
Abstract
The progression of melanoma toward the metastatic phenotype occurs in a defined stepwise manner. While many molecular changes take place early in melanoma development, progression toward the malignant phenotype, most notably during the transition from the radial growth phase (RGP) to the vertical growth phase (VGP) involves deregulated expression of several transcription factors. For example, the switch from RGP to VGP is associated with the loss of the transcription factor AP2α and gain of transcriptional activity of cAMP-responsive element binding protein. Together with the upregulation of microphthalmia-associated transcription factor, activating transcription factor 2, nuclear factor kappa B, and other transcription factors, these changes lead to dysregulated expression or function of important cellular adhesion molecules, matrix degrading enzymes, survival factors, as well as other factors leading to metastatic melanoma. Additionally, recent evidence suggests that microRNAs and RNA editing machinery influence the expression of transcription factors or are regulated themselves by transcription factors. Many of the downstream signaling molecules regulated by transcription factors, such as protease activated receptor-1, interleukin-8, and MCAM/MUC18 represent new treatment prospects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Malignant melanoma is a highly aggressive type of skin cancer for which a successful treatment has not yet been established, albeit some success with V600E BRAF inhibitors. Treatment of metastatic melanoma is a challenge, as most therapeutic agents have failed to demonstrate an improved survival rate in phase III trials. Therefore, it is important to determine the molecular events leading to melanoma metastasis to identify novel therapeutic molecular targets to cure the disease. For the last four decades, the incidence of malignant melanoma continues to rise. In the USA, melanoma is the fifth and sixth most common cancer in men and women, respectively [1]. In the year 2012, it is estimated that more than 76,000 new cases will be diagnosed, and the estimated number of deaths from melanoma will be more than 9,000 patients [1]. When melanoma is detected early (stage I), patients have a 97 % 5-year survival rate after surgical removal of nonulcerated, thin (<1 mm) primary melanomas [2]. On the other hand, advanced melanoma patients, with regional lymph nodes or metastasis in other organs, have a 5-year survival rate of <10 % [2].
Melanoma has become an excellent model for identifying molecular changes that are associated with the metastatic phenotype as the disease progresses through five very defined sequential steps based on clinical and histopathological features [3]. These steps include a variety of genetic and epigenetic changes as well as changes in microenvironmental factors [4]. First, genetic alterations in normal melanocytes, such as BRAF mutations, lead to transformation into benign nevi [5]. Benign nevi are defined by having normal melanocytic morphology and are characterized by an initial phase of melanocytic proliferation in the basal layer of the epidermis. Next, dysplastic nevi begin to show structural and architectural atypia, brought on by NRAS mutations or loss of PTEN [5]. Further molecular alterations lead to the third step, which is characterized by growth and spread horizontally through the epidermis by the premalignant lesions. This step is termed the radial growth phase (RGP) [6]. A small population of cells acquire the necessary molecular changes, such as increased extracellular matrix degrading enzymes and loss of adhesive molecules, needed to invade vertically through the dermis into the subcutaneous tissue, termed the vertical growth phase (VGP) [5, 7]. The transition from the radial growth phase to the vertical growth phase is the key step in the progression to metastatic melanoma, which subsequently leads to poor clinical outcome [8]. Finally, only if melanoma cells have gained the ability to survive in the circulation, arrest in the capillary bed of a distant organ, enter the parenchyma, grow within the organ microenvironment, and further initiate angiogenesis, a metastatic lesion can be formed [9]. Common metastatic sites include the lymph nodes, lung, liver, and brain [10, 11].
Transcriptional regulation of the genes, which lead to enhanced invasiveness, mobility, and homing of melanoma cells to distant organs, is altered during these progressive steps [12]. The deregulation of transcription factors during the transition from RGP to VGP, along with their downstream target genes, can regulate the metastatic phenotype of melanoma. In this review, we will summarize the data on the roles of the transcription factors that play major roles in the acquisition of the malignant melanoma phenotype.
2 Activator protein-2α
The transcription factor activator protein-2α (AP2α) is a 52-kD protein regulated by retinoic acid and cyclic AMP [13, 14]. The gene can be alternatively spliced to form the AP2B isoform, which acts as a dominant negative, inhibiting AP2α transcriptional activity [15]. AP2α binds to the consensus DNA-binding motif, CCCCAGGC, and regulates a variety of cell processes, including cell growth, apoptosis, and differentiation both in embryonic development and in adult homeostasis [16–19]. In many tumors, including melanoma, AP2α acts as a tumor suppressor through enhanced expression of p21WAF1/Cip1 and stimulation of p53-dependent transcription, thereby inducing cell cycle arrest and apoptosis [20–22]. We have shown through progressive tissue microarray analysis from a cohort of melanoma patients that a high cytoplasmic to nuclear expression ratio of AP2α correlates with a poor patient prognosis and that loss of expression of nuclear AP2α is connected to melanoma progression [23–25]. A strong inverse correlation between loss of AP2α and upregulation of cAMP-responsive element binding (CREB) in melanoma progression has also been observed, leading our group to identify a mechanism in which CREB binds to the promoter region of AP2α, directly repressing its transcriptional activity during the transition to the metastatic phenotype [26].
Loss of AP2α has been shown to play a key role in progression from the radial growth phase to the vertical growth phase in melanoma (Fig. 1). Re-expression of AP2α in highly metastatic melanoma cell lines significantly reduced tumor growth and decreased experimental lung metastasis in vivo [27]. Conversely, inactivation of AP2α in nonmetastatic melanoma cell lines using the dominant negative AP-2B isoform increased tumorigenicity in mice [28]. AP2α repression has been linked to several genes, including the loss of c-KIT expression as well as the gain of the melanoma cell adhesion molecule (MCAM/MUC18), protease activated receptor-1 (PAR-1), and vascular endothelial growth factor (VEGF) leading to dysregulation of cell proliferation, cell cycle, and apoptosis as well as enhanced tumor cell adhesion and invasion, and increased angiogenesis [23, 27, 29–32].

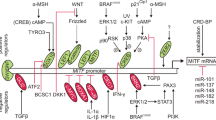
Molecular changes associated with the transition from RGP to VGP. a In primary melanomas, AP2α directly inhibits the expression of multiple genes such as PAR-1, BCL-2, and VEGF, which contribute to melanoma progression. Additionally, AP2α drives the expression of genes such as p21 and c-KIT. As in the case of PAR-1, AP2α directly competes with SP1, and for VEGF, AP2α competes with SP3 to suppress their expression. b During the transition from RGP to VGP, the activation of CREB, through its phosphorylation at ser133, transcriptionally reduces AP2α expression, thereby affecting the expression of genes regulated by AP2α. Repression of AP2α leads to PAR-1 expression, which in turn enhances the expression of PAFR and connexin-43. PAR-1 also reduces the expression of the tumor suppressor Maspin. CREB itself transcriptionally regulates several genes such as MCAM/MUC18, MITF, and CYR61. Since CREB functions upstream of AP2α, it may act as the master switch in melanoma progression. Additionally, NF-κB activation leads to the upregulation of proinflammatory genes such as IL-6. Phosphorylation of ATF-2 by PKCε causes nuclear translocation, which is associated with melanoma progression. Collectively, these events lead to increased tumor migration, invasion, and angiogenesis, leading to increased tumor growth and metastasis
The tyrosine kinase receptor, c-KIT, plays an important role in normal growth and differentiation of embryonic melanoblasts. However, approximately 70 % of metastatic melanoma lesions do not express c-KIT. Re-expression of c-KIT in highly metastatic melanoma cells resulted in decreased tumor growth and metastasis, indicating the importance of c-KIT in melanoma metastasis [33]. Promoter analysis of the c-KIT gene identified three putative binding sites for AP2α [34]. Additionally, re-expression of AP2α in highly metastatic cell lines leads to increased expression of c-KIT, indicating a direct effect on c-KIT expression through AP2α [27].
The adhesion molecule MCAM/MUC18 is over expressed in metastatic melanoma. Its expression is commonly used as an independent prognostic indicator for decreased patient survival [35]. MCAM/MUC18 functions to mediate homotypic adhesion between melanoma cells, as well as heterotypic adhesion between melanoma cells and endothelial cells during tumor progression [36, 37]. The introduction of MCAM/MUC18 in primary melanoma cells led to a highly tumorigenic and metastatic phenotype in nude mice [38]. This phenotype included an increase in adhesion between melanoma cells, increased binding to endothelial cells, decreased ability to adhere to laminin, and increased matrix metallopeptidase 2 (MMP2) activity [38, 39]. Promoter analysis of the MCAM/MUC18 gene revealed four AP2α binding sites. Our group has established that AP2α binds directly to the MCAM/MUC18 promoter and inhibits its transcription [30]. Additionally, when AP2α is expressed in highly metastatic melanoma cells, a significant decrease in MCAM/MUC18 expression was observed, further establishing the regulation of MCAM/MUC18 by AP2α [30]. These data indicate that the loss of AP2α during melanoma progression results in increased MCAM/MUC18 expression.
Another gene that is upregulated in the transition from RGP to VGP is the G-protein-coupled receptor, protease activated receptor-1 (PAR-1) [23, 29]. Interestingly, there are two AP2α/SP1 binding complexes in the promoter region of PAR-1. In nonmetastatic melanoma cell lines, AP2α binds to the PAR-1 promoter and inhibits PAR-1 transcription. However, in highly metastatic melanoma cells, AP2α no longer competitively inhibits the binding of SP1 to the promoter, which allows for SP1-driven transcription of PAR-1 [5, 29].
PAR-1 is activated through cleavage at the N-terminal domain by thrombin, thus allowing the new N-terminal end, acting as a tethered ligand, to bind to the body of the receptor and induce downstream signaling events. These events include activation of RAS, PI3K, and MAP kinase pathways, which are involved in cell growth and tumor promotion [40–44]. We studied the role of PAR-1 in melanoma by silencing its expression in highly metastatic cell lines. After PAR-1 silencing, in vivo studies revealed a significant reduction in tumor growth and metastatic potential [45]. PAR-1 induces several genes, which are important for cell adhesion, invasion, and angiogenesis; these include αvβ3 and αvβ5 integrins, MMP2, urokinase-type plasminogen activator, platelet-derived growth factor, interleukin-8 (IL-8), VEGF, and basic fibroblast growth factor [29, 44, 46, 47].
Recently, we have shown that another G-protein-coupled receptor, platelet-activating factor receptor (PAFR) is regulated by PAR-1 [48]. PAFR is overexpressed during melanoma progression and stimulation by its ligand, platelet-activating factor (PAF), plays an important role in stimulating endothelial cells and angiogenesis. Signaling through PAFR leads to upregulation of several inflammatory molecules, including IL-6, IL-8, IL-10, COX2, and VEGF [40]. Inhibition of PAFR with the antagonist PCA4248 leads to growth inhibition of highly metastatic melanoma cell lines in vitro and an IP injection of PAF into murine B16F10 melanoma tumors enhanced pulmonary metastasis, while a PAFR antagonist decreased lung colonization [49]. Interestingly, treatment with the nonhydrolyzable PAF analog (cPAF) in the A375SM cell line resulted in increased phosphorylation of the CREB transcription factor [50]. Additionally, cPAF upregulated the invasive molecules MMP2 and MT1-MMP [50, 51]. These data indicate the importance of secreted molecules in the tumor microenvironment, such as PAF, in melanoma progression. Both PAF and thrombin are abundant within the tumor microenvironment. Since both receptors, PAR-1 and PAFR, are over expressed in metastatic melanoma cells, these cells are better equipped to respond to these ligands and use them for their growth advantage.
Complementary DNA microarray analysis comparing PAR-1-silenced cells to nontargeting cells revealed differential expression of several novel downstream target genes regulated by PAR-1 (Fig. 1b). Among these differentially expressed genes are connexin-43 and the tumor suppressor gene Maspin [48, 52]. The gap junction protein connexin-43 is downregulated following silencing of PAR-1 in metastatic melanoma cells [52]. Connexin-43 has been reported to be expressed in several tumor types, including melanoma, and has been shown to mediate tumor cell adhesion to endothelial cells and extravasation into the site of metastasis [52–54]. Decreased invasive potential through matrigel was observed in metastatic melanoma cells after connexin-43 silencing [52]. Our group has shown that expression of connexin-43 is transcriptionally regulated through binding of SP1, c-Fos, and c-Jun to the connexin-43 promoter. Our data show that PAR-1 signaling increases the binding of these transcription factors to the connexin-43 promoter resulting in its over expression [52].
The tumor suppressor Maspin was increased following PAR-1 silencing in our gene expression analysis by ∼46-fold. This observation was further validated at the protein levels. In highly metastatic melanoma cell lines, very little Maspin is expressed, indicating a loss of its tumor suppressive function. At the transcriptional level, Maspin is regulated by Ets-1 and c-Jun after PAR-1 silencing. However, PAR-1 had no effect on Ets-1 or c-Jun expression, but rather increased expression of CBP-300 and decreased activity of p38, leading to increased binding of Ets-1 and c-Jun to the Maspin promoter [55]. Expression of Maspin in these cell lines reduced tumor growth and metastasis in nude mice. Additionally, silencing Maspin in PAR-1-silenced cells reverted the inhibition of tumor growth and metastasis observed in PAR-1-silenced cells [55]. These data indicate that PAR-1 negatively regulates Maspin during melanoma progression. These downstream events, which enhance melanoma metastasis, however, only occur following the loss of the AP2α transcription factor.
3 cAMP-responsive element binding/activating transcription factor-1
The progression of melanoma from RGP to VGP leads to the upregulation of activating transcription factor-1 (ATF-1) and cAMP-responsive element (CRE)-binding protein. Both ATF-1 and CREB are known to be involved in cAMP and Ca2+-induced signaling [56, 57]. CREB belongs to the bZIP superfamily and binds to CREs located in gene promoter sequences. Activation of CREB occurs through phosphorylation at Ser133 and through binding to its coactivators CBP and p300 [58, 59]. After phosphorylation, CREB can either homodimerize or heterodimerize with other members of its subfamily, including ATF-1 and CREM, to become transcriptionally active [60]. CREB mostly heterodimerizes, but it is unclear if the specific dimers are connected to specific targets. Several stimuli can lead to increased cAMP or Ca2+ levels, including growth factors, neurotransmitters, inflammatory biolipids, stress signals, and other factors that lead to activated CREB [50, 59, 61, 62]. The method in which CREB is activated can vary greatly and allows CREB to selectively regulate a certain set of genes, depending upon the stimulus used to activate it [63]. Our group has shown specifically that one biolipid, platelet activating factor stimulates phosphorylation of CREB, through phosphorylation of intermediate signaling proteins PKA and p38. This phosphorylation of CREB leads to induction of MMP2 and MT1-MMP [50].
In melanoma, CREB activation regulates the expression of many genes important for invasion, inflammation, and survival including MCAM/MUC18, MMP2, IL-8, and BCL2 [64–66]. Quenching of CREB activity in highly metastatic melanoma cells using a dominant-negative form of CREB (KCREB) or the addition of anti-ATF-1 single chain antibody fragment (ScFv) led to decreased tumorigenicity and metastatic potential in in vivo studies [50, 67–70]. Two distinct roles have been shown for the overexpression of CREB and ATF-1 in metastatic melanoma. First, CREB and ATF-1 regulate invasion through activating MMP2 and MCAM/MUC18 [67]. Secondly, CREB and ATF-1 act as survival factors for melanoma cells. Metastatic melanoma cells were shown to become sensitive to thapsigargin-induced apoptosis after the expression of the dominant-negative form of CREB (KCREB) or with the addition of the single chain antibody fragment (ScFv) against ATF-1 [70, 71].
Silencing of CREB in highly metastatic melanoma cell lines identified the cysteine-rich protein 61 (CYR61) to act as a tumor suppressor in melanoma [72]. CYR61 belongs to the growth factor-inducible immediate-early gene family. Members of this family have been shown to regulate cell proliferation, migration, adhesion, survival, and extracellular matrix formation [73]. In highly metastatic melanoma, expression of CYR61 specifically inhibits tumor growth and metastasis through decreasing angiogenesis and inducing apoptosis in nude mice [72]. We have shown that CYR61 is transcriptionally repressed by CREB binding to the promoter region at two different binding sites [72].
Additionally, CREB can positively or negatively regulate the expression of other transcription factors, which are important for melanoma progression. One that is positively regulated by CREB is the microphthalmia-associated transcription factor (MITF). CREB binds to two CREs located at −140 and −147 bp from the transcription start site in the MITF promoter, leading to increased transcription of MITF [74, 75]. Our group recently identified that CREB negatively regulates the transcription of AP2α during melanoma progression [26].
Several studies had identified an inverse correlation between upregulation of CREB and the loss of AP2α during melanoma progression [5, 76, 77]. We found that blocking the phosphorylation of CREB in highly metastatic cell lines, using the inhibitor H-89, led to increased expression of AP2α. This finding led our lab to further investigate the correlation between these two important transcription factors to determine the mechanism. We identified three CREB binding sites in the promoter of AP2α and, using CHIP and luciferase promoter analyses, showed that CREB is indeed binding to and repressing transcription of AP2α (Fig. 1b) [26]. Furthermore, we identified that CREB is regulating E2F-1, which can also bind to the AP2α promoter. Analysis of downstream targets of AP2α after CREB silencing revealed an upregulation of p21 and downregulation of MCAM/MUC18 [26]. These data indicate that the downregulation of AP2α in melanoma progression can be attributed to CREB activity, suggesting that CREB may act as the master switch in melanoma progression.
4 Microphthalmia-associated transcription factor
The MITF is a basic–helix–loop–helix–leucine–zipper family member that binds to a conical E-Box and is transcriptionally active during melanocyte development and melanoma progression. Although multiple isoforms of MITF exist, which vary at the 5′ messenger RNA (mRNA) region, the isoform MITF-M is predominantly transcribed in melanocytes [78]. Its role in melanocyte development was first realized in mice as MITF mutants lose pigmentation and histologically represent a loss of melanocytes within the epidermis [79]. The loss of pigmentation is due to the transcriptional ablation of melanin-producing enzymes, such as tyrosinase, which are controlled by MITF [80]. Its expression has been positively associated with multiple pathways involved in melanocyte cell fate such as Wnt/β-catenin signaling, factors SOX10 and PAX3 [74, 81–83]. Mutations in MITF, SOX10, PAX3, or EdnrB can result in Waardenburg syndromes, which phenotypically present irregular pigmentation [79].
During melanoma development MITF expression is maintained at high levels in benign and primary tumors [77, 84]. It has also been observed that MITF is amplified at the genomic level in approximately 10–20 % of melanoma specimens. Interestingly, this decreased the 5-year survival rate of melanoma patients [85]. In the majority of melanoma specimens, MITF expression is transcriptionally regulated by the same genes that regulate MITF during melanocyte development. In B16 mouse melanoma cells, β-catenin expression enhances MITF expression by interacting with the TCF/LEF DNA-binding site within the MITF promoter. MITF expression in these cells increased colony formation, and a dominant negative MITF reduces this phenotype [86]. However, others have shown that WNT3A reduces melanoma proliferation even though WNT3A increases MITF expression [87, 88]. This could be contributed to varied activity of MITF or that MITF expression was minimally induced by WNT3A and might be induced more strongly by other WNTs such as WNT1 [87]. Other deregulated pathways in melanoma can also induce MITF. The endothelin receptor B is highly expressed in primary and metastatic melanomas, and targeting EdnrB with monoclonal antibodies in vivo reduces subcutaneous tumor growth [89]. Interestingly EdnrB and MITF levels are highly correlated in melanoma cell lines and patient tumors [89]. EdnrB is a G-protein-coupled receptor that is activated by its ligands endothelin 1 and 3. This induces downstream effectors such as ERK activation and increased cAMP levels [90, 91] cAMP enhances another prominent gene that is activated during melanoma progression, CREB. CREB has been identified to increase MITF expression during melanoma development [92], and we have previously shown that CREB activity is critical for melanoma proliferation, survival, and metastasis [26, 63]. Its regulation of MITF and the downstream genes, such as HIF1-α, Bcl2, CDK2, and p27Kip1 [93–96], which MITF controls, can contribute to this observed phenotype.
Although genomic amplification of MITF in select melanomas maintains its expression in metastatic lesions, it is variably expressed in metastatic lesions that do not have MITF gene amplification [77]. Others have shown that downregulation of MITF and other melanoma development genes lead to a more invasive phenotype in vitro [97]. For example, it has been shown that ablation of MITF increases tumor growth of B16 melanoma cells in vivo [98]. Hypoxic conditions have been reported to reduce MITF expression while inducing HIF1-α, mesenchymal markers, and experimental lung metastasis [99]. Nevertheless, others have shown that cAMP increases the binding of MITF to the promoter of HIF1-α and induces its expression [93]. MITF also functions to induce progression of melanoma by transcribing the antiapoptotic Bcl2, the cell cycle driver CDK2, and represses the growth arrest protein p27Kip1. [94–96]. These data suggest that although MITF is expressed in primary melanomas, its expression and role in metastasis is yet to be determined.
5 Activating transcription factor 2
Another ATF/CREB family basic-region leucine zipper (bZIP) transcription factor is activating transcription factor 2 (ATF-2) [100, 101]. Its activity is regulated by p38 and mitogen-activated protein kinases which heterodimerizes with other transcription factors such as CREB, nuclear factor kappa B (NF-κB), and c-Jun [102, 103]. It is known to regulate multiple genes involved in cell cycle, inflammation, and stress such as c-Jun, cyclin-A, transforming growth factor beta, and tumor necrosis factor alpha (TNF-α) [104–107]. ATF-2 expression has been correlated with melanoma progression. More interestingly, in two separate studies, a higher ratio of cytoplasmic ATF-2 compared to active, nuclear expression predicts better survival for patients. High levels of nuclear ATF-2 predict poor survival, and metastatic lesions stain higher for nuclear ATF-2 as compared to primary tumors [103, 108]. It has recently been reported that high nuclear ATF-2 is attributed to the activity of PKCε, which is highly expressed in melanoma (Fig. 1b). This maintains the transcriptional role of ATF-2 to promote melanoma growth and metastasis, as compared to its more tumor suppressive and apoptotic functions when sublocalized in the cytoplasm [109]. Gene silencing of ATF-2 in B-RAFV600E mutant mouse melanocytes significantly decreases soft agar colony formation in vitro [110]. Others have shown that injecting SW1 mouse melanoma cells that express an ATF-2 blocking peptide (ATF-250-100) significantly decreases tumor growth and metastasis and increased the survival rate of mice injected with B16F10 ATF-250–100 melanoma cells [101].
6 Nuclear factor kappa B
NF-κB is a member of the RelA/NF-κB family of transcription factors which all contain a similar DNA-binding domain, the REL homology domain [111]. NF-κB acts as a first line of defense in inflammation as proinflammatory cytokines induce activation of IKK, which then phosphorylates the inhibitor of NF-κB, IκB. This leads to proteosomal degradation of IkB and nuclear localization of Rel/NF-κB heterodimers, followed by the upregulation of inflammatory genes such as COX2, TNF, and IL-6 [111, 112]. In solid tumors, including melanoma, activation of NF-κB enhances cancer progression. NF-κB can have a proinvasive effect on melanoma cells. This is mediated through αvβ3 integrin signaling induced by HABP1, which activates NF-κB and increases MT1-MMP. MT1-MMP then proteolytically cleaves pro-MMP-2, which generates the active form of MMP-2 [113]. Furthermore, the cytosolic interacting protein of integrins, integrin-linked kinase (ILK), has been shown to induce migration and invasion [114]. ILK activates the NF-κB p65 (RelA), which binds to the IL-6 promoter and induces its expression in melanoma cells (Fig. 1b). This leads to further downstream signaling of STAT3 activation, which increases VEGF expression [115]. This loop highlights the scaffolding of multiple genes that originate from extracellular signals, to IL-6 secretion, and then to VEGF production. Conditioned media from melanoma cells treated with small-interfering RNA (siRNA) against IL-6 or STAT3 reduced endothelial cell tube formation. Additionally, targeting NF-κB with the inhibitor BI-69A11 significantly reduced tumor growth of melanoma cells in vivo. Furthermore, melanoma cells that are resistant to B-RAF inhibitors are still sensitized by the inhibition of NF-κB [116]. Inhibition of IKKb or inhibiting IkB proteosomal degradation reduces the number of pulmonary metastasis of B16F10 melanoma cells [117]. This also results in an increase in p21 expression and increased cytochrome C within the cytoplasm and induced cell cycle arrest and apoptosis after the inhibition of NF-κB [117].
7 MicroRNA regulation of melanoma metastasis
MicroRNAs are small noncoding RNAs, which repress gene expression through binding to complementary mRNA sequences. A single microRNA can regulate hundreds of mRNA sequences [118]. MicroRNAs were first discovered in Caenorhabditis elegans when a noncoding RNA, lin-4, was reported to bind at the 3′ untranslated region (UTR) of lin-14 mRNA, which decreased its protein expression [119, 120]. They are transcribed by RNA polymerase II and are further modified by drosha to form a hairpin pre-microRNA (miRNA). This is then exported to the cytoplasm and processed into a single-strand miRNA by dicer, followed by binding to the 3′ UTR of target mRNA via the RNA-induced silencing complex [121, 122]. Currently, there are over 1,000 discovered microRNAs in humans [121, 123].
Since miRNAs regulate the expression of many cellular genes leading to various biological functions, it can be hypothesized that the deregulation of these genes can lead to the progression of many different cancers. Expression profiles of miRNAs in cancer have clustered mostly based on tissue of origin, indicating that the deregulation of most miRNAs might be specific for each type of tumor [124–126]. Expression profiling of miRNA during the development of melanoma have identified the up- and downregulation of several miRNAs during the progression from melanocytes to malignant melanoma [127].
A number of these microRNAs have been reported to affect the expression of genes involved in melanoma progression. miRs 137, 148, and 182 can bind to and inhibit the translation of the transcription factor MITF, a regulator of melanocyte growth, maturation, and pigmentation [122, 128–130]. The loss of functional miR-137 and miR-148 might play a role in progression of melanoma; however, overexpression of miR-182 in established melanoma cell lines increased the metastatic phenotype and decreased expression of FOXO3 and MITF [128–130]. Other miRNAs such as miR-221 and miR-222 are over expressed in melanoma and have been identified to repress c-Kit and p27 expression [131]. Downregulation of miR-193b in melanoma leads to increased cyclin D1 expression [132]. The expression of miR-532-5p is increased in metastatic tumors and leads to the downregulation of RUNX3 [133]. Furthermore, the let-7 family has been observed in melanoma progression. The loss of let-7a leads to increased expression of integrin beta-3 to enhance migration and invasion [134]. Cell-cycle control can be regulated by let-7b as let-7b mimics reduce the expression of cyclins and decreases cell proliferation [135]. Recently, the overexpression of miR-214 in metastatic melanomas led to the identification of the transcription factors AP2γ (directly) and AP2α (indirectly) as novel targets [136, 137]. As previously mentioned, AP2α plays a major role in melanoma metastasis through regulation of genes involved in angiogenesis, extravasation, and invasion. The identification of these microRNAs and their functions in melanoma indicate that microRNA can play an epigenetic role in regulating melanoma progression. The deregulation of other microRNAs in melanoma is most likely capable of contributing to various functions such as invasion, survival, angiogenesis, and metastasis.
8 A link between CREB and RNA editing in melanoma
RNA transcripts undergo numerous post-transcriptional modifications including alternative splicing and RNA editing, which are important for increasing the variability of the transcriptome in a cell [138]. The most common type of RNA editing is the deamination of adenosine (A) to inosine (I) in double-stranded RNAs, through the action of the adenosine deaminase acting on double-stranded RNA (ADAR) enzymes [139]. Inosine is read as a guanosine by the cellular translation machinery, which can lead to codon changes, alternative splicing, or introduction or removal of stop codons. Additionally, ADAR editing can modify regulatory RNAs, such as microRNAs, which can affect their processing or their binding specificity [140]. These events are highly regulated during development and homeostasis, but many cancers are associated with an imbalance in RNA editing events. Global hypoediting in Alu repeats in brain, prostate, lung, kidney, and testis tumors were shown using a bioinformatic approach [141]. Several of these findings were further validated, including a reduction of ADAR enzymes in brain tumors [141, 142]. Our lab has recently identified the ADAR1 enzyme to be transcriptionally regulated by CREB in highly metastatic melanoma cell lines (unpublished data). During melanoma progression, ADAR1 expression is reduced, indicating potentially altered editing events that are contributing to melanoma metastasis.
9 Therapy
Recently, two new drugs, ipilimumab, an anti-CTLA-4 antibody, and vemurafenib, a V600E BRAF inhibitor, also known as PLX4032, have been approved by the Food and Drug Administration. These drugs offer an emerging new hope for patients bearing unresectable metastatic melanoma. Several phase III clinical trials have shown that metastatic melanoma patients treated with ipilimumab or vemurafenib have a significantly longer overall survival rate when compared to the control groups [143, 144]. However, treatment with ipilimumab causes severe immune-related side effects [144, 145]. In addition, many patients treated with vemurafenib acquired a resistance to the treatment, and it is only effective in 50–60 % of patients, those that harbor the V600E BRAF mutation [146, 147]. Therefore, other alternative molecular-targeted therapeutic modalities are still needed for melanoma patients who are not eligible to be treated with these drugs.
The vital function of transcription factors contributing to metastatic progression has been highlighted in melanoma, which makes them an attractive therapeutic target. Phosphorylation of CREB has been shown to act as a master regulator of the metastatic phenotype in melanoma [148]. However, targeting CREB for treatment is challenging, as it has been previously shown in a CREB knockout mouse model that complete deletion of CREB is lethal perinatally, due to respiratory distress and dysfunctional fetal T cell development [149]. These results indicate that CREB is important for normal cell homeostasis and development. Therefore, important downstream molecules regulated by AP2-α and CREB, such as PAR-1, MCAM/MUC18, and IL-8, may be ideal therapeutic targets in metastatic melanoma patients.
We have shown that PAR-1 repression through delivery of PAR-1 siRNA-incorporated neutral liposomes to tumors and surrounding tissues inhibited tumor growth and metastasis in nude mice through modulating the tumor microenvironment. Additionally, significant decreases in expression of MMP-2, IL-8, VEGF, and CD31 were observed in tumors treated with PAR-1 siRNA when compared to the control group [45]. A PAR-1 pepducin inhibitor, P1pal-7, has also been shown to inhibit tumor growth in a breast carcinoma model [150]. One potential disadvantage of utilizing liposomes is that nanoparticles are not only delivered to tumor cells but also to normal tissue. An important function of PAR-1 on platelets is hemostasis and thrombosis, and due to this nanoparticle characteristic, an anticipated adverse side effect could be bleeding caused by the repression of PAR-1 on platelets. However, in a recent clinical trial, no increased risk of bleeding was observed when a PAR-1 inhibitor was applied as an antiplatelet agent [151]. We have also developed a fully humanized monoclonal antibody (ABX-MA1) targeting MCAM/MUC18, which is constitutively expressed in metastatic melanoma cell lines. Treatment with ABX-MA1 in highly metastatic melanoma cell lines inhibited tumor growth and decreased the formation of lung metastasis in nude mice. Furthermore, a significant reduction of tumor vessel formation and decreased MMP-2 expression was seen when compared to the control group [152]. We have also generated a fully human anti-IL-8 antibody. Interestingly, IL-8 is regulated by PAR-1 and acts as a major angiogenic factor in melanoma. Treatment with ABX-IL-8 inhibited melanoma tumor growth and metastasis in vivo [153]. Taken together, PAR-1, MCAM/MUC18, and IL-8 represent novel alternative therapeutic targets for patients with metastatic melanoma.
10 Concluding remarks
We have summarized the evidence indicating that altered transcriptional activity of each of the transcription factors discussed leads to enhanced tumorigenicity and/or progression toward the metastatic phenotype. Altered activities of CREB and AP2α leads to downregulation of c-KIT and overexpression of MMP2, MCAM/MUC18, PAR-1, and PAFR (Fig. 1). Signaling events through these molecules lead to increased invasion, angiogenesis, and survival, all of which contribute to melanoma progression. We are only just beginning to understand the role of microRNAs and RNA editing in regulation of genes, and further studies are needed to identify potential roles in transcriptional regulation of microRNAs as well as microRNA regulation of transcription factors in melanoma. Taken together, these transcription factors, and their downstream signaling effectors, represent novel therapeutic targets for the treatment of melanoma.
References
Siegel, R., Naishadham, D., & Jemal, A. (2012). Cancer statistics, 2012. CA: A Cancer Journal for Clinicians, 62(1), 10–29.
Balch, C. M., Gershenwald, J. E., Soong, S. J., Thompson, J. F., Atkins, M. B., Byrd, D. R., et al. (2009). Final version of 2009 AJCC melanoma staging and classification. Jounal Clinical Oncology, 27(36), 6199–6206.
Clark, W. H., Jr., Elder, D. E., Guerry, D. T., Epstein, M. N., Greene, M. H., & Van Horn, M. (1984). A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Human Pathology, 15(12), 1147–1165.
Satyamoorthy, K., & Herlyn, M. (2002). Cellular and molecular biology of human melanoma. Cancer Biology & Therapy, 1(1), 14–17.
Leslie, M. C., & Bar-Eli, M. (2005). Regulation of gene expression in melanoma: new approaches for treatment. Journal Cell Biochemistry, 94(1), 25–38.
Greene, V. R., Johnson, M. M., Grimm, E. A., & Ellerhorst, J. A. (2009). Frequencies of NRAS and BRAF mutations increase from the radial to the vertical growth phase in cutaneous melanoma. Journal Investigation Dermatology, 129(6), 1483–1488.
Villanueva, J., & Herlyn, M. (2008). Melanoma and the tumor microenvironment. Current Oncology Reports, 10(5), 439–446.
Chin, L. (2003). The genetics of malignant melanoma: Lessons from mouse and man. Nature Revista Cancer, 3(8), 559–570.
Gray-Schopfer, V., Wellbrock, C., & Marais, R. (2007). Melanoma biology and new targeted therapy. Nature, 445(7130), 851–857.
Leiter, U., Meier, F., Schittek, B., & Garbe, C. (2004). The natural course of cutaneous melanoma. Journal Surgery Oncology, 86(4), 172–178.
Patel, J. K., Didolkar, M. S., Pickren, J. W., & Moore, R. H. (1978). Metastatic pattern of malignant melanoma. A study of 216 autopsy cases. The American Journal of Surgery, 135(6), 807–810.
Fidler, I. J. (2002). Critical determinants of metastasis. Seminars in Cancer Biology, 12(2), 89–96.
Williams, T., Admon, A., Luscher, B., & Tjian, R. (1988). Cloning and expression of AP-2, a cell-type-specific transcription factor that activates inducible enhancer elements. Genes Developments, 2(12A), 1557–1569.
Gravel, M., Gao, E., Hervouet-Zeiber, C., Parsons, V., & Braun, P. E. (2000). Transcriptional regulation of 2′,3′-cyclic nucleotide 3′-phosphodiesterase gene expression by cyclic AMP in C6 cells. Journal of Neurochemistry, 75(5), 1940–1950.
Buettner, R., Kannan, P., Imhof, A., Bauer, R., Yim, S. O., Glockshuber, R., et al. (1993). An alternatively spliced mRNA from the AP-2 gene encodes a negative regulator of transcriptional activation by AP-2. Molecular Cell Biology, 13(7), 4174–4185.
Imagawa, M., Chiu, R., & Karin, M. (1987). Transcription factor AP-2 mediates induction by two different signal-transduction pathways: Protein kinase C and cAMP. Cell, 51(2), 251–260.
Schorle, H., Meier, P., Buchert, M., Jaenisch, R., & Mitchell, P. J. (1996). Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature, 381(6579), 235–238.
Luscher, B., Mitchell, P. J., Williams, T., & Tjian, R. (1989). Regulation of transcription factor AP-2 by the morphogen retinoic acid and by second messengers. Genes & Development, 3(10), 1507–1517.
Zhang, J., & Williams, T. (2003). Identification and regulation of tissue-specific cis-acting elements associated with the human AP-2alpha gene. Development Dynamics, 228(2), 194–207.
Zeng, Y. X., Somasundaram, K., & el-Deiry, W. S. (1997). AP2 inhibits cancer cell growth and activates p21WAF1/CIP1 expression. Nature Genetics, 15(1), 78–82.
McPherson, L. A., Loktev, A. V., & Weigel, R. J. (2002). Tumor suppressor activity of AP2alpha mediated through a direct interaction with p53. Journal Biology Chemistry, 277(47), 45028–45033.
Wajapeyee, N., & Somasundaram, K. (2003). Cell cycle arrest and apoptosis induction by activator protein 2alpha (AP-2alpha) and the role of p53 and p21WAF1/CIP1 in AP-2alpha-mediated growth inhibition. Journal Biology Chemistry, 278(52), 52093–52101.
Tellez, C. S., Davis, D. W., Prieto, V. G., Gershenwald, J. E., Johnson, M. M., McCarty, M. F., et al. (2007). Quantitative analysis of melanocytic tissue array reveals inverse correlation between activator protein-2alpha and protease-activated receptor-1 expression during melanoma progression. Journal Investment Dermatology, 127(2), 387–393.
Berger, A. J., Davis, D. W., Tellez, C., Prieto, V. G., Gershenwald, J. E., Johnson, M. M., et al. (2005). Automated quantitative analysis of activator protein-2alpha subcellular expression in melanoma tissue microarrays correlates with survival prediction. Cancer Research, 65(23), 11185–11192.
Karjalainen, J. M., Kellokoski, J. K., Eskelinen, M. J., Alhava, E. M., & Kosma, V. M. (1998). Downregulation of transcription factor AP-2 predicts poor survival in stage I cutaneous malignant melanoma. Journal of Clinical Oncology, 16(11), 3584–3591.
Melnikova, V. O., Dobroff, A. S., Zigler, M., Villares, G. J., Braeuer, R. R., Wang, H., et al. (2010). CREB inhibits AP-2alpha expression to regulate the malignant phenotype of melanoma. PloS One, 5(8), e12452.
Huang, S., Jean, D., Luca, M., Tainsky, M. A., & Bar-Eli, M. (1998). Loss of AP-2 results in downregulation of c-KIT and enhancement of melanoma tumorigenicity and metastasis. The EMBO Journal, 17(15), 4358–4369.
Gershenwald, J. E., Sumner, W., Calderone, T., Wang, Z., Huang, S., & Bar-Eli, M. (2001). Dominant-negative transcription factor AP-2 augments SB-2 melanoma tumor growth in vivo. Oncogene, 20(26), 3363–3375.
Tellez, C., McCarty, M., Ruiz, M., & Bar-Eli, M. (2003). Loss of activator protein-2alpha results in overexpression of protease-activated receptor-1 and correlates with the malignant phenotype of human melanoma. Journal Biology Chemistry, 278(47), 46632–46642.
Jean, D., Gershenwald, J. E., Huang, S., Luca, M., Hudson, M. J., Tainsky, M. A., et al. (1998). Loss of AP-2 results in up-regulation of MCAM/MUC18 and an increase in tumor growth and metastasis of human melanoma cells. Journal Biology Chemistry, 273(26), 16501–16508.
Bar-Eli, M. (1997). Molecular mechanisms of melanoma metastasis. Journal Cell Physiology, 173(2), 275–278.
Ruiz, M., Pettaway, C., Song, R., Stoeltzing, O., Ellis, L., & Bar-Eli, M. (2004). Activator protein 2alpha inhibits tumorigenicity and represses vascular endothelial growth factor transcription in prostate cancer cells. Cancer Research, 64(2), 631–638.
Lassam, N., & Bickford, S. (1992). Loss of c-kit expression in cultured melanoma cells. Oncogene, 7(1), 51–56.
Yamamoto, K., Tojo, A., Aoki, N., & Shibuya, M. (1993). Characterization of the promoter region of the human c-kit proto-oncogene. Japan Journal Cancer Research, 84(11), 1136–1144.
Pacifico, M. D., Grover, R., Richman, P. I., Daley, F. M., Buffa, F., & Wilson, G. D. (2005). Development of a tissue array for primary melanoma with long-term follow-up: Discovering melanoma cell adhesion molecule as an important prognostic marker. Plast Reconstruction Surgery, 115(2), 367–375.
Shih, I. M., Elder, D. E., Speicher, D., Johnson, J. P., & Herlyn, M. (1994). Isolation and functional characterization of the A32 melanoma-associated antigen. Cancer Research, 54(9), 2514–2520.
Ellis, H. A., & Peart, K. M. (1976). Iliac bone marrow mast cells in relation to the renal osteodystrophy of patients treated by haemodialysis. Journal of Clinical Pathology, 29(6), 502–516.
Xie, S., Luca, M., Huang, S., Gutman, M., Reich, R., Johnson, J. P., et al. (1997). Expression of MCAM/MUC18 by human melanoma cells leads to increased tumor growth and metastasis. Cancer Research, 57(11), 2295–2303.
Natali, P. G., Nicotra, M. R., Digiesi, G., Cavaliere, R., Bigotti, A., Trizio, D., et al. (1994). Expression of gp185HER-2 in human cutaneous melanoma: Implications for experimental immunotherapeutics. International Journal of Cancer, 56(3), 341–346.
Melnikova, V. O., Villares, G. J., & Bar-Eli, M. (2008). Emerging roles of PAR-1 and PAFR in melanoma metastasis. Cancer Microenvironment, 1(1), 103–111.
Fischer, E. G., Ruf, W., & Mueller, B. M. (1995). Tissue factor-initiated thrombin generation activates the signaling thrombin receptor on malignant melanoma cells. Cancer Research, 55(8), 1629–1632.
Vu, T. K., Hung, D. T., Wheaton, V. I., & Coughlin, S. R. (1991). Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell, 64(6), 1057–1068.
Macfarlane, S. R., Seatter, M. J., Kanke, T., Hunter, G. D., & Plevin, R. (2001). Proteinase-activated receptors. Pharmacology Review, 53(2), 245–282.
Grand, R. J., Turnell, A. S., & Grabham, P. W. (1996). Cellular consequences of thrombin-receptor activation. Biochemistry Journal, 313(Pt 2), 353–368.
Villares, G. J., Zigler, M., Wang, H., Melnikova, V. O., Wu, H., Friedman, R., et al. (2008). Targeting melanoma growth and metastasis with systemic delivery of liposome-incorporated protease-activated receptor-1 small interfering RNA. Cancer Research, 68(21), 9078–9086.
Zucker, S., Conner, C., DiMassmo, B. I., Ende, H., Drews, M., Seiki, M., et al. (1995). Thrombin induces the activation of progelatinase A in vascular endothelial cells. Physiologic regulation of angiogenesis. Journal Biology Chemistry, 270(40), 23730–23738.
Yoshida, E., Verrusio, E. N., Mihara, H., Oh, D., & Kwaan, H. C. (1994). Enhancement of the expression of urokinase-type plasminogen activator from PC-3 human prostate cancer cells by thrombin. Cancer Research, 54(12), 3300–3304.
Melnikova, V. O., Balasubramanian, K., Villares, G. J., Dobroff, A. S., Zigler, M., Wang, H., et al. (2009). Crosstalk between protease-activated receptor 1 and platelet-activating factor receptor regulates melanoma cell adhesion molecule (MCAM/MUC18) expression and melanoma metastasis. Journal Biology Chemistry, 284(42), 28845–28855.
Im, S. Y., Ko, H. M., Kim, J. W., Lee, H. K., Ha, T. Y., Lee, H. B., et al. (1996). Augmentation of tumor metastasis by platelet-activating factor. Cancer Research, 56(11), 2662–2665.
Melnikova, V. O., Mourad-Zeidan, A. A., Lev, D. C., & Bar-Eli, M. (2006). Platelet-activating factor mediates MMP-2 expression and activation via phosphorylation of cAMP-response element-binding protein and contributes to melanoma metastasis. Journal Biology Chemistry, 281(5), 2911–2922.
Axelrad, T. W., Deo, D. D., Ottino, P., Van Kirk, J., Bazan, N. G., Bazan, H. E., et al. (2004). Platelet-activating factor (PAF) induces activation of matrix metalloproteinase 2 activity and vascular endothelial cell invasion and migration. The FASEB Journal, 18(3), 568–570.
Villares, G. J., Dobroff, A. S., Wang, H., Zigler, M., Melnikova, V. O., Huang, L., et al. (2009). Overexpression of protease-activated receptor-1 contributes to melanoma metastasis via regulation of connexin 43. Cancer Research, 69(16), 6730–6737.
Pollmann, M. A., Shao, Q., Laird, D. W., & Sandig, M. (2005). Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Research, 7(4), R522–R534.
el-Sabban, M. E., & Pauli, B. U. (1994). Adhesion-mediated gap junctional communication between lung-metastatatic cancer cells and endothelium. Invasion & Metastasis, 14(1-6), 164–176.
Villares, G. J., Zigler, M., Dobroff, A. S., Wang, H., Song, R., Melnikova, V. O., et al. (2011). Protease activated receptor-1 inhibits the Maspin tumor-suppressor gene to determine the melanoma metastatic phenotype. Processes National Academy Science U S A, 108(2), 626–631.
Bohm, M., Moellmann, G., Cheng, E., Alvarez-Franco, M., Wagner, S., Sassone-Corsi, P., et al. (1995). Identification of p90RSK as the probable CREB-Ser133 kinase in human melanocytes. Cell Growth Differences, 6(3), 291–302.
Rutberg, S. E., Goldstein, I. M., Yang, Y. M., Stackpole, C. W., & Ronai, Z. (1994). Expression and transcriptional activity of AP-1, CRE, and URE binding proteins in B16 mouse melanoma subclones. Molecular Carcinogenesis, 10(2), 82–87.
Ravnskjaer, K., Kester, H., Liu, Y., Zhang, X., Lee, D., Yates, J. R., 3rd, et al. (2007). Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. The EMBO Journal, 26(12), 2880–2889.
Shaywitz, A. J., & Greenberg, M. E. (1999). CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annual Review of Biochemistry, 68, 821–861.
Meyer, T. E., & Habener, J. F. (1993). Cyclic adenosine 3′,5′-monophosphate response element binding protein (CREB) and related transcription-activating deoxyribonucleic acid-binding proteins. Endocrine Review, 14(3), 269–290.
Mayr, B., & Montminy, M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nature Review Molecular Cell Biology, 2(8), 599–609.
Iourgenko, V., Zhang, W., Mickanin, C., Daly, I., Jiang, C., Hexham, J. M., et al. (2003). Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Process National Academy Science U S A, 100(21), 12147–12152.
Montminy, M. (1997). Transcriptional regulation by cyclic AMP. Annual Review of Biochemistry, 66, 807–822.
Melnikova, V. O., & Bar-Eli, M. (2008). Transcriptional control of the melanoma malignant phenotype. Cancer Biology & Therapy, 7(7), 997–1003.
White, P. C., Shore, A. M., Clement, M., McLaren, J., Soeiro, I., Lam, E. W., et al. (2006). Regulation of cyclin D2 and the cyclin D2 promoter by protein kinase A and CREB in lymphocytes. Oncogene, 25(15), 2170–2180.
Zhang, X., Odom, D. T., Koo, S. H., Conkright, M. D., Canettieri, G., Best, J., et al. (2005). Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Process National Academy Science U S A, 102(12), 4459–4464.
Xie, S., Price, J. E., Luca, M., Jean, D., Ronai, Z., & Bar-Eli, M. (1997). Dominant-negative CREB inhibits tumor growth and metastasis of human melanoma cells. Oncogene, 15(17), 2069–2075.
Jean, D., & Bar-Eli, M. (2000). Regulation of tumor growth and metastasis of human melanoma by the CREB transcription factor family. Molecular Cell Biochemistry, 212(1–2), 19–28.
Jean, D., & Bar-Eli, M. (2001). Targeting the ATF-1/CREB transcription factors by single chain Fv fragment in human melanoma: potential modality for cancer therapy. Criticism Review Immunology, 21(1–3), 275–286.
Jean, D., Tellez, C., Huang, S., Davis, D. W., Bruns, C. J., McConkey, D. J., et al. (2000). Inhibition of tumor growth and metastasis of human melanoma by intracellular anti-ATF-1 single chain Fv fragment. Oncogene, 19(22), 2721–2730.
Jean, D., Harbison, M., McConkey, D. J., Ronai, Z., & Bar-Eli, M. (1998). CREB and its associated proteins act as survival factors for human melanoma cells. Journal Biology Chemistry, 273(38), 24884–24890.
Dobroff, A. S., Wang, H., Melnikova, V. O., Villares, G. J., Zigler, M., Huang, L., et al. (2009). Silencing cAMP-response element-binding protein (CREB) identifies CYR61 as a tumor suppressor gene in melanoma. Journal Biology Chemistry, 284(38), 26194–26206.
Brigstock, D. R. (2003). The CCN family: A new stimulus package. The Journal of Endocrinology, 178(2), 169–175.
Bertolotto, C., Abbe, P., Hemesath, T. J., Bille, K., Fisher, D. E., Ortonne, J. P., et al. (1998). Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. The Journal of Cell Biology, 142(3), 827–835.
Price, E. R., Horstmann, M. A., Wells, A. G., Weilbaecher, K. N., Takemoto, C. M., Landis, M. W., et al. (1998). alpha-Melanocyte-stimulating hormone signaling regulates expression of microphthalmia, a gene deficient in Waardenburg syndrome. Journal Biology Chemistry, 273(49), 33042–33047.
Nyormoi, O., & Bar-Eli, M. (2003). Transcriptional regulation of metastasis-related genes in human melanoma. Clinical & Experimental Metastasis, 20(3), 251–263.
Zhuang, L., Lee, C. S., Scolyer, R. A., McCarthy, S. W., Zhang, X. D., Thompson, J. F., et al. (2007). Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Modern Pathology, 20(4), 416–426.
Fuse, N., Yasumoto, K., Suzuki, H., Takahashi, K., & Shibahara, S. (1996). Identification of a melanocyte-type promoter of the microphthalmia-associated transcription factor gene. Biochemistry Biophysics Research Communication, 219(3), 702–707.
Widlund, H. R., & Fisher, D. E. (2003). Microphthalamia-associated transcription factor: A critical regulator of pigment cell development and survival. Oncogene, 22(20), 3035–3041.
Ganss, R., Schutz, G., & Beermann, F. (1994). The mouse tyrosinase gene. Promoter modulation by positive and negative regulatory elements. Journal Biology Chemistry, 269(47), 29808–29816.
Dorsky, R. I., Moon, R. T., & Raible, D. W. (1998). Control of neural crest cell fate by the Wnt signalling pathway. Nature, 396(6709), 370–373.
McCallion, A. S., & Chakravarti, A. (2001). EDNRB/EDN3 and Hirschsprung disease type II. Pigments Cell Research, 14(3), 161–169.
Lang, D., & Epstein, J. A. (2003). Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Human Molecular Genetics, 12(8), 937–945.
King, R., Googe, P. B., Weilbaecher, K. N., Mihm, M. C., Jr., & Fisher, D. E. (2001). Microphthalmia transcription factor expression in cutaneous benign, malignant melanocytic, and nonmelanocytic tumors. American Journal Surgery Pathology, 25(1), 51–57.
Garraway, L. A., Widlund, H. R., Rubin, M. A., Getz, G., Berger, A. J., Ramaswamy, S., et al. (2005). Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature, 436(7047), 117–122.
Widlund, H. R., Horstmann, M. A., Price, E. R., Cui, J., Lessnick, S. L., Wu, M., et al. (2002). Beta-catenin-induced melanoma growth requires the downstream target microphthalmia-associated transcription factor. The Journal of Cell Biology, 158(6), 1079–1087.
Chien, A. J., Moore, E. C., Lonsdorf, A. S., Kulikauskas, R. M., Rothberg, B. G., Berger, A. J., et al. (2009). Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proceedings National Academy Science U S A, 106(4), 1193–1198.
Takeda, K., Yasumoto, K., Takada, R., Takada, S., Watanabe, K., Udono, T., et al. (2000). Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. Journal Biology Chemistry, 275(19), 14013–14016.
Asundi, J., Reed, C., Arca, J., McCutcheon, K., Ferrando, R., Clark, S., et al. (2011). An antibody–drug conjugate targeting the endothelin B receptor for the treatment of melanoma. Clin Cancer Res, 17(5), 965–975.
Fuchs, S., Amiel, J., Claudel, S., Lyonnet, S., Corvol, P., & Pinet, F. (2001). Functional characterization of three mutations of the endothelin B receptor gene in patients with Hirschsprung’s disease: Evidence for selective loss of Gi coupling. Molecular Medical, 7(2), 115–124.
Imokawa, G., Yada, Y., & Kimura, M. (1996). Signalling mechanisms of endothelin-induced mitogenesis and melanogenesis in human melanocytes. Biochemistry Journal, 314(Pt 1), 305–312.
Ji, M., & Andrisani, O. M. (2005). High-level activation of cyclic AMP signaling attenuates bone morphogenetic protein 2-induced sympathoadrenal lineage development and promotes melanogenesis in neural crest cultures. Molecular Cell Biology, 25(12), 5134–5145.
Busca, R., Berra, E., Gaggioli, C., Khaled, M., Bille, K., Marchetti, B., et al. (2005). Hypoxia-inducible factor 1{alpha} is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. The Journal of Cell Biology, 170(1), 49–59.
McGill, G. G., Horstmann, M., Widlund, H. R., Du, J., Motyckova, G., Nishimura, E. K., et al. (2002). Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell, 109(6), 707–718.
Du, J., Widlund, H. R., Horstmann, M. A., Ramaswamy, S., Ross, K., Huber, W. E., et al. (2004). Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell, 6(6), 565–576.
Carreira, S., Goodall, J., Denat, L., Rodriguez, M., Nuciforo, P., Hoek, K. S., et al. (2006). Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes & Development, 20(24), 3426–3439.
Jeffs, A. R., Glover, A. C., Slobbe, L. J., Wang, L., He, S., Hazlett, J. A., et al. (2009). A gene expression signature of invasive potential in metastatic melanoma cells. PloS One, 4(12), e8461.
Cheli, Y., Giuliano, S., Botton, T., Rocchi, S., Hofman, V., Hofman, P., et al. (2011). Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene, 30(20), 2307-2318.
Cheli, Y., Giuliano, S., Fenouille, N., Allegra, M., Hofman, V., Hofman, P., et al. (2012). Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene, 31, 2461–2470.
Hai, T. W., Liu, F., Coukos, W. J., & Green, M. R. (1989). Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes & Development, 3(12B), 2083–2090.
Bhoumik, A., Huang, T. G., Ivanov, V., Gangi, L., Qiao, R. F., Woo, S. L., et al. (2002). An ATF2-derived peptide sensitizes melanomas to apoptosis and inhibits their growth and metastasis. Journal Clinical Investment, 110(5), 643–650.
Kaszubska, W., Hooft van Huijsduijnen, R., Ghersa, P., DeRaemy-Schenk, A. M., Chen, B. P., Hai, T., et al. (1993). Cyclic AMP-independent ATF family members interact with NF-kappa B and function in the activation of the E-selectin promoter in response to cytokines. Molecular Cell Biology, 13(11), 7180–7190.
Berger, A. J., Kluger, H. M., Li, N., Kielhorn, E., Halaban, R., Ronai, Z., et al. (2003). Subcellular localization of activating transcription factor 2 in melanoma specimens predicts patient survival. Cancer Research, 63(23), 8103–8107.
Gupta, S., Campbell, D., Derijard, B., & Davis, R. J. (1995). Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science, 267(5196), 389–393.
Kim, S. J., Wagner, S., Liu, F., O’Reilly, M. A., Robbins, P. D., & Green, M. R. (1992). Retinoblastoma gene product activates expression of the human TGF-beta 2 gene through transcription factor ATF-2. Nature, 358(6384), 331–334.
Shimizu, M., Nomura, Y., Suzuki, H., Ichikawa, E., Takeuchi, A., Suzuki, M., et al. (1998). Activation of the rat cyclin A promoter by ATF2 and Jun family members and its suppression by ATF4. Experimental Cell Research, 239(1), 93–103.
Tsai, E. Y., Jain, J., Pesavento, P. A., Rao, A., & Goldfeld, A. E. (1996). Tumor necrosis factor alpha gene regulation in activated T cells involves ATF-2/Jun and NFATp. Molecular Cell Biology, 16(2), 459–467.
Gould Rothberg, B. E., Berger, A. J., Molinaro, A. M., Subtil, A., Krauthammer, M. O., Camp, R. L., et al. (2009). Melanoma prognostic model using tissue microarrays and genetic algorithms. Journal of Clinical Oncology, 27(34), 5772–5780.
Lau, E., Kluger, H., Varsano, T., Lee, K., Scheffler, I., Rimm, D. L., et al. (2012) PKCepsilon promotes oncogenic functions of ATF2 in the nucleus while blocking its apoptotic function at mitochondria. Cell, 148(3), 543–555.
Shah, M., Bhoumik, A., Goel, V., Dewing, A., Breitwieser, W., Kluger, H., et al. (2010). A role for ATF2 in regulating MITF and melanoma development. PLoS Genet, 6(12), e1001258.
Perkins, N. D. (2012). The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer, 12(2), 121–132.
Karin, M., & Greten, F. R. (2005). NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nature Review Immunology, 5(10), 749–759.
Prakash, M., Kale, S., Ghosh, I., Kundu, G. C., & Datta, K. (2011). Hyaluronan-binding protein 1 (HABP1/p32/gC1qR) induces melanoma cell migration and tumor growth by NF-kappa B dependent MMP-2 activation through integrin alpha(v)beta(3) interaction. Cell Signal, 23(10), 1563–1577.
Persad, S., & Dedhar, S. (2003). The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metastasis Review, 22(4), 375–384.
Wani, A. A., Jafarnejad, S. M., Zhou, J., & Li, G. (2011). Integrin-linked kinase regulates melanoma angiogenesis by activating NF-kappaB/interleukin-6 signaling pathway. Oncogene, 30(24), 2778–2788.
Feng, Y., Barile, E., De, S. K., Stebbins, J. L., Cortez, A., Aza-Blanc, P., et al. (2011) Effective inhibition of melanoma by BI-69A11 is mediated by dual targeting of the AKT and NF-kappaB pathways. Pigment Cell Melanoma Res, 24(4), 703–713.
Amschler, K., Schon, M. P., Pletz, N., Wallbrecht, K., Erpenbeck, L., & Schon, M. (2010) NF-kappaB inhibition through proteasome inhibition or IKKbeta blockade increases the susceptibility of melanoma cells to cytostatic treatment through distinct pathways. J Invest Dermatol, 130(4), 1073-1086.
Brennecke, J., Stark, A., Russell, R. B., & Cohen, S. M. (2005). Principles of microRNA-target recognition. PLoS Biology, 3(3), e85.
Lee, R. C., Feinbaum, R. L., & Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75(5), 843–854.
Wightman, B., Ha, I., & Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell, 75(5), 855–862.
Krol, J., Loedige, I., & Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nature Reviews Genetics, 11(9), 597–610.
Mueller, D. W., & Bosserhoff, A. K. (2009). Role of miRNAs in the progression of malignant melanoma. British Journal of Cancer, 101(4), 551–556.
Griffiths-Jones, S., Saini, H. K., van Dongen, S., & Enright, A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Research, 36(Database issue), D154–D158.
Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D., et al. (2005). MicroRNA expression profiles classify human cancers. Nature, 435(7043), 834–838.
Gaur, A., Jewell, D. A., Liang, Y., Ridzon, D., Moore, J. H., Chen, C., et al. (2007). Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Research, 67(6), 2456–2468.
Blower, P. E., Verducci, J. S., Lin, S., Zhou, J., Chung, J. H., Dai, Z., et al. (2007). MicroRNA expression profiles for the NCI-60 cancer cell panel. Molecular Cancer Therapy, 6(5), 1483–1491.
Mueller, D. W., Rehli, M., & Bosserhoff, A. K. (2009). miRNA expression profiling in melanocytes and melanoma cell lines reveals miRNAs associated with formation and progression of malignant melanoma. Journal Investment Dermatology, 129(7), 1740–1751.
Haflidadottir, B. S., Bergsteinsdottir, K., Praetorius, C., & Steingrimsson, E. (2010). miR-148 regulates Mitf in melanoma cells. PLoS One, 5(7), e11574.
Bemis, L. T., Chen, R., Amato, C. M., Classen, E. H., Robinson, S. E., Coffey, D. G., et al. (2008). MicroRNA-137 targets microphthalmia-associated transcription factor in melanoma cell lines. Cancer Research, 68(5), 1362–1368.
Segura, M. F., Hanniford, D., Menendez, S., Reavie, L., Zou, X., Alvarez-Diaz, S., et al. (2009). Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proceeding National Academy Science U S A, 106(6), 1814–1819.
Felicetti, F., Errico, M. C., Bottero, L., Segnalini, P., Stoppacciaro, A., Biffoni, M., et al. (2008). The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Research, 68(8), 2745–2754.
Chen, J., Feilotter, H. E., Pare, G. C., Zhang, X., Pemberton, J. G., Garady, C., et al. (2010). MicroRNA-193b represses cell proliferation and regulates cyclin D1 in melanoma. American Journal of Pathology, 176(5), 2520–2529.
Kitago, M., Martinez, S. R., Nakamura, T., Sim, M. S., & Hoon, D. S. (2009). Regulation of RUNX3 tumor suppressor gene expression in cutaneous melanoma. Clinical Cancer Research, 15(9), 2988–2994.
Muller, D. W., & Bosserhoff, A. K. (2008). Integrin beta 3 expression is regulated by let-7a miRNA in malignant melanoma. Oncogene, 27(52), 6698–6706.
Schultz, J., Lorenz, P., Gross, G., Ibrahim, S., & Kunz, M. (2008). MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Research, 18(5), 549–557.
Bar-Eli, M. (2011). Searching for the ‘melano-miRs’: miR-214 drives melanoma metastasis. The EMBO Journal, 30(10), 1880–1881.
Penna, E., Orso, F., Cimino, D., Tenaglia, E., Lembo, A., Quaglino, E., et al. (2011). microRNA-214 contributes to melanoma tumour progression through suppression of TFAP2C. The EMBO Journal, 30(10), 1990–2007.
Keegan, L. P., Gallo, A., & O’Connell, M. A. (2001). The many roles of an RNA editor. Nature Review Genet, 2(11), 869–878.
Valente, L., & Nishikura, K. (2005). ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Progress in Nucleic Acid Research and Molecular Biology, 79, 299–338.
Nishikura, K. (2006). Editor meets silencer: crosstalk between RNA editing and RNA interference. Nature Review Molecular Cell Biology, 7(12), 919–931.
Paz, N., Levanon, E. Y., Amariglio, N., Heimberger, A. B., Ram, Z., Constantini, S., et al. (2007). Altered adenosine-to-inosine RNA editing in human cancer. Genome Research, 17(11), 1586–1595.
Maas, S., Patt, S., Schrey, M., & Rich, A. (2001). Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proceeding National Academy Science U S A, 98(25), 14687–14692.
Chapman, P. B., Hauschild, A., Robert, C., Haanen, J. B., Ascierto, P., Larkin, J., et al. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England Journal of Medicine, 364(26), 2507–2516.
Hodi, F. S., O’Day, S. J., McDermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. The New England Journal of Medicine, 363(8), 711–723.
Robert, C., Thomas, L., Bondarenko, I., O’Day, S., M, D. J., Garbe, C., et al. (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. The New England Journal of Medicine, 364(26), 2517–2526.
Flaherty, K. T., Puzanov, I., Kim, K. B., Ribas, A., McArthur, G. A., Sosman, J. A., et al. Inhibition of mutated, activated BRAF in metastatic melanoma. The New England Journal of Medicine, 363(9), 809-819.
Davies, H., Bignell, G. R., Cox, C., Stephens, P., Edkins, S., Clegg, S., et al. (2002). Mutations of the BRAF gene in human cancer. Nature, 417(6892), 949–954.
Melnikova, V. O., Dobroff, A. S., Zigler, M., Villares, G. J., Braeuer, R. R., Wang, H., et al. (2010). CREB inhibits AP-2alpha expression to regulate the malignant phenotype of melanoma. PLoS One, 5(8), e12452.
Rudolph, D., Tafuri, A., Gass, P., Hammerling, G. J., Arnold, B., & Schutz, G. (1998). Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proceeding National Academy Science U S A, 95(8), 4481–4486.
Yang, E., Boire, A., Agarwal, A., Nguyen, N., O’Callaghan, K., Tu, P., et al. (2009). Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Research, 69(15), 6223–6231.
Becker, R. C., Moliterno, D. J., Jennings, L. K., Pieper, K. S., Pei, J., Niederman, A., et al. (2009). Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet, 373(9667), 919–928.
Mills, L., Tellez, C., Huang, S., Baker, C., McCarty, M., Green, L., et al. (2002). Fully human antibodies to MCAM/MUC18 inhibit tumor growth and metastasis of human melanoma. Cancer Research, 62(17), 5106–5114.
Huang, S., Mills, L., Mian, B., Tellez, C., McCarty, M., Yang, X. D., et al. (2002). Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. The American Journal of Pathology, 161(1), 125–134.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mobley, A.K., Braeuer, R.R., Kamiya, T. et al. Driving transcriptional regulators in melanoma metastasis. Cancer Metastasis Rev 31, 621–632 (2012). https://doi.org/10.1007/s10555-012-9358-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-012-9358-8