
Abstract
Both immune checkpoint inhibitors and molecularly targeted agents have dramatically improved clinical outcomes for patients with metastatic melanoma. These two therapeutic approaches harness distinct mechanistic pathways—on the one hand, monoclonal antibodies against the immune checkpoints CTLA-4 and PD-1/PD-L1 stimulate the T cell mediated host immune response, while targeted inhibitors of the proto-oncogenes BRAF and MEK disrupt constitutive kinase activity responsible for tumor growth. The prospect of combining these two treatment modalities has been proposed as a potential way to increase overall response rate, extend durability of the anti-tumor response, and circumvent the immune-mediated resistance to targeted therapy. This review explores the preclinical rationale—building upon a wealth of in vitro and in vivo studies—for improved anti-tumor efficacy from combined immune checkpoint inhibition and targeted therapy. In the process, we detail the early clinical trials that have assessed the compatibility of combining these two therapies and the unexpected challenges faced from studies showing increased toxicity from these regimens. Ultimately, with more clinical data expected to mature and accrue in the near future, we elucidate a potentially novel and promising strategy for patients with advanced melanoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
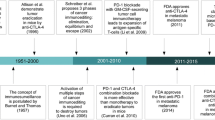
Melanoma is the leading cause of skin cancer mortality and morbidity worldwide, with an estimated 232,000 new cases diagnosed each year and approximately 55,000 deaths annually [1]. Until only recently, the standard first-line treatment for disseminated disease was the alkylating agent dacarbazine, for which there was no proven overall survival (OS) benefit and an objective response rate (ORR) in the 10% range [2]. While a modest survival benefit was observed with high dose IL-2 monotherapy and interferon alfa-2b adjuvant therapy in subgroups of patients, the significant toxicity of these treatments in conjunction with the modest clinical efficacy have limited their clinical utility [3, 4]. However, following the regulatory approval of novel immunomodulatory and molecularly targeted therapies in 2011, a paradigm shift occurred in the clinical management and course of this historically “incurable” disease [5].
The first immune checkpoint inhibitor (ICI) ipilimumab, an antibody directed against the inhibitory surface T cell immune checkpoint receptor cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), demonstrated improved OS compared to standard therapy and, for some melanoma patients, durable clinical benefit without the limiting toxicity previously associated with immunotherapy [6, 7]. The success of this new immune-based treatment led the way for a host of novel agents designed to disrupt the receptor-ligand interaction between tumors and T cell inhibitory regulators [8]. Subsequent studies of ICIs directed at programmed cell death protein 1 (PD-1), a receptor expressed on NK, B, and T cells [9], resulted in further improved anti-tumor activity in melanoma, with both nivolumab and pembrolizumab demonstrating improved ORR and OS in previously treated and untreated patients compared to chemotherapy as well as compared to ipilimumab [10–12]. Furthermore, a trial of combination ipilimumab and nivolumab revealed further improved progression-free survival (PFS) and ORR compared to the monotherapy regimens of ipilimumab or nivolumab [13].
Concurrent with these exciting advances in the realm of immunotherapy, substantial progress in our understanding of the molecular pathways and biological processes of melanoma have led to new genomic-based targeted treatment strategies. After the discovery of activating BRAF V600 mutations—most commonly a valine to glutamic acid substitution at amino acid position 600 (V600E)—present in approximately 50% of melanomas [14], new agents were developed that specifically target the constitutive kinase activity of these oncogene products found to be critical in the initiation of melanocytic neoplasia [9]. Early in vitro studies of BRAF protein kinase inhibition induced apoptosis and tumor regression in BRAF mutant cell lines and tumor specimens and led the way for clinical trials [15]. Follow-up large phase III trials of selective BRAF V600E inhibitors vemurafenib and dabrafenib demonstrated significant clinical benefit in patients with BRAF mutant metastatic melanoma [16, 17]. More recently, combined inhibition of BRAF V600 and mitogen-activated protein kinase (MEK), another key downstream kinase in the RAS-RAF-MEK-ERK mitogen-activated protein kinase (MAPK) cell proliferation pathway, using dabrafenib and trametinib or vemurafenib and cobimetinib have shown superior OS over BRAF inhibition alone and have replaced BRAF directed monotherapy [18, 19].
Collectively, both BRAF V600 kinase inhibitors and PD-1 and/or CTLA-4 blockade have extended the life expectancy of many patients with advanced melanoma; over 70% of patients are alive at 12 months on current standard of care treatment regimens [20]. However, despite improved patient outcomes, each class of therapy has notable shortcomings. Objective tumor responses with combined BRAF/MEK inhibition are in the 70–80% range; however, they are not typically durable [17, 21], with acquired resistance developing in the majority of patients on combination MAPK inhibitor therapy by approximately 12 months [22]. Alternatively, tumor responses achieved with ICIs are more frequently durable [23], but response rates are lower and the onset of responses is slower on average [24]. Notably, recent studies have positioned the MAPK pathway at the intersection of targeted and immunotherapy in melanoma, as demonstrated by evidence of immune-mediated resistance mechanisms to MAPK inhibition on one end and of enhanced immune responses mediated by MEK and BRAF inhibition on the other. Therefore, with the goal of achieving both durable and frequent responses, as well as to prevent the emergence of immune-mediated resistance mechanisms to MAPK inhibition [25], the combination of ICI and kinase-targeted therapy is being assessed as a potential synergistic treatment for advanced BRAF V600 mutant melanoma. In this article, we will review preclinical and clinical studies that have recently brought this critical aspect of melanoma therapy to the forefront.
2 Preclinical and human correlative evidence
There is emerging evidence that MAPK pathway dysregulation in melanoma cells can lead to tumor immune evasion [26]. A number of in vitro and in vivo studies suggest that BRAF and/or MEK inhibition mediate an intratumoral immune milieu that may be more susceptible to the effects of ICIs. While accurate biomarkers of clinical response to checkpoint inhibition remain elusive [27], several features of the tumor microenvironment have been identified that can potentially predict clinical efficacy of ICIs. For example, upregulation of PD-L1 on tumor and/or immune cells [28], high mutational burden and neoantigen load [29], microsatellite instability [30], T cell receptor clonality [9], an adaptive tumoral immune signature [27], and the presence of intratumoral CD8+ lymphocytes [31] have all been associated with improved efficacy to ICIs. In addition, preliminary evidence from early trials of combination radiotherapy and checkpoint inhibition suggests improved anti-tumor activity mediated by the tumoricidal and immunomodulatory effects of tissue radiation [32]. With improved knowledge of the factors that determine tumor responsiveness to ICIs, a thorough understanding of the biological changes induced by targeted therapies and their effects in combination with immunotherapy is critical to evaluating the potential efficacy of combined ICI and targeted therapy.
The following is an overview of the in vitro cell-line and human tumor tissue experiments, as well as the in vivo murine studies that explore immune-features of MAPK-directed therapy and assess the potential for targeted therapy and ICI synergy (schematic of immune-related effects of MAPK inhibition in Table 1).
2.1 Studies of single agent BRAF or MEK inhibition
An early study of BRAF mutant melanoma cell lines exposed to the highly selective MEK inhibitor U0126 or a lentivirus-mediated RNA interference targeting BRAF found significant reductions in cellular mRNA expression of the immunosuppressive cytokines IL-6, IL-10, and VEGF following treatment with either agent [26]. In addition to downregulation of these suppressive factors, inhibition of BRAF in this study also resulted in reversal of the inhibitory effects of TNF-α and IL-12 on monocyte-derived dendritic cell (moDC) maturation. Further investigation into the immune response to BRAF inhibition in melanoma cell lines and human tumor biopsies treated with vemurafenib additionally implicated downregulation of IL-1α/β—proteins capable of impairing T cell functionality through their effects on tumor-associated stromal fibroblast cells—as an important contributor to the immune milieu following targeted therapy [38].
Likewise, changes in the quantity and quality of tumor-infiltrating lymphocytes have been described post-BRAF inhibition. For example, immunohistochemical analysis of tumor tissue from 15 patients with metastatic melanoma who were treated with the BRAF inhibitors dabrafenib or vemurafenib revealed an increase in CD4+ and CD8+ T cells [39]. In this study, the extent of CD8+ tumor influx was inversely correlated with tumor size, highlighting the preserved effector function of these lymphocytes. Moreover, subsequent investigation of response to vemurafenib in a syngenic mouse BRAF V600E-resistant melanoma model revealed an increased intratumoral CD8+ to T regulatory cell ratio, as well as recruitment of natural killer cells and decreased tumor chemokine CCL2 expression post-treatment [33]. Interestingly, sequencing of the complementarity determining region 3 (CDR3) of rearranged T cell receptor (TCR) β chain-coding genes of intratumoral CD8+ lymphocytes from a small cohort of BRAF mutant melanoma patients found efficacy of MAPK inhibitors closely linked to the degree of expansion of pre-existing T cell clones, suggesting a coordinated immune response of antigen-driven T cells underlying BRAF inhibition [35].
In addition to the tumor-specific changes noted above, there has been interest in understanding the systemic immune response to MAPK inhibition. In one study, no significant changes in serum cytokine levels, blood count, or leukocyte subset frequencies were detected before and after either one or two 21-day cycles of dabrafenib in 13 patients with BRAF V600 mutant tumors [34]. However, an increase in TNF-α was noted over the duration of therapy and elevations in circulating CD8+ T cells were seen in some patients. Moreover, a similar study found increases in serum TNF-α, CCL4, and IFN-γ, and a concomitant decrease in CXCL8 after treatment with BRAF inhibition [36]. Reductions in serum CXCL8 levels significantly correlated with tumoral CD8+ infiltration, proliferation marker Ki-67, and decreased OS.
2.2 Studies of combined MEK/BRAF inhibition
Given that combined BRAF MEK inhibition is now the standard treatment for BRAFV600 mutant advanced melanoma (rather than BRAF inhibition alone), understanding the role of the combination on the immune milieu is critical to assessing their synergistic potential with ICIs. An initial pivotal study of BRAF mutant melanoma cell lines and human tumor digests treated with vemurafenib or the two MEK inhibitors U0126 and PD0325901 reported upregulation of melanocyte differentiation antigens (MDAs) in response to either of these agents [40]. Melanocyte differentiation antigens play a key role in T cell tumor sensitization and are vital to ensuring anti-tumor immunity. While T cell function and proliferation in vitro were not negatively impacted by BRAF inhibition, there was noticeable impairment in T cell functionality from MEK inhibition. In a different study on isolated T lymphocytes and moDC, these suppressive in vitro effects were also observed after administration of trametinib, both alone and in combination with BRAF inhibitor dabrafenib [41].
While such in vitro studies would suggest that MEK inhibitors may not be able to effectively synergize with checkpoint inhibitors, other work has shown that these negative effects may be, at most, transient and often difficult to reproduce in vivo. In a study of human CD4+/CD8+ T cells isolated from healthy individuals and stimulated with anti-CD3/anti-CD28 antibodies, exposure to simultaneous and independent administration of dabrafenib and trametinib revealed that only trametinib by itself induced partial and transient inhibition of CD4+ T cell proliferation after 3 days with decreased caspase-3/7 activity for 24 h following T cell activation [37]. Further in vivo analysis of trametinib in the CT26 murine syngenic colon cancer model found that the MEK inhibitor actually reduced immunosuppressive factors (including CCL2, IL-1 β, and TGF-β1) and significantly increased intratumoral CD4+ T cells without compromising their functionality. Moreover, in line with these beneficial in vivo immune effects, dabrafenib and trametinib either alone or in combination had effects on BRAF V600 mutant melanoma cells lines that may render them more susceptible to an attack by immune cells. Specifically, expression of various tumor suppressive factors and molecules (i.e., IL1A, IL8, VEGFA, PD-L1) was reduced and cellular expression of HLA-class I/II molecules, MDAs as well as apoptotic factors was increased.
Another study assessing changes in post-treatment biopsies from 16 patients with BRAF V600 mutant metastatic melanoma who were treated with either vemurafenib or dabrafenib and trametinib documented prominent immune-sensitizing effects [42]. These changes, apparent 10–14 days post-dosing, included decreased levels of the immunosuppressive cytokines IL-6 and IL-8, intratumoral recruitment of CD8+ T cells, and increased MDAs and markers of cytotoxic activity (granzyme B, perforin). Despite these results indicating a beneficial effect on the anti-melanoma immune response, immunohistochemical analysis of T cells also revealed upregulation of the exhaustion markers TIM3, PD1, and PD-L1 potentially suppressing the anti-tumor immune response. Nonetheless, given that PD-L1 upregulation has been associated with improved clinical benefit with PD-1 and PD-L1 inhibition in melanoma [43], it is reasonable to conclude that PD-L1 upregulation by MAPK inhibitors may facilitate a more potent ICI response.
2.3 Studies of MAPK inhibition combined with immunotherapy
The data gathered from experimental models combining immunotherapy and MAPK inhibition have substantially enhanced our understanding of the synergistic potential for these the two therapeutic classes. Early in vivo studies of concomitant BRAF and/or MEK inhibition with adoptive T cell therapy, an immune cell-based approach involving reinfusion of autologous reengineered lymphocytes into lymphodepleted subjects [8], demonstrated combined potentiated efficacy in several trials. In a murine xenograft human BRAF V600E tumor melanoma model engineered to constitutively express gp100/H-2Db recognized by gp100-specific transgenic T cells (pmel-1), vemurafenib lead to increased infiltration of tumors with adoptively transferred T cells and resulted in significant tumor size reduction [44]. The enhanced tumor response was attributed to decreased VEGF production mediated by BRAF inhibition. Similarly, increased numbers of tumor infiltrating lymphocytes (TILs) and significant decrease in tumor size including complete regression was noted in a study of in vivo SM1 murine syngenic BRAF V600E tumors treated with “triple combination” dabrafenib, trametinib, and pmel-1 adoptive cell transfer [45]. In addition, this triple therapy led to upregulation of immune-activating genes (IFN-γ, granzyme B, CD8, etc.) and increased expression of MHC. Interestingly, the addition of trametinib to dabrafenib did not impair the functionality of adoptively transferred pmel-1 cells as assessed by cytokine release and antigen-specific lytic activity in vivo. Furthermore, the addition of trametinib reversed some aspects of suppression in the tumor microenvironment when only single agent dabrafenib was combined with adoptive transfer of pmel-1 T cells, including increased infiltration of T regulatory cells and macrophages. In this study, PD-1 inhibition combined with BRAF and/or MEK inhibition was also more effective. Specifically, dabrafenib, trametinib, and the mouse anti-PD1 antibody DX400 mediated superior anti-tumor activity compared with either therapy separately on SM1 murine melanoma tumor lines in vivo. Another study, in which mouse CT26 colon carcinoma lines were treated with trametinib in combination with PD-1, PD-L1, or CTLA-4 inhibition, also demonstrated superior efficacy compared to single agent therapy [37]. The addition of trametinib to anti-PD-1 therapy leads to increased tumor infiltration with CD8+ cells. Notably, in these in vitro studies, sequential treatment of trametinib followed by PD-1 inhibition demonstrated superior efficacy compared to PD-1 inhibition followed by trametinib, suggesting that MEK inhibitors may prime the immune environment to enhance ICIs. Furthermore, an in vivo study of the effects of anti-CTLA-4 therapy combined with the pan-RAF inhibitor BMS908662 on the CT26 and SA1N murine sygnenic, BRAF wildtype tumor strain found that this combination led to a more significant reduction in tumor growth in both tumor subsets [46]. The rationale for this improved efficacy was paradoxical activation of T cells in the tumor microenvironment that utilize RAS-activation and can be manipulated by BMS908662.
While the majority of preclinical studies have identified beneficial effects of MAPK-directed therapy and synergistic effects with immunotherapy, some have not. The combination of CTLA-4 inhibition and vemurafenib in a murine melanoma model with induced primary tumors deficient in PTEN expression and also positive for the BRAF V600E mutation did not lead to improvements in survival or tumor growth [47]. In this study, TIL frequencies decreased post-BRAF inhibition, which is contrary to what has been reported in most other studies and in contrast to another in vivo study of a similar BRAF V600E, PTEN deficient subcutaneous implantable murine melanoma model, which found PD-1 or PD-L1 inhibitors to be synergistic with vemurafenib as evident by increased TIL, improved response and survival [48]. In particular, CD3+ T cells in the tumor increased at least 7.5-fold in combination over monotherapy, and CD8+ T cells and CD8+/Treg ratio increased significantly as well. Additionally, CD8+ cells expressed significantly greater amounts of granzyme B, IFN-γ, and TNFα in mice receiving both therapies.
3 Clinical studies
Early phase clinical trials combining ICI and BRAF targeted therapy revealed unexpected toxicity associated with this novel combination. In a phase 1 dose safety study, patients with previously treated BRAF V600E metastatic melanoma received either 960 or 720 mg of vemurafenib twice daily for a month followed by four concurrent infusions of ipilimumab (3 mg/kg every 3 weeks) and daily vemurafenib [49]. Within the first 2 to 5 weeks following the first infusion of ipilimumab, 6 of the initial 10 patients developed grade 3 transaminitis (reversible with either drug discontinuation or glucocorticoid therapy) leading to discontinuation of the study.
While the etiology for this hepatotoxicity has not been fully elucidated, hepatotoxicity has not been consistently seen with the combination of vemurafenib and ipilimumab. For example, in a case series of 10 patients with advanced stage melanoma receiving combination vemurafenib and ipilimumab upon clinical response to vemurafenib, asymptomatic, reversible grade >3 transaminitis was found in only 2 patients [50]. In addition to vemurafenib and ipilimumab, an early dose-expansion safety study explored the safety of combination dabrafenib and/or trametinib with ipilimumab in BRAF V600 metastatic melanoma patients [51]. In one study arm, four patients underwent a 2-week run-in period of dabrafenib (150 mg BID) followed by concurrent treatment with dabrafenib and ipilimumab (3 mg/kg every 3 weeks for four doses). Among these patients, no grade 3/4 transaminitis or dose-limiting toxicities were observed. Two patients discontinued treatment due to disease progression and two remained on therapy at data cutoff. In another study arm, patients received 2-week run-ins of dabrafenib (100 mg BID) and trametinib (1 mg QD) followed by ipilimumab. Two of the first seven patients enrolled, however, developed grade 3 colitis associated with colonic perforation soon after initiation of ipilimumab. Patient accrual was subsequently suspended. Follow-up pathology of resected bowel in one patient showed ipilimumab-induced colitis [52].
The recognition of substantial toxicity with combined CTLA-4 blockade and BRAF/MEK inhibition and the availability of PD-1/PD-L1 inhibitors lead to clinical trials exploring BRAF/MEK inhibition in combination with PD-1 pathway blockade. In a phase 1 study, the safety of adding the PD-L1 inhibitor MEDI4736 to dabrafenib and/or trametinib in patients with BRAF mutant and wildtype metastatic melanoma was investigated [53]. In this study, 26 patients received triple combination dabrafenib (150 mg BID), trametinib (2 mg qD), and MEDI4736 (3 or 10 mg/kg IV every 2 weeks); 20 patients received combination trametinib and MEDI4736 and 19 received a sequential 4-week run-in of trametinib followed by 2 weeks of trametinib and MEDI4736 and then MED14736 monotherapy until disease progression. At 16 weeks after the initial dose, ORR for the three groups were 69, 21, and 13%, respectively, and disease control rates (including complete/partial response and stable disease) were 100, 79, and 80%, respectively. Even at 50 weeks follow-up, 90% of patients on triple combination dabrafenib, trametinib, and MEDI4736 continued to derive clinical benefit. Additionally, no significantly increased toxicity or adverse immuno-related events were reported and the treatment appeared to be well tolerated.
Results from a phase 1B dose-escalation study of combination vemurafenib and PD-L1 inhibitor atezolizumab were presented at the Society for Melanoma Research meeting in 2015 [54]. In this study, three patients received concurrent vemurafenib (720 mg BID) and atezolizumab (20 or 15 mg/kg or 1200 mg fixed intravenously every 3 weeks). In addition, 14 patients were treated with this combination therapy after a run-in period of vemurafenib (960 mg BID) for either 28 (six patients) or 56 days (eight patients). Patients without a run-in of vemurafenib had an ORR of 33% with one complete response noted. On the other hand, for those with a run-in of either 28 or 56 days with vemurafenib, ORR was 100 and 75%, respectively. For the entire cohort, ORR was 76% (95% CI, 50.1 to 93.2%) with a median duration of response of 20.9 months and a median PFS of 10.9 months. Overall, this combination was well tolerated with no dose-limiting toxicities or atezolizumab-related treatment discontinuations.
The anti-PD-1 antibody pembrolizumab is currently being tested in combination with dabrafenib and trametinib in advanced BRAFV600 mutant melanoma patients. In the phase 1 dose escalation part, the toxicity was found to be manageable and included grade 4 neutropenia and grade 4 elevated transaminases that occurred in 3 of the first 15 patients, all of which resolved. The study has therefore moved to phase 2; all three drugs are given at the approved standard of care doses (Ribas ASCO 2016). Initial efficacy and safety data were also reported on a phase 1b trial combining atezolizumab, cobimetinib, and trametinib. About 40% of the first 30 patients treated on the study experienced grade ¾ toxicities, most of which were attributed to atezolizumab and resolved with dose interruptions and or reductions. The overall response rate was 83% and only one patient experienced primary progressive disease (Sullivan, SMR 2016).
While much of the data on these combination regimens is relatively early, several studies are underway to further our understanding of this combination treatment approach (Table 2). As new combinations are proposed, it is important to understand not only the correct dosing of therapies but the optimal clinical sequence of BRAF/MEK inhibitors and ICIs. At this time, data from many trials suggest no significant difference in PFS or response rates depending on the order in which ICIs or BRAF/MEK inhibitors are given [55, 56]. A recent retrospective analysis of 114 patients with BRAF V600 mutant melanoma found a similar median OS irrespective of whether patients received a BRAF and/or MEK inhibitor or an anti-PD-1 agent first [57]. However, clinical outcome to front-line therapy was superior and those who benefited from BRAF inhibition for greater than 6 months showed significantly higher response rates to anti-PD-1 therapy. Alternatively, other studies suggest that immunotherapy before targeted therapy may be the preferred sequence due to the potential in about 40% for rapid progression from BRAF inhibition that may hamper alternative therapy efforts [58]. Nonetheless, only through randomized clinical trials, like NCT02224781 comparing ipilimumab and nivolumab with dabrafenib and trametinib with crossover at progression, will more definitive answers emerge to this question.
Additionally, since specific molecular subclasses of melanoma have differing responses to immunotherapy—for example, NRAS mutant melanoma may have a better response to PD-1/PD-L1 inhibition than BRAF or wildtype tumors [59]—more investigation is needed into the interplay of genomics and the immune response to combination therapy. Moreover, given the potential for acquired resistance to both MAPK inhibition [59] and immune checkpoint inhibition [60], downstream mutational changes may create an evolving immune milieu as new genetic changes occur with time. Also, since only patients that harbor a V600E mutation can benefit from vemurafenib and/or dabrafenib, alternative molecular-targeted agents—such as those targeting transcriptional regulators important for melanoma progression [61] or driver pathways of resistance implicated such as the PI3K/AKT/mTOR [15]—should be tested in combination with immunotherapy as well. Moreover, as more clinical trials ensue, it is critical to optimize biomarkers to help design rational combination therapies and help with patient stratification and treatment selection. As more data from clinical trials becomes available, our knowledge in this important area of melanoma investigation will aid future efforts for optimal synergistic combinations.
4 Conclusion
As new strategies emerge to extend survival benefit in metastatic melanoma patients, the combination of both targeted therapy and ICIs offers the potential for a both durable and potent anti-tumor response. Preclinical studies have demonstrated favorable effects of MAPK-directed therapy on the anti-tumor immune response, providing a rationale for combining these therapies with ICIs. Immune-modulating effects of MAPK inhibition include increased TILs, upregulated expression of melanoma tumor antigens and pro-apoptotic factors, and decreased immunosuppressive cytokines. These changes may account for the improved survival and enhanced anti-tumor response of in vivo studies that combine immunotherapy with BRAF/MEK inhibition. Initial clinical studies of ipilimumab with either vemurafenib or dabrafenib and trametinib have exposed some unexpected toxicity with combination ICI and MEK/BRAF inhibition. However, recent BRAF/MEK inhibition plus ICI combination studies demonstrated a safe toxicity profile and promising clinical activity. Future studies with novel combinations and extended follow-through of recent studies are needed to better evaluate this potential treatment strategy.
References
Ferlay, J., Soerjomataram, I., Dikshit, R., et al. (2015). Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International Journal of Cancer, 136, E359–E386.
Eggermont, A. M., & Kirkwood, J. M. (2004). Re-evaluating the role of dacarbazine in metastatic melanoma: what have we learned in 30 years? European Journal of Cancer, 40, 1825–1836.
Davey, R. J., van der Westhuizen, A., & Bowden, N. A. (2016). Metastatic melanoma treatment: combining old and new therapies. Critical Reviews in Oncology/Hematology, 98, 242–253.
Malczewski, A., Marshall, A., Payne, M. J., et al. (2016). Intravenous high-dose interferon with or without maintenance treatment in melanoma at high risk of recurrence: meta-analysis of three trials. Cancer Medicine, 5, 17–23.
Girotti, M. R., Saturno, G., Lorigan, P., & Marais, R. (2014). No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Molecular Oncology, 8, 1140–1158.
Hodi, F. S., O'Day, S. J., McDermott, D. F., et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. The New England Journal of Medicine, 363, 711–723.
Robert, C., Thomas, L., Bondarenko, I., et al. (2011). Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med cdex, 364, 2517–2526.
Rotte, A., Bhandaru, M., Zhou, Y., & McElwee, K. J. (2015). Immunotherapy of melanoma: present options and future promises. Cancer Metastasis Reviews, 34, 115–128.
Khagi, Y., Kurzrock, R., & Patel, S. P. (2016). Cancer Metastasis Reviews. doi:10.1007/s10555-016-9652-y.
Weber, J. S., D'Angelo, S. P., Minor, D., et al. (2015). Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. The Lancet Oncology, 16, 375–384.
Robert, C., Long, G. V., Brady, B., et al. (2015). Nivolumab in previously untreated melanoma without BRAF mutation. The New England Journal of Medicine, 372, 320–330.
Robert, C., Schachter, J., Long, G. V., et al. (2015). Pembrolizumab versus ipilimumab in advanced melanoma. The New England Journal of Medicine, 372, 2521–2532.
Larkin, J., Chiarion-Sileni, V., Gonzalez, R., et al. (2015). Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. The New England Journal of Medicine, 373, 23–34.
Davies, H., Bignell, G. R., Cox, C., et al. (2002). Mutations of the BRAF gene in human cancer. Nature, 417, 949–954.
Cheng, Y., Zhang, G., & Li, G. (2013). Targeting MAPK pathway in melanoma therapy. Cancer Metastasis Reviews, 32, 567–584.
McArthur, G. A., Chapman, P. B., Robert, C., et al. (2014). Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. The Lancet Oncology, 15, 323–332.
Hauschild, A., Grob, J. J., Demidov, L. V., et al. (2012). Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet, 380, 358–365.
Larkin, J., Ascierto, P. A., Dreno, B., et al. (2014). Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. The New England Journal of Medicine, 371, 1867–1876.
Robert, C., Karaszewska, B., Schachter, J., et al. (2015). Improved overall survival in melanoma with combined dabrafenib and trametinib. The New England Journal of Medicine, 372, 30–39.
Ugurel, S., Rohmel, J., Ascierto, P. A., et al. (2016). Survival of patients with advanced metastatic melanoma: the impact of novel therapies. European Journal of Cancer, 53, 125–134.
Long, G. V., Stroyakovskiy, D., Gogas, H., et al. (2015). Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet, 386, 444–451.
Welsh, S. J., Rizos, H., Scolyer, R. A., & Long, G. V. (2016). Resistance to combination BRAF and MEK inhibition in metastatic melanoma: where to next? European Journal of Cancer, 62, 76–85.
Schadendorf, D., Hodi, F. S., Robert, C., et al. (2015). Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. Journal of Clinical Oncology, 33, 1889–1894.
Buzaid, A. C., Agarwala, S. S., Hauschild, A., & Atkins, M. (2014). Algorithm for the management of metastatic cutaneous melanoma. Chinese Clinical Oncology, 3, 32,3865.2014.07.01.
Coffelt, S. B., & de Visser, K. E. (2015). Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends in Immunology, 36, 198–216.
Sumimoto, H., Imabayashi, F., Iwata, T., & Kawakami, Y. (2006). The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. The Journal of Experimental Medicine, 203, 1651–1656.
Chen PL, Roh W, Reuben A, et al. (2016). Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov.
Festino, L., Botti, G., Lorigan, P., et al. (2016). Cancer treatment with anti-PD-1/PD-L1 agents: is PD-L1 expression a biomarker for patient selection? Drugs, 76, 925–945.
McGranahan, N., Furness, A. J., Rosenthal, R., et al. (2016). Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science, 351, 1463–1469.
Hermel, D., & Sigal, D. (2016). “Check”-ing the data: a review of immune checkpoint inhibitor biomarkers. Personalized Medicine in Oncology., 5(6), 234–240.
Tumeh, P. C., Harview, C. L., Yearley, J. H., et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 515, 568–571.
Salama, A. K., Postow, M. A., & Salama, J. K. (2016). Irradiation and immunotherapy: from concept to the clinic. Cancer, 122, 1659–1671.
Knight, D. A., Ngiow, S. F., Li, M., et al. (2013). Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. The Journal of Clinical Investigation, 123, 1371–1381.
Hong, D. S., Vence, L., Falchook, G., et al. (2012). BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clinical Cancer Research, 18, 2326–2335.
Cooper, Z. A., Frederick, D. T., Juneja, V. R., et al. (2013). BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology, 2, e26615.
Wilmott, J. S., Haydu, L. E., Menzies, A. M., et al. (2014). Dynamics of chemokine, cytokine, and growth factor serum levels in BRAF-mutant melanoma patients during BRAF inhibitor treatment. Journal of Immunology, 192, 2505–2513.
Liu, L., Mayes, P. A., Eastman, S., et al. (2015). The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clinical Cancer Research, 21, 1639–1651.
Khalili, J. S., Liu, S., Rodriguez-Cruz, T. G., et al. (2012). Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clinical Cancer Research, 18, 5329–5340.
Wilmott, J. S., Long, G. V., Howle, J. R., et al. (2012). Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clinical Cancer Research, 18, 1386–1394.
Boni, A., Cogdill, A. P., Dang, P., et al. (2010). Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Research, 70, 5213–5219.
Vella, L. J., Pasam, A., Dimopoulos, N., et al. (2014). MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunology Research, 2, 351–360.
Frederick, D. T., Piris, A., Cogdill, A. P., et al. (2013). BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical Cancer Research, 19, 1225–1231.
Manson, G., Norwood, J., Marabelle, A., Kohrt, H., & Houot, R. (2016). Biomarkers associated with checkpoint inhibitors. Annals of Oncology, 27, 1199–1206.
Liu, C., Peng, W., Xu, C., et al. (2013). BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clinical Cancer Research, 19, 393–403.
Hu-Lieskovan, S., Mok, S., Homet Moreno, B., et al. (2015). Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Science Translational Medicine, 7, 279ra41.
Callahan, M. K., Masters, G., Pratilas, C. A., et al. (2014). Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunology Research, 2, 70–79.
Hooijkaas, A., Gadiot, J., Morrow, M., Stewart, R., Schumacher, T., & Blank, C. U. (2012). Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology, 1, 609–617.
Cooper, Z. A., Juneja, V. R., Sage, P. T., et al. (2014). Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunology Research, 2, 643.
Ribas, A., Hodi, F. S., Callahan, M., Konto, C., & Wolchok, J. (2013). Hepatotoxicity with combination of vemurafenib and ipilimumab. The New England Journal of Medicine, 368, 1365–1366.
Hassel, J. C., Lee, S. B., Meiss, F., et al. (2016). Vemurafenib and ipilimumab: a promising combination? Results of a case series. Oncoimmunology, 5, e1101207.
Puzanov, I. (2015). Combining targeted and immunotherapy: BRAF inhibitor dabrafenib (D) ± the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation-positive unresectable or metastatic melanoma (MM). Journal of Translational Medicine., 13(Suppl 1), K8. doi:10.1186/1479-5876-13-S1-K8.
Gonzalez-Cao, M., Boada, A., Teixidó, C., Fernandez-Figueras, M., et al. (2016). Fatal gastrointestinal toxicity with ipilimumab after BRAF/MEK inhibitor combination in a melanoma patient achieving pathological complete response. Oncotarget, 7(35), 56619–56627.
Ribas, A., Butler, M., Lutzky, J., et al. (2015). Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. Journal Clinical Oncology, 33, 3003.
Hamid O, et al. (2015). Preliminary clinical safety, tolerability and activity of atezolizumab (anti-PD-L1) combined with Zelboraf in BRAFv600 metastatic melanoma (pp. 18–21). Presented at the Society for Melanoma Research 2015 International Congress; San Francisco, CA.
Ackerman, A., Klein, O., McDermott, D. F., et al. (2014). Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer, 120, 1695–1701.
Aya F, Fernandez-Martinez A, Gaba L, et al. (2016). Sequential treatment with immunotherapy and BRAF inhibitors in BRAF-mutant advanced melanoma. Clin Transl Oncol.
Johnson, D. B., Pectasides, E., Feld, E., et al. (2017). Sequencing treatment in BRAFV600 mutant melanoma: anti-PD-1 before and after BRAF inhibition. J Immunotherapy, 40(1), 31–35.
Ascierto, P. A., & Margolin, K. (2014). Ipilimumab before BRAF inhibitor treatment may be more beneficial than vice versa for the majority of patients with advanced melanoma. Cancer, 120, 1617–1619.
Timar, J., Vizkeleti, L., Doma, V., Barbai, T., & Raso, E. (2016). Genetic progression of malignant melanoma. Cancer Metastasis Reviews, 35, 93–107.
O’Donnell, J. S., Long, G. V., Scolyer, R. A., et al. (2016). Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treatment Reviews, 52, 71–81.
Mobley, A. K., Braeuer, R. R., Kamiya, T., Shoshan, E., & Bar-Eli, M. (2012). Driving transcriptional regulators in melanoma metastasis. Cancer Metastasis Reviews, 31, 621–632.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original version of this article was revised: The middle initial of Patrick A. Ott was not captured.
An erratum to this article is available at http://dx.doi.org/10.1007/s10555-017-9665-1.
Rights and permissions
About this article

Cite this article
Hermel, D.J., Ott, P.A. Combining forces: the promise and peril of synergistic immune checkpoint blockade and targeted therapy in metastatic melanoma. Cancer Metastasis Rev 36, 43–50 (2017). https://doi.org/10.1007/s10555-017-9656-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-017-9656-2