3 decreased metastatic activity in the mouse lung cancer model. The authors concluded that high levels of caspase 3 were correlated with high activity of the extracellular signal regulated kinases (ERK), which is needed for lung cancer metastasis. The authors also showed that overexpression of caspase 3 in MCF7 cells (low malignancy breast cancer cells deficient in caspase 3) increased their metastatic potential [67]. Zhou et al. further supported this notion by showing that colon cancer caspase 3–knockout cells were more sensitive to IR and less invasive. They also showed that the caspase 3–knockouts were less likely to form pulmonary metastases, with increased E-cadherin expression and reduced N-cadherin, Snail, Slug, and ZEB1 expression, which are known markers of metastasis [68]. Gydnia et al. reported that glioblastomas had constant activation of caspases and that caspase inhibition resulted in decreased migration and invasiveness of tumor cells. Their use of siRNA to knockdown caspases 3 and 8 led to a decrease in migration of tumor cells. Furthermore, they showed that the caspase-mediated migration was due to cleavage of the motility-associated gelsolin protein [69]. Lastly, Chang et al. provide a cautionary tale against IAP inhibitor drugs; IAPs are anti-apoptotic as they inhibit caspases and upregulate pro-survival proteins; however, they provide evidence of serious, deadly side effects of these drugs such as bone metastasis [70]. Thus, in light of caspase promoting metastasis, it is possible that IAP inhibition could lead to this side effect.
Targeting Caspases for Therapeutic Benefits
The conventional assumption that activating apoptosis is beneficial in cancer treatment has resulted in a plethora of research looking into drugs that can activate apoptotic caspases. One target is Tp53, as the gene encoding this protein is usually mutated in cancers, without which the intrinsic apoptosis cannot be effectively initiated. However, despite many drugs that have been tested, no Tp53-targeting drug has yet been approved for use in cancer treatment. Another potential target is the inhibitor of IAP—the thought being that if the inhibitors of apoptosis is blocked, more apoptosis will occur. But unfortunately, IAP inhibitors have not yet been able to find their way into the clinic—as outlined above, as such an approach has the potential to increase metastatic potential. Instead, perhaps we should be looking at inhibition of caspases to block their proliferative roles in tumor growth? Caspase inhibitors have been investigated to treat diseases like Alzheimer’s, Huntington’s, and Parkinson’s [71,72,73]. Using a lung cancer xenograft model, Kim et al. studied the effects of caspase 3 inhibition in response to IR using the inhibitor M867. They found that M867 in conjunction with IR decreased the overall survival of lung cancer cells and suggested that caspase inhibition could be used to enhance the effects of IR during radiotherapy [74].
Caspase inhibitors have also been used to regulate the inflammatory response [58]. Flanagan et al. demonstrated that inhibition of caspase 3 reduced the expression of proliferative inflammatory markers in colorectal tumors. Additionally, metastatic CRC patients with a low level of active caspase 3 had increased disease-free survival, especially in patients who received 5-FU chemotherapy [75]. These studies provide the rationale to use caspase inhibitors for the treatment of cancer in the clinic. However, to date, caspase inhibitors have been understudied clinically. Caspases play a diverse role in many proliferation, differentiation and cell death, and pro-survival pathways, where inhibition in one scenario (e.g., cell or tissue type) can be beneficial but detrimental in another. Further studies are needed to better identify cellular and molecular pathways responsible for the regulation of stem cell properties by caspases in order to identify promising pharmacological candidates that target these pathways specifically. We propose that the role of caspases in cancer treatment should be further studied in depth at both preclinical and clinical level in the near future.
Conclusion
Recent studies have dramatically shifted the paradigm of caspases being solely the “angels of cell death.” Many reports demonstrate they also play important roles in promoting carcinogenesis, tumor growth, metastasis, and epigenetic reprogramming. However, despite this evidence, the idea of inhibiting caspases to enhance cancer therapy has yet to be translated into the clinic. It is proposed that with more studies demonstrating the additive or synergistic roles caspase activity modulation may play in existing and emerging cancer treatment modalities, caspase inhibitors may one day be evaluated for clinical treatment of cancers. Such an idea was taboo not too long ago but may eventually prove to be correct and a promising way to treat cancers in the future.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656.
Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274(17):11549–56.
Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999;33:29–55.
Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10(1):26–35.
Zakeri Z, Penaloza CG, Smith K, Ye Y, Lockshin RA. What cell death does in development. Int J Dev Biol. 2015;59(1–3):11–22.
Dyche WJ. A comparative study of the differentiation and involution of the Mullerian duct and Wolffian duct in the male and female fetal mouse. J Morphol. 1979;162(2):175–209.
Zuzarte-Luis V, Hurle JM. Programmed cell death in the developing limb. Int J Dev Biol. 2002;46(7):871–6.
Iversen OH. Cell death in vivo: terminal maturation, necrosis and apoptosis. East Afr Med J. 1996;73(5 Suppl):S5–6.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386(6625):623–7.
Taghiyev AF, Rokhlin OW, Glover RB. Caspase-2-based regulation of the androgen receptor and cell cycle in the prostate cancer cell line LNCaP. Genes Cancer. 2011;2(7):745–52.
King D, Pringle JH, Hutchinson M, Cohen GM. Processing/activation of caspases, -3 and -7 and -8 but not caspase-2, in the induction of apoptosis in B-chronic lymphocytic leukemia cells. Leukemia. 1998;12(10):1553–60.
Liang G, Zhang Y. Embryonic stem cell and induced pluripotent stem cell: an epigenetic perspective. Cell Res. 2013;23(1):49–69.
Baena-Lopez, L.A., et al., Non-apoptotic caspase regulation of stem cell properties. Semin Cell Dev Biol. 2018;82:118-126.
Chagastelles PC, Nardi NB. Biology of stem cells: an overview. Kidney Int Suppl. 2011;1(3):63–7.
Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840(8):2506–19.
Kiefer JC. Primer and interviews: the dynamic stem cell niche. Dev Dyn. 2011;240(3):737–43.
Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132(4):598–611.
Schmidt R, Plath K. The roles of the reprogramming factors Oct4, Sox2 and Klf4 in resetting the somatic cell epigenome during induced pluripotent stem cell generation. Genome Biol. 2012;13(10):251.
Young RA. Control of the embryonic stem cell state. Cell. 2011;144(6):940–54.
Li F, He Z, Shen J, Huang Q, Li W, Liu X, et al. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell. 2010;7(4):508–20.
Fujita J, Crane AM, Souza MK, Dejosez M, Kyba M, Flavell RA, et al. Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell. 2008;2(6):595–601.
Bell RAV, Megeney LA. Evolution of caspase-mediated cell death and differentiation: twins separated at birth. Cell Death Differ. 2017;24(8):1359–68.
Jeong J, Conboy IM. Phosphatidylserine directly and positively regulates fusion of myoblasts into myotubes. Biochem Biophys Res Commun. 2011;414(1):9–13.
Marzesco AM, et al. Release of extracellular membrane particles carrying the stem cell marker prominin-1 (CD133) from neural progenitors and other epithelial cells. J Cell Sci. 2005;118(Pt 13):2849–58.
Hossain MS, Kurokawa K, Sekimizu K. Induction of fusion-competent myoblast-specific gene expression during myogenic differentiation of Drosophila Schneider cells by DNA double-strand breaks or replication inhibition. Biochim Biophys Acta. 2005;1743(1–2):176–86.
Johnstone AP, Williams GT. Role of DNA breaks and ADP-ribosyl transferase activity in eukaryotic differentiation demonstrated in human lymphocytes. Nature. 1982;300(5890):368–70.
Hartley JA, Gibson NW, Zwelling LA, Yuspa SH. Association of DNA strand breaks with accelerated terminal differentiation in mouse epidermal cells exposed to tumor promoters. Cancer Res. 1985;45(10):4864–70.
Khan Z, Francis GE. Contrasting patterns of DNA strand breakage and ADP-ribosylation-dependent DNA ligation during granulocyte and monocyte differentiation. Blood. 1987;69(4):1114–9.
Gunji H, Hass R, Kufe D. Internucleosomal DNA fragmentation during phorbol ester-induced monocytic differentiation and G0/G1 arrest. J Clin Invest. 1992;89(3):954–60.
Ishizaki Y, Jacobson MD, Raff MC. A role for caspases in lens fiber differentiation. J Cell Biol. 1998;140(1):153–8.
Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2002;99(17):11025–30.
Abdul-Ghani M, Megeney LA. Rehabilitation of a contract killer: caspase-3 directs stem cell differentiation. Cell Stem Cell. 2008;2(6):515–6.
Fernando P, Megeney LA. Is caspase-dependent apoptosis only cell differentiation taken to the extreme? FASEB J. 2007;21(1):8–17.
Fernando P, Brunette S, Megeney LA. Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J. 2005;19(12):1671–3.
Janzen V, Fleming HE, Riedt T, Karlsson G, Riese MJ, Lo Celso C, et al. Hematopoietic stem cell responsiveness to exogenous signals is limited by caspase-3. Cell Stem Cell. 2008;2(6):584–94.
Bulatovic I, Ibarra C, Österholm C, Wang H, Beltrán-Rodríguez A, Varas-Godoy M, et al. Sublethal caspase activation promotes generation of cardiomyocytes from embryonic stem cells. PLoS One. 2015;10(3):e0120176.
Cardona M, López JA, Serafín A, Rongvaux A, Inserte J, García-Dorado D, et al. Executioner caspase-3 and 7 deficiency reduces myocyte number in the developing mouse heart. PLoS One. 2015;10(6):e0131411.
• Cartwright, I.M., et al., Essential roles of caspase-3 in facilitating Myc-induced genetic instability and carcinogenesis. Elife, 2017;6:e26371. https://doi.org/10.7554/eLife.26371. This report provides strong evience for the importance of Casp3-mediatd double strand breaks in Myc induced carcinogenesis.
• Ding, A.X., et al., CasExpress reveals widespread and diverse patterns of cell survival of caspase-3 activation during development in vivo. Elife, 2016;5:e10936. https://doi.org/10.7554/eLife.10936. This report elegantly demonstrate cellular surival after Casp3 activation in vivo and implicating its imporatnt roles in cellular differentiation.
Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–38.
Fulda S. Regulation of apoptosis pathways in cancer stem cells. Cancer Lett. 2013;338(1):168–73.
Watabe T, Miyazono K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Res. 2009;19(1):103–15.
Hernandez-Vargas H, et al. Methylome analysis reveals Jak-STAT pathway deregulation in putative breast cancer stem cells. Epigenetics. 2011;6(4):428–39.
Karamboulas C, Ailles L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim Biophys Acta. 2013;1830(2):2481–95.
• Liu X, et al. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res. 2017;27(6):764–83. This report provides evidence for low level leakage of mitochondria, which leads to caspase activation and DNA doule strand breaks, and enhanced tumorigenicity in cancer cells.
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–8.
Sun M, Guo X, Qian X, Wang H, Yang C, Brinkman KL, et al. Activation of the ATM-snail pathway promotes breast cancer metastasis. J Mol Cell Biol. 2012;4(5):304–15.
Liu MJ, Chen LH, Hu KF, Yang XL, Dong JY, Liu J. Study on the effect of overexpression of miR-18a on cellular proliferation and migration by targeting ATM in human colorectal cancer cells. Sichuan Da Xue Xue Bao Yi Xue Ban. 2016;47(4):451–7.
Coppe JP, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
Li F, et al. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3(110):ra13.
•• Huang Q, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17(7):860–6. This report provides strong evidence for a paradoxical role for Casp3 in promoting PGE 2 production and tumor repopulation after radiotherapy.
Feng X, Yu Y, He S, Cheng J, Gong Y, Zhang Z, et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 2017;385:12–20.
• Kurtova AV, et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517(7533):209–13. This report suggests that chemotherapy of bladder induced apoptosis promotes PGE2 secretion from dying cells, which stimulates cancer stem cell activation and tumor resistance to therapy.
Khalil H, Peltzer N, Walicki J, Yang JY, Dubuis G, Gardiol N, et al. Caspase-3 protects stressed organs against cell death. Mol Cell Biol. 2012;32(22):4523–33.
Oficjalska K, Raverdeau M, Aviello G, Wade SC, Hickey A, Sheehan KM, et al. Protective role for caspase-11 during acute experimental murine colitis. J Immunol. 2015;194(3):1252–60.
Bonner C, Bacon S, Concannon CG, Rizvi SR, Baquie M, Farrelly AM, et al. INS-1 cells undergoing caspase-dependent apoptosis enhance the regenerative capacity of neighboring cells. Diabetes. 2010;59(11):2799–808.
Zhao R, Kaakati R, Lee AK, Liu X, Li F, Li CY. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018;37:227–36.
Tseng AS, Adams DS, Qiu D, Koustubhan P, Levin M. Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev Biol. 2007;301(1):62–9.
Chera S, Ghila L, Dobretz K, Wenger Y, Bauer C, Buzgariu W, et al. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell. 2009;17(2):279–89.
Brockes JP, Kumar A. Comparative aspects of animal regeneration. Annu Rev Cell Dev Biol. 2008;24:525–49.
Kang TB, Jeong JS, Yang SH, Kovalenko A, Wallach D. Caspase-8 deficiency in mouse embryos triggers chronic RIPK1-dependent activation of inflammatory genes, independently of RIPK3. Cell Death Differ. 2018;25(6):1107–17.
Geisbrecht ER, Montell DJ. A role for Drosophila IAP1-mediated caspase inhibition in Rac-dependent cell migration. Cell. 2004;118(1):111–25.
Rudrapatna VA, Bangi E, Cagan RL. Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep. 2013;14(2):172–7.
Oh YT, Yue P, Wang D, Tong JS, Chen ZG, Khuri FR, et al. Suppression of death receptor 5 enhances cancer cell invasion and metastasis through activation of caspase-8/TRAF2-mediated signaling. Oncotarget. 2015;6(38):41324–38.
Torres VA, Mielgo A, Barbero S, Hsiao R, Wilkins JA, Stupack DG. Rab5 mediates caspase-8-promoted cell motility and metastasis. Mol Biol Cell. 2010;21(2):369–76.
Cheng YJ, Lee CH, Lin YP, Huang JY, Su CC, Chang WT, et al. Caspase-3 enhances lung metastasis and cell migration in a protease-independent mechanism through the ERK pathway. Int J Cancer. 2008;123(6):1278–85.
Zhou M, Liu X, Li Z, Huang Q, Li F, Li CY. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int J Cancer. 2018;143(4):921–30.
Gdynia G, Grund K, Eckert A, Bock BC, Funke B, Macher-Goeppinger S, et al. Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol Cancer Res. 2007;5(12):1232–40.
Yang C, Novack DV. Anti-cancer IAP antagonists promote bone metastasis: a cautionary tale. J Bone Miner Metab. 2013;31(5):496–506.
Rohn TT, Head E. Caspase activation in Alzheimer’s disease: early to rise and late to bed. Rev Neurosci. 2008;19(6):383–93.
Rideout HJ, Stefanis L. Caspase inhibition: a potential therapeutic strategy in neurological diseases. Histol Histopathol. 2001;16(3):895–908.
Sanchez Mejia RO, Friedlander RM. Caspases in Huntington's disease. Neuroscientist. 2001;7(6):480–9.
Kim KW, Moretti L, Lu B. M867, a novel selective inhibitor of caspase-3 enhances cell death and extends tumor growth delay in irradiated lung cancer models. PLoS One. 2008;3(5):e2275.
Flanagan L, Meyer M, Fay J, Curry S, Bacon O, Duessmann H, et al. Low levels of caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016;7:e2087.
Funding
Work in our laboratory is supported by grants CA208852, CA216876, and ES024015 from the US National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
Data described in this study is approved by the Duke University Institutional Animal Care and Use Committee.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Radiation Biology and Stem Cells
Rights and permissions
About this article

Cite this article
Kaakati, R., Zhao, R., Bao, X. et al. Non-apoptotic Roles of Caspases in Stem Cell Biology, Carcinogenesis, and Radiotherapy. Curr Stem Cell Rep 5, 31–37 (2019). https://doi.org/10.1007/s40778-019-0151-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-019-0151-2