
Abstract
Angiogenesis is a necessary step in tumor growth and metastasis. It is well established that the metabolites of omega-6 and omega-3 fatty acids, which must be obtained through the diet and cannot be synthesized de novo in mammals, have differential effects on cellular processes. Omega-6 fatty acid (n−6 FA)-derived metabolites promote angiogenesis by increasing growth factor expression whereas omega-3 fatty acids (n−3 FA) have anti-angiogenic and antitumor properties. However, most studies thus far have failed to account for the role of the n−6 FA/n−3 FA ratio in angiogenesis and instead examined the absolute levels of n−6 and n−3 FA. This review highlights the biochemical interactions between n−6 and n−3 FA and focuses on how the n−6/n−3 FA ratio in tissues modulates tumor angiogenesis. We suggest that future work should consider the n−6/n−3 FA ratio to be a key element in experimental design and analysis. Furthermore, we recommend that clinical interventions should aim to both reduce n−6 metabolites and simultaneously increase n−3 FA intake.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Angiogenesis, defined as the formation of new blood vessels, is controlled by a balance of pro-angiogenic growth factors and anti-angiogenic growth inhibitors. Although angiogenesis is a natural process that occurs during wound healing, placental growth, and exercise, abnormal angiogenesis underlies many chronic diseases [1, 2]. Excessive angiogenesis is associated with cancer, obesity, arthritis, and diabetic retinopathy whereas insufficient angiogenesis is associated with heart attack, stroke, osteoporosis, and preeclampsia [3].
Understanding the factors that regulate angiogenesis is important for interventions across a multitude of diseases that share malfunctions in angiogenesis. Research has demonstrated pro-angiogenic effects of the metabolites of omega-6 fatty acids (n−6 FA) and anti-angiogenic effects of the metabolites of omega-3 fatty acids (n−3 FA). This review will examine how manipulating the tissue ratio of n−6 FA/n−3 FA may be a promising approach to modulating tumor angiogenesis as a part of cancer therapy.
1.1 Angiogenesis and cancer
Angiogenesis is maintained by endothelial cells, which line the inner membrane of the vasculature and act as receptors for a variety of chemical stimuli. The general mechanism of angiogenesis can be explained as follows: tissue secretes an angiogenic signal that binds to receptors on pre-existing vessels, causing (a) existing vessels to dilate, vascular permeability to increase, and the extracellular matrix to degrade; (b) endothelial cells to proliferate and migrate towards the angiogenic signal; (c) endothelial cells to aggregate and begin tube formation, connecting the tissue-secreting growth factor to the pre-existing vasculature; and (d) capillaries to stabilize through pericyte recruitment and remodeling of the vasculature [4].
One of the hallmarks of cancer is the body’s ability to promote sustained angiogenesis because the rapid rate of tumor expansion necessitates a constant and expanding blood supply [5, 6]. Microscopic breast cancer cells can be found in approximately 50 % of middle-aged women, but these cells are typically dormant and asymptomatic; however, in some of these women, the microscopic cancer cells become active and proliferate uncontrollably until they begin to metastasize [7]. It has been hypothesized that the ability to induce angiogenesis underlies the switch between dormant, microscopic cancer cells, and an active tumor [8, 9].
Various molecules are responsible for promoting and sustaining angiogenesis. Common growth factors and their functions include vascular endothelial growth factor (VEGF), which causes dilation of blood vessels and endothelial cell proliferation/migration, platelet-derived growth factor (PDGF), which recruits smooth muscle cells to stabilize new vessels, fibroblast growth factors (FGF), which promote endothelial cell proliferation and the physical organization of endothelial cells intro tube-like structures, matrix metalloproteinase (MMP), which causes breakdown of the basement membrane, and angiopoietin, which mediates vascular remodeling and maintains vascular integrity [10].
Most growth factors, including VEGF, PDGF, and angiopoietins, are receptor tyrosine kinases. Binding of the growth factor to its endothelial receptor activates signal transduction, ultimately causing transcription of the growth factor in the nucleus [11]. Some pro-angiogenic molecules regulate the production of angiogenic growth factors. Tumor necrosis factor-alpha (TNF-α), an inflammatory cytokine, increases expression of VEGF and MMP production [12]. Another macrophage-derived chemokine, interleukin (IL)-8, upregulates MMP-2 and MMP-9 [13]. Therefore, molecules that regulate growth factor expression, production, or downstream effects will likewise impact angiogenesis.
1.2 The biochemistry of omega-6 and omega-3 fatty acids
Both n−6 and n−3 FA are long-chain, polyunsaturated fats. n−6 FA contain two or more double bonds, with the first double bond on the sixth carbon from the methyl end of the molecule; n−3 FA contain three or more double bonds, with the first double bond on the third carbon atom from the methyl end. n−3 FA and n−6 FA play crucial biological roles that include altering the properties of cell membranes, providing substrates for the production of signaling molecules or functioning mediators, and modulating gene expression [14]. These fatty acids are considered “essential” because they cannot be produced de novo by the body and must be obtained from the diet.
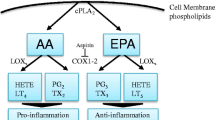
The primary n−6 FA is linoleic acid (LA), which can be converted to arachidonic acid (AA). The three main n−3 FAs are α-linolenic acid (ALA), docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA). Through the same desaturase and elongase enzymes, the n−3 FA ALA can be converted into EPA and DHA (Fig. 1). However, because these conversions are inefficient, consuming preformed EPA and DHA is necessary to meet dietary requirements [15].

The metabolic pathways of n−6 FA and n−3 FA. Cyclooxygenases (COX), lipooxygenases (LOX), and cytochrome P450 (CYP 450) convert n−6 and n−3 FA into bioactive lipid mediators. The n−6-derived eicosanoids, which include the prostaglandins (PGH2, PGE2, PGI2), leukotrienes (four-series LTs), thromboxanes (TXA2), and hydroxyeicosatraenoic acids (12-HETE, 15-HETE), promote tumor angiogenesis. In contrast, n−3 FA metabolism produces eicosanoids that attenuate excess vascularization, including leukotrienes and prostaglandins
The enzymes responsible for the metabolism of both n−6 FA and n−3 FA are the cyclooxygenases (COX), lipooxygenases (LOX), and cytochrome P450 (CYP 450; Fig. 1). Several n−6 FA-derived eicosanoids promote tumor angiogenesis, such as the prostaglandins (PGH2, PGE2, PGI2), leukotrienes (4-series LTs), thromboxanes (TXA2), and hydroxyeicosatraenoic acids (12-HETE, 15-HETE) [16–24]. These eicosanoids make the tumor microenvironment more favorable for neoplasms and metastasis by encouraging the transcription of angiogenic growth factors, increasing the rate of endothelial cell migration and proliferation, and increasing the rate of vascularization. In contrast, n−3 FA metabolism produces leukotrienes and prostaglandins that attenuate excess vascularization. n−3 FA and n−6 FA compete for incorporation into the cell membrane in addition to enzymes for eicosanoid production, including COX-2 and 5-LOX (Fig. 1). Thus, high levels of tissue n−3 FA can reduce angiogenesis through decreased production of pro-angiogenic AA-derived eicosanoids and through n−3 FA’s intrinsic antitumor properties [16–27]. Furthermore, n−3 FA have been found to downregulate expression of angiogenic growth factors VEGF, PDGF, IL-6, and MMP-2 [28, 29].
Unlike other fatty acids that are widely available in foodstuffs, n−3 FA are primarily found in fatty fish and certain vegetables and nuts. Single-celled algae and phytoplankton produce n−3 FA and because these organisms enter our food web through various routes (e.g., fish that have eaten phytoplankton), they are considered to be the primary origin of most of the n−3 FAs in the human diet [30]. Currently, fish populations are diminishing and the price of seafood is increasing, rendering dietary n−3 FA increasingly scarce. However, whereas n−3 FA are present in few foods, n−6 FA are abundant in the Western diet. Corn and soybean oils, processed foods containing these oils, and grain-fed meat are all high in n−6 FA.
Throughout our evolution as hunters and gatherers, humans thrived on a meat and fish diet that was high in n−3 FA and low in n−6 FA; the ratio of n−6 FA/n−3 FA was likely close to 1:1 [28]. In more recent human history, this ratio has drastically changed. During the last century, the emergence of agribusiness alongside processed foods, grain-fattened livestock, and hydrogenation of vegetable fats have reduced the content of n−3 fatty acids and increased n−6 fatty acids. Now, the ratio of n−6 FA/n−3 FA is approximately 15:1 or higher [29]; our bodies may not be accustomed to utilizing such high levels of n−6 FA [31]. This is considered to be one of many factors responsible for the relatively recent rise in chronic diseases, predominantly those associated with inflammation including cancer, heart disease, arthritis, and diabetes [32].
2 The effects of omega-6 fatty acid metabolites on angiogenesis
2.1 COX-2 metabolites
COX-2 is the enzyme that catalyzes the formation of PGE2, PGI2, PGH2, and TXA2 from AA, which have been shown to promote angiogenesis by encouraging the expression of growth factors and creating favorable conditions for growth in the tumor microenvironment [33]. Downstream products of COX-2 induce tumor vascularization by regulating multiple steps of angiogenesis including the production of VEGF and matrix metalloproteinases, promoting vascular sprouting, migration, and tube formation, and enhancing endothelial cell survival [34].
COX-2 is upregulated in tumor tissue and leads to an increased production of pro-angiogenic eicosanoids. Moderate to strong levels of COX-2 have been found in pulmonary, colonic, and mammary tumors as compared to negligible levels of COX-2 in non-neoplastic epithelium [35]. Similarly, COX-2 mRNA and protein expression are elevated in the tumor cells of head and neck cancer patients [36]. Furthermore, patients with metastases have higher COX-2 expression, PGE2 levels, and microvessel densities than cancer patients without metastases [36]. COX-2 levels are also higher in the tumor front zone, which is characterized by active proliferation, than in the tumor core. These results indicate increased levels of COX-2 are linked to the degree of tumor progression and angiogenesis.
Moreover, high COX-2 activity in tumors leads to increased expression of VEGF. Under hypoxic conditions, COX-2/PGE2 enhances HIF-1 transcriptional activity and increases VEGF levels, facilitating the angiogenesis that enables tumor cell survival in this hostile environment [37, 38]. COX-2 activity has also been positively associated with lymphoangiogenesis, which is a key step in metastatic progression. Lymphoangiogenesis is stimulated by COX-2 upregulation of VEGF-C through the Her/Neu pathway, which activates NFκβ [39, 40]. In leukemic cells, COX-2 induction of VEGF can be mediated through the JNK pathway in which C-jun binds to CRE [41]. The wide range of signal transduction pathways affected by COX-2 illustrates the diverse mechanisms through which COX-2 metabolites can facilitate angiogenesis.
A convincing line of research shows that inhibition of COX-2 reduces the synthesis of pro-angiogenic prostaglandins and thromboxane A2, effectively mitigating tumor growth and excess vascularization. Indomethacin, a nonsteroidal anti-inflammatory drug that inhibits COX-1 and COX-2, has been shown to induce cell death in tumors and reduce tumor size and vascularization in mice with mammary tumors [42]. Furthermore, NS-398, a COX-2 inhibitor, reduces the expression of VEGF mRNA in mouse carcinoma cells [43]. Gene knockdown of COX-2 also influences growth factor production; siRNA of COX-2 in MDA-MB-231 (breast cancer) cells reduces VEGF-C production [44]. These findings suggest a link between COX-2 and angiogenesis.
2.2 LOX and cytochrome P-450 metabolites
Relative to COX, less research has focused on the roles of 12-LOX, 5-LOX, and CYP in angiogenesis. 12-LOX is an enzyme that catalyzes the formation of 12-HETE from AA and 5-LOX catalyzes the formation of HPETE and subsequently several inflammatory leukotrienes. 12-HETE is produced by tumor cells and induces tumor metastasis by activating growth factors such as bFGF and PDGF [45]. When prostate cancer cells (PC3) with or without 12-LOX were injected into mice, 12-LOX PC3 mice had larger tumors, increased vascularization, and decreased tumor necrosis compared to PC3 control mice [46]. Similarly, the inhibition of 12-LOX blocks the stages necessary for angiogenesis. BHPP, a 12-LOX inhibitor, reduces VEGF-induced cell migration whereas overexpression of 12-LOX stimulates endothelial cell motility [22]. In rodent tumor models, inflammation, upregulation of 5-LOX, and increases in MMP-2 and VEGF have been found to occur concomitantly [47, 48]. Overall, an anti-angiogenic cancer treatment that inhibits both COX and LOX classes of enzymes may be more effective than blocking either COX or LOX separately because the production of AA derived pro-angiogenic metabolites would be minimized [49].
Cytochrome P450 transforms AA into epoxyeicosatrienoic acids (EETs) [50]. Cells that over-express CYP show increased angiogenesis in vitro and enhanced matrix metalloproteinase activity [51]. Furthermore, EET incubation causes endothelial cells to proliferate in a dose-dependent manner in addition to increasing endothelial cell migration, with EET signaling through the MAPK and PI3/kinase pathways [52]. Moreover, EETs may function as crucial second messengers in response to VEGF; EET antagonists abrogate VEGF-induced endothelial tube cell formation [50].
To summarize, two lines of evidence support the pro-angiogenic effects of AA metabolites; first, high eicosanoid concentration due to high activity of COX-2, 12-LOX, 5-LOX, and CYP 450, upregulates growth factors through a variety of mechanisms. In vivo, tumor tissue shows increased vascularization in response to high levels of AA metabolites. Second, inhibition of COX-2, 12-LOX and 5-LOX attenuates angiogenesis through downregulating growth factor expression.
3 The effects of omega-3 fatty acid metabolites on angiogenesis
3.1 Decreased production of pro-angiogenic n−6 eicosanoids
n−3 FA modulate angiogenesis by suppressing downstream metabolites of AA. Two critical elements of AA metabolism are the availabilities of substrate and enzyme. n−3 FA target both factors related to AA metabolism, displacing n−6 FA from the cell membrane and competing for COX, LOX, desaturases, and elongases [53]. Supplementation with fish oil lowers production of the pro-angiogenic, AA-derived metabolites PGE2 and LTB4 [54]. n−3 FA work as natural inhibitors of n−6 eicosanoids and have the added benefit of antitumor properties, which include increased cancer cell apoptosis [55], altered estrogen metabolism, and decreased production of free radicals or reactive oxygen species [56].
3.2 Transcriptional downregulation of growth factors
Population studies have demonstrated a strong chemopreventive effect of n−3 FA. Populations that ingest high levels of fatty fish have a lower risk of developing cancer. Moreover, the VEGF levels of individuals with lower serum n−6 FA/n−3 FA ratios show lower circulating blood levels of VEGF [57]. In vitro, in vivo, and human experiments show that EPA and DHA inhibit angiogenesis growth factors including VEGF, PDGF, COX2 and PGE 2, and MMP 2 [58].
The n−3 FA metabolites resolvin D1 (RvD1) and E1 (RvE1) are potent downregulators of VEGF and cytokines. In one study, corneal neovascularization was induced in mice and n−3 resolvins were then subconjunctivally injected with aspirin-triggered lipoxin A4 analog (ATLa), RvD1 or RvE1 at 48-h intervals. The mice treated with lipid mediators had reduced mRNA expression of VEGF (A, C and R2), TNF-α, IL-1 alpha, IL-1 beta, and suppressed hemoangiogenesis due to reductions in the production of pro-angiogenic interleukins [20]. Similarly, in a mouse model of pathological retinal angiogenesis, n−3 supplementation was found to be effective in reducing pathologic neovascularization; this effect that was largely mediated by the ability of n−3 metabolites to block the production of pro-inflammatory cytokine TNF-α [59]. A proposed mechanism by which n−3 FA regulate pro-angiogenic and inflammatory cytokines is through the suppression of transcription factor NFκB [60]. Thus, n−3 FA have robust and varied nutrigenomic effects and are a natural substance that counteracts the pro-angiogenic actions of a high n−6 FA diet [61].
3.3 Suppression of inflammation
Inflammation and angiogenesis have several mechanisms and pathways in common, so it is important to understand that n−3 FA inhibit inflammation in several ways; omega-3 FA (1) displace inflammatory omega-6 FA and prevent them from making pro-inflammatory eicosanoids; (2) they downregulate gene transcription of the enzymes (e.g., COX-2) that catalyze the synthesis of lipid mediators; (3) they decrease the generation of inflammatory cytokines (e.g., TNF-2, IL-6, IL-1β); and (4) they generate novel anti-inflammatory compounds, including the resolvins and protectins [62]. In fact, low n−3 FA levels in the Western diet are associated with chronic inflammatory disorders such as heart disease, cancer, Crohn’s disease, lupus, psoriasis colitis, pancreatitis, asthma, hepatitis, liver disease, and supplementation with n−3 may decrease these diseases [31, 63–72].
The relationship between inflammation and angiogenesis is multidirectional: inflammation can stimulate angiogenesis, angiogenesis can induce inflammation [73], and inflammatory cytokines, macrophages, and leukocytes can release angiogenic growth factors [74]. For instance, IL-1 induces inflammation and also contributes to tumor invasiveness via the production of VEGF and TNFα [75]. One disease that has benefitted from adopting both anti-inflammatory and anti-angiogenic approaches to treatment is rheumatoid arthritis. The inflammation of the rheumatoid synovium can be likened to a tumor; blood vessels provide nutrients to the inflamed synovium and are a portal for chemokines, which exacerbate and increase inflammation [76]. An αv/β3 integrin inhibitor was shown to both reduce symptom severity and increase apoptosis of angiogenic blood vessels in a rabbit model of arthritis [77]. Thus, a reduction in inflammation is one of the ways that n−3 FA modulate angiogenesis.
4 Perspectives
To summarize, thus far this review has emphasized two main points. The first is that n−6 FA metabolites promote tumor angiogenesis through a variety of signaling pathways, encouraging epithelial cell proliferation and migration, and decreasing tumor apoptosis. The second is that n−3 FA and their metabolites can reverse the pro-angiogenic consequences of high n−6 FA metabolite levels. The n−3 FA lower production of n−6 FA-derived prostaglandins and leukotrienes and downregulate growth factor expression (Fig. 2). In this final section, we hope to integrate these two ideas by highlighting a potentially important factor in regulating angiogenesis and tumor growth: the n−6 FA/n−3 FA ratio (Fig. 2).

The effects of n−6 FA and n−3 FA and their metabolites on angiogenesis. Phospholipase A2 releases n−6 and n−3 from the phospholipid cell membrane. The n−6-derived eicosanoids, which include the prostaglandins (PGs), leukotrienes (LTs), and epoxyeicosatrienoic acids (EETs) promote tumor angiogenesis and inflammatory factors. In contrast, n−3 FA and their metabolites suppress the activity of angiogenic and inflammatory factors and ultimately inhibit excess vascularization. Overall, n−6 FA promote pro-angiogenic conditions whereas n−3 FA discourage them
Given that n−6 FA and n−3 FA have opposing effects and compete for the same enzymes, lowering the ratio of n−6/n−3 FA in tissues shows great potential for controlling dysregulated angiogenesis and reducing cancer risk. However, few epidemiological studies have directed attention towards the n−6/n−3 FA ratio as it relates to their findings, instead favoring absolute levels of n−6 or n−3 FA. This lack of attention may be one factor contributing to the disparity between epidemiological and lab-based studies on the anticancer effects of n−3 FA. Only approximately1/3 of epidemiological studies have found an association between n−3 FA and cancer risk although in vivo and in vitro experiments have consistently demonstrated that n−3 FA can modulate angiogenesis, tumor growth, and cancer risk.
Several epidemiological studies have concluded that there is no significant effect of n−3 FA on cancer risk, but these studies have only accounted for absolute as opposed to relative levels of n−3 and n−6 FA. For example, in a meta-analysis that reviewed 38 different prospective cancer cohorts, only 10 out of 65 measures concerning n−3 intake and cancer risk showed positive statistical relationships [78]. While the authors admitted a large discrepancy between their research and lab studies, they concluded that, “Dietary supplementation with omega-3 fatty acids is unlikely to reduce the risk of cancer.” Although most of the studies included in the analysis represented large cohorts, many of the studies that found no beneficial effect of n−3 FA on cancer risk only examined n−3 FA levels but not the n−6/n−3 ratio. For example, a study of Swedish women used the absolute intake of ALA, LA, EPA, and DHA to conclude that no relationship existed between n−3 intake and cancer [79]. Several studies have found that absolute ALA levels are positively correlated with prostate cancer but have not accounted for the n−6/n−3 FA ratio in their analyses [80, 81].
The Health Professionals Follow-Up Study chose the absolute intake of ALA, AA, LA, EPA, and DHA as the basis for their conclusion that ALA intake is positively associated with advanced prostate cancer [82]. However, when looking at the highest and lowest quintiles of relative risk, there was one statistic “LA: EPA + DHA” that served as a rough estimate of the n−6n−6 FA/n−3 FA ratio. For the lowest quintile of relative risk, this value was <23.57 whereas for the highest quintile of relative risk the LA/EPA + DHA ratio was >102.34. This indicates that the n−6 FA/n−3 FA ratio may be important and that accounting for AA and LA intake along with ALA levels may have shifted the authors’ conclusions. It would be interesting to reanalyze these data and those from other epidemiological studies using the n−6 FA/n−3 FA ratio.
Several studies have found significant correlations between the n−6 FA/n−3 FA ratio and cancer risk. The EURAMIC study showed a decrease in the risk for developing breast cancer when the ratio of n−6 FA/n−3 FA was decreased, but no association when n−6 FA intake and n−3 FA intake were considered separately [83]. After examining adipose tissue in breast cancer patients and in patients with benign breast disease, the ratio of n−6 FA/n−3 FA was significant; women in the highest tertile of the long-chain n−3/total n−6 ratio had an odds ratio of 0.33 [84]. In addition, a high ratio of n−6 FA/n−3 FA in Caucasian men has been found to be associated with an increased risk of prostate cancer [85]. Standardizing analysis methods using the n−6 FA/n−3 FA ratio may lead to more consistent results regarding the anticancer effects of n−3 FA.
4.1 The fat-1 mouse model
In both animal and human studies, there are technical difficulties in generating accurate dietary ratios of n−6 FA/n−3 FA for research and other dietary nutrients may interact with fatty acid metabolism and its actions. Thus, both epidemiological and in vivo animal studies may not give a full picture. The fat-1 mouse is a transgenic animal that has the ability to endogenously convert n−6 FA to n−3 FA because it has a copy of the Caenorhabditis elegans fat-1 gene, making it an ideal model for controlling the tissue ratio of n−6 FA/n−3 FA and eliminating the confounding factors associated with dietary supplementation [86]. Two different n−6 FA/n−3 FA ratios can be established in fat-1 mice from the same litter when they are fed an identical, high n−6 FA diet; the wild type mice exhibit a high tissue n−6/n−3 ratio whereas the fat-1 mice will have a lower n−6/n−3 ratio.
Experiments using the fat-1 mouse have shown a reduction in pro-angiogenic molecules, such as downregulation of the pro-angiogenic factors NFκB, TNFα, and IL-1β [87]. Furthermore, fat-1 mice with melanoma exhibit high levels of n−3 prostaglandins in tumor and surrounding tissues accompanied by decelerated melanoma progression [88]. Studies from our and other laboratories have consistently demonstrated that decreasing the ratio of n−6 FA/n−3 FA conveys antitumor effects [88–96].
4.2 Clinical applications and implications for cancer therapies
Given that n−6 FA increase angiogenesis, n−3 FA decrease angiogenesis, and tumor progression and metastasis depend on angiogenesis, reducing the tissue n−6 FA/n−3 FA would appear to be a new strategy for cancer treatment. The two distinct approaches to addressing angiogenesis in cancer are either (1) using drugs that block the production of AA metabolites or (2) dietary supplementation with n−3 FA. Both treatments only regulate absolute levels of AA-derived eicosanoids or n−3 FA-derived eicosanoids. The ideal therapy would be to combine a reduction in tissue levels of n−6-derived eicosanoids with an increase in tissue levels of n−3-derived lipid mediators.
Cancer drugs that target AA eicosanoid production focus on COX-2 and LOX-5 inhibition. For example, in a prospective cohort study of nearly 48,000 men, those who took aspirin more than two times per week had a lower risk for colorectal cancer [97]. However, direct COX-2 inhibition is not a sound approach because this also inhibits production of antitumor n−3-derived eicosanoids and can be associated with an increased risk of heart attack/stroke and other undesirable side effects. Furthermore, a drug-based approach overlooks other methods of regulating n−6 eicosanoid production, such as increasing dietary intake of n−3 FA [98]. Coupling these inhibitors of AA metabolites with increased n−3 FA intake to slow the growth of metastatic cells not only increases treatment efficacy by further targeting COX-2 and LOX 5 enzymes but is also a natural method with few, if any, harmful side effects. Furthermore, n−3 FA convey benefits to cancer patients beyond modulating n−6 metabolites, including prolonging survival, increasing immune function, decreasing postsurgery complications, and possibly reversing cachexia [99–101].
In conclusion, the upregulation or overproduction of AA-derived metabolites encourages angiogenesis, and this effect is suppressed by the n−3 FA-derived eicosanoids. Overall, epidemiological studies have failed to account for the n−6/n−3 ratio in their analyses but from a biochemical perspective it is clear that a high tissue n−6/n−3 FA ratio promotes inflammation and angiogenesis. One practical clinical intervention would be lowering the n−6/n−3 FA ratio to reduce tissue levels of AA-derived pro-angiogenic eicosanoids by increasing intake of n−3 and decreasing intake of n−6 FA.
References
Arnold, F., & West, D. (1991). Angiogenesis in wound healing. Pharmacology & Theraputics, 52, 407–422.
Kleim, J. A., Cooper, N. R., & VandenBerg, P. M. (2002). Exercise induces angiogenesis but does not alter movement representations within rat motor cortex. Brain Research, 934(1), 1–6.
Carmeliet, P. (2003). Angiogenesis in health and disease. Nature Medicine, 9(6), 653–660.
Conway, E. M., Collen, D., & Carmeliet, P. (2001). Molecular mechanisms of blood vessel growth. Cardiovascular Research, 49(3), 507–521.
Folkman, J., Cole, P., & Zimmerman, S. (1966). Tumor behavior in isolated perfused organs: in vitro growth and metastases of biopsy material in rabbit thyroid and canine intestinal segment. Annals of Surgery, 164(3), 491–502.
Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100(1), 57–70.
Folkman, J., & Kalluri, R. (2004). Cancer without disease. Nature, 427(6977), 787.
Blood, C. H., & Zetter, B. R. (1990). Tumor interactions with the vasculature: angiogenesis and tumor metastasis. Biochimica et Biophysica Acta, 1032(1), 89–118.
Naumov, G. N., Akslen, L. A., & Folkman, J. (2006). Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle, 5(16), 1779–1787.
Morisada, T., Kubota, Y., Urano, T., Suda, T., & Oike, Y. (2006). Angiopoietins and angiopoietin-like proteins in angiogenesis. Endothelium, 13(2), 71–79.
Rajkumar, T. (2001). Growth factors and growth factor receptors in cancer. Current Science, 81, 535–541.
Wu, Y., & Zhou, B. P. (2010). TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. British Journal of Cancer, 102(4), 639–644.
Li, A., Dubey, S., Varney, M. L., Dave, B. J., & Singh, R. K. (2003). IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. Journal of Immunology, 170(6), 3369–3376.
Kang, J. X. (2005). From fat to fat-1: a tale of omega-3 fatty acids. Journal of Membrane Biology, 206(2), 165–172.
Burdge, G. C., & Calder, P. C. (2005). Conversion of alpha-linolenic acid to longer-chain polyunsaturated fatty acids in human adults. Reproduction, Nutrition, Development, 45(5), 581–597.
Hoagland, K. M., Maier, K. G., Moreno, C., Yu, M., & Roman, R. J. (2001). Cytochrome P450 metabolites of arachidonic acid: novel regulators of renal function. Nephrology, Dialysis, Transplantation, 16(12), 2283–2285.
Smith, W. L. (1989). The eicosanoids and their biochemical mechanisms of action. Biochemical Journal, 259(2), 315–324.
Arita, M., Yoshida, M., Hong, S., Tjonahen, E., Glickman, J. N., Petasis, N. A., et al. (2005). Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proceedings of the National Academy of Sciences of the United States of America, 102(21), 7671–7676.
Bagga, D., Wang, L., Farias-Eisner, R., Glaspy, J. A., & Reddy, S. T. (2003). Differential effects of prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proceedings of the National Academy of Sciences of the United States of America, 100(4), 1751–1756.
Jin, Y., Arita, M., Zhang, Q., Saban, D. R., Chauhan, S. K., Chiang, N., et al. (2009). Anti-angiogenesis effect of the novel anti-inflammatory and pro-resolving lipid mediators. Investigative Ophthalmology and Visual Science, 50(10), 4743–4752.
Kamiyama, M., Pozzi, A., Yang, L., DeBusk, L. M., Breyer, R. M., & Lin, P. C. (2006). EP2, a receptor for PGE2, regulates tumor angiogenesis through direct effects on endothelial cell motility and survival. Oncogene, 25(53), 7019–7028.
Nie, D., Lamberti, M., Zacharek, A., Li, L., Szekeres, K., Tang, K., et al. (2000). Thromboxane A(2) regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochemical and Biophysical Research Communications, 267(1), 245–251.
Pai, R., Szabo, I. L., Soreghan, B. A., Atay, S., Kawanaka, H., & Tarnawski, A. S. (2001). PGE(2) stimulates VEGF expression in endothelial cells via ERK2/JNK1 signaling pathways. Biochemical and Biophysical Research Communications, 286(5), 923–928.
Pola, R., Gaetani, E., Flex, A., Aprahamian, T. R., Bosch-Marce, M., Losordo, D. W., et al. (2004). Comparative analysis of the in vivo angiogenic properties of stable prostacyclin analogs: a possible role for peroxisome proliferator-activated receptors. Journal of Molecular and Cellular Cardiology, 36(3), 363–370.
Grimminger, F., Wahn, H., Mayer, K., Kiss, L., Walmrath, D., & Seeger, W. (1997). Impact of arachidonic versus eicosapentaenoic acid on exotonin-induced lung vascular leakage: relation to 4-series versus 5-series leukotriene generation. American Journal of Respiratory and Critical Care Medicine, 155(2), 513–519.
Samuelsson, B. (1983). Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science, 220(4597), 568–575.
Yuan, Y. M., Fang, S. H., Qian, X. D., Liu, L. Y., Xu, L. H., Shi, W. Z., et al. (2009). Leukotriene D4 stimulates the migration but not proliferation of endothelial cells mediated by the cysteinyl leukotriene cyslt(1) receptor via the extracellular signal-regulated kinase pathway. Journal of Pharmacological Sciences, 109(2), 285–292.
Spencer, L., Mann, C., Metcalfe, M., Webb, M. B., Pollard, C., Spencer, D., et al. (2009). The effect of omega-3 FAs on tumour angiogenesis and their therapeutic potential. European Journal of Cancer, 45(19493674), 2077–2086.
Kang, J. X., & Weylandt, K. H. (2008). Modulation of inflammatory cytokines by omega-3 fatty acids. Sub-Cellular Biochemistry, 49(18751910), 133–143.
Kang, J. X. (2011). Omega-3: a link between global climate change and human health. Biotechnology Advances, 29(4), 388–390.
Simopoulos, A. P. (2002). Omega-3 fatty acids in inflammation and autoimmune diseases. Journal of the American College of Nutrition, 21(6), 495–505.
Simopoulos, A. P. (2006). Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: nutritional implications for chronic diseases. Biomedicine and Pharmacotherapy, 60(9), 502–507.
Wang, D., & Dubois, R. N. (2010). Eicosanoids and cancer. Nature Reviews. Cancer, 10(3), 181–193.
Gately, S., & Li, W. W. (2004). Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Seminars in Oncology, 31(2 Suppl 7), 2–11.
Soslow, R. A., Dannenberg, A. J., Rush, D., Woerner, B. M., Khan, K. N., Masferrer, J., et al. (2000). COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer, 89(12), 2637–2645.
Gallo, O., Franchi, A., Magnelli, L., Sardi, I., Vannacci, A., Boddi, V., et al. (2001). Cyclooxygenase-2 pathway correlates with VEGF expression in head and neck cancer. Implications for tumor angiogenesis and metastasis. Neoplasia, 3(1), 53–61.
Greenhough, A., Smartt, H. J., Moore, A. E., Roberts, H. R., Williams, A. C., Paraskeva, C., et al. (2009). The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis, 30(3), 377–386.
Kaidi, A., Qualtrough, D., Williams, A. C., & Paraskeva, C. (2006). Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Research, 66(13), 6683–6691.
Kyzas, P. A., Stefanou, D., & Agnantis, N. J. (2005). COX-2 expression correlates with VEGF-C and lymph node metastases in patients with head and neck squamous cell carcinoma. Modern Pathology, 18(1), 153–160.
Su, J. L., Shih, J. Y., Yen, M. L., Jeng, Y. M., Chang, C. C., Hsieh, C. Y., et al. (2004). Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Research, 64(2), 554–564.
Chien, M. H., Ku, C. C., Johansson, G., Chen, M. W., Hsiao, M., Su, J. L., et al. (2009). Vascular endothelial growth factor-C (VEGF-C) promotes angiogenesis by induction of COX-2 in leukemic cells via the VEGF-R3/JNK/AP-1 pathway. Carcinogenesis, 30(12), 2005–2013.
Lala, P. K., Al-Mutter, N., & Orucevic, A. (1997). Effects of chronic indomethacin therapy on the development and progression of spontaneous mammary tumors in C3H/HEJ mice. International Journal of Cancer, 73(3), 371–380.
Yoshida, S., Amano, H., Hayashi, I., Kitasato, H., Kamata, M., Inukai, M., et al. (2003). COX-2/VEGF-dependent facilitation of tumor-associated angiogenesis and tumor growth in vivo. Laboratory Investigation, 83(10), 1385–1394.
Timoshenko, A. V., Chakraborty, C., Wagner, G. F., & Lala, P. K. (2006). COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. British Journal of Cancer, 94(8), 1154–1163.
Honn, K. V., Tang, D. G., Gao, X., Butovich, I. A., Liu, B., Timar, J., et al. (1994). 12-lipoxygenases and 12(S)-HETE: role in cancer metastasis. Cancer and Metastasis Reviews, 13(3–4), 365–396.
Nie, D., Hillman, G. G., Geddes, T., Tang, K., Pierson, C., Grignon, D. J., et al. (1998). Platelet-type 12-lipoxygenase in a human prostate carcinoma stimulates angiogenesis and tumor growth. Cancer Research, 58(18), 4047–4051.
Chatterjee, M., Das, S., Roy, K., & Chatterjee, M. (2011). Overexpression of 5-lipoxygenase and its relation with cell proliferation and angiogenesis in 7,12-dimethylbenz(alpha)anthracene-induced rat mammary carcinogenesis. Molecular Carcinogenesis. doi:10.1002/mc.21858.
Ye, Y. N., Liu, E. S., Shin, V. Y., Wu, W. K., & Cho, C. H. (2004). Contributory role of 5-lipoxygenase and its association with angiogenesis in the promotion of inflammation-associated colonic tumorigenesis by cigarette smoking. Toxicology, 203(1–3), 179–188.
Romano, M., & Claria, J. (2003). Cyclooxygenase-2 and 5-lipoxygenase converging functions on cell proliferation and tumor angiogenesis: implications for cancer therapy. The FASEB Journal, 17(14), 1986–1995.
Webler, A. C., Michaelis, U. R., Popp, R., Barbosa-Sicard, E., Murugan, A., Falck, J. R., et al. (2008). Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis. American Journal of Physiology. Cell Physiology, 295(5), C1292–C1301.
Michaelis, U. R., Fisslthaler, B., Barbosa-Sicard, E., Falck, J. R., Fleming, I., & Busse, R. (2005). Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. Journal of Cell Science, 118(Pt 23), 5489–5498.
Wang, Y., Wei, X., Xiao, X., Hui, R., Card, J. W., Carey, M. A., et al. (2005). Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. Journal of Pharmacology and Experimental Therapeutics, 314(2), 522–532.
Hyde, C. A., & Missailidis, S. (2009). Inhibition of arachidonic acid metabolism and its implication on cell proliferation and tumour-angiogenesis. International Immunopharmacology, 9(6), 701–715.
James, M. J., Gibson, R. A., & Cleland, L. G. (2000). Dietary polyunsaturated fatty acids and inflammatory mediator production. American Journal of Clinical Nutrition, 71(1 Suppl), 343S–348S.
Chen, Z. Y., & Istfan, N. W. (2000). Docosahexaenoic acid is a potent inducer of apoptosis in HT-29 colon cancer cells. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 63(5), 301–308.
Larsson, S. C., Kumlin, M., Ingelman-Sundberg, M., & Wolk, A. (2004). Dietary long-chain n−3 fatty acids for the prevention of cancer: a review of potential mechanisms. American Journal of Clinical Nutrition, 79(6), 935–945.
Ambring, A., Johansson, M., Axelsen, M., Gan, L., Strandvik, B., & Friberg, P. (2006). Mediterranean-inspired diet lowers the ratio of serum phospholipid n−6 to n−3 fatty acids, the number of leukocytes and platelets, and vascular endothelial growth factor in healthy subjects. American Journal of Clinical Nutrition, 83(3), 575–581.
Spencer, L., Mann, C., Metcalfe, M., Webb, M., Pollard, C., Spencer, D., et al. (2009). The effect of omega-3 FAs on tumour angiogenesis and their therapeutic potential. European Journal of Cancer, 45(12), 2077–2086.
Connor, K. M., SanGiovanni, J. P., Lofqvist, C., Aderman, C. M., Chen, J., Higuchi, A., et al. (2007). Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nature Medicine, 13(17589522), 868–873.
Kang, J. X., & Weylandt, K. H. (2008). Modulation of inflammatory cytokines by omega-3 fatty acids. Sub-Cellular Biochemistry, 49, 133–143.
De Caterina, R., & Massaro, M. (2005). Omega-3 fatty acids and the regulation of expression of endothelial pro-atherogenic and pro-inflammatory genes. Journal of Membrane Biology, 206(2), 103–116.
Weylandt, K. H., & Kang, J. X. (2005). Rethinking lipid mediators. The Lancet, 366(9486), 618–620.
Wan, J. B., Huang, L. L., Rong, R., Tan, R., Wang, J., & Kang, J. X. (2010). Endogenously decreasing tissue n−6/n−3 fatty acid ratio reduces atherosclerotic lesions in apolipoprotein E-deficient mice by inhibiting systemic and vascular inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology, 30(12), 2487–2494.
Schmocker, C., Weylandt, K. H., Kahlke, L., Wang, J., Lobeck, H., Tiegs, G., et al. (2007). Omega-3 fatty acids alleviate chemically induced acute hepatitis by suppression of cytokines. Hepatology, 45(4), 864–869.
Bilal, S., Haworth, O., Wu, L., Weylandt, K. H., Levy, B. D., & Kang, J. X. (2011). Fat-1 transgenic mice with elevated omega-3 fatty acids are protected from allergic airway responses. Biochimica et Biophysica Acta, 1812(9), 1164–1169.
Weylandt, K. H., Nadolny, A., Kahlke, L., Kohnke, T., Schmocker, C., Wang, J., et al. (2008). Reduction of inflammation and chronic tissue damage by omega-3 fatty acids in fat-1 transgenic mice with pancreatitis. Biochimica et Biophysica Acta, 1782(11), 634–641.
Jia, Q., Lupton, J. R., Smith, R., Weeks, B. R., Callaway, E., Davidson, L. A., et al. (2008). Reduced colitis-associated colon cancer in fat-1 (n−3 fatty acid desaturase) transgenic mice. Cancer Research, 68(18483285), 3985–3991.
White, P. J., Arita, M., Taguchi, R., Kang, J. X., & Marette, A. (2010). Transgenic restoration of long-chain n−3 fatty acids in insulin target tissues improves resolution capacity and alleviates obesity-linked inflammation and insulin resistance in high-fat-fed mice. Diabetes, 59(12), 3066–3073.
Wada, S., Yamazaki, T., Kawano, Y., Miura, S., & Ezaki, O. (2008). Fish oil fed prior to ethanol administration prevents acute ethanol-induced fatty liver in mice. Journal of Hepatology, 49(3), 441–450.
McCusker, M. M., & Grant-Kels, J. M. (2010). Healing fats of the skin: the structural and immunologic roles of the omega-6 and omega-3 fatty acids. Clinics in Dermatology, 28(4), 440–451.
Wright, S. A., O’Prey, F. M., McHenry, M. T., Leahey, W. J., Devine, A. B., Duffy, E. M., et al. (2008). A randomised interventional trial of omega-3-polyunsaturated fatty acids on endothelial function and disease activity in systemic lupus erythematosus. Annals of the Rheumatic Diseases, 67(6), 841–848.
Wang, D., & Dubois, R. N. (2010). Eicosanoids and cancer. Nature Reviews. Cancer, 10(20168319), 181–193.
Jackson, J. R., Seed, M. P., Kircher, C. H., Willoughby, D. A., & Winkler, J. D. (1997). The codependence of angiogenesis and chronic inflammation. The FASEB Journal, 11(6), 457–465.
Carmeliet, P., & Jain, R. K. (2000). Angiogenesis in cancer and other diseases. Nature, 407(6801), 249–257.
Voronov, E., Shouval, D. S., Krelin, Y., Cagnano, E., Benharroch, D., Iwakura, Y., et al. (2003). IL-1 is required for tumor invasiveness and angiogenesis. Proceedings of the National Academy of Sciences of the United States of America, 100(5), 2645–2650.
Firestein, G. S. (1999). Starving the synovium: angiogenesis and inflammation in rheumatoid arthritis. Journal of Clinical Investigation, 103(1), 3–4.
Storgard, C. M., Stupack, D. G., Jonczyk, A., Goodman, S. L., Fox, R. I., & Cheresh, D. A. (1999). Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist. Journal of Clinical Investigation, 103(1), 47–54.
MacLean, C. H., Newberry, S. J., Mojica, W. A., Khanna, P., Issa, A. M., Suttorp, M. J., et al. (2006). Effects of omega-3 fatty acids on cancer risk: a systematic review. JAMA : The Journal of the American Medical Association, 295(4), 403–415.
Terry, P., Bergkvist, L., Holmberg, L., & Wolk, A. (2001). No association between fat and fatty acids intake and risk of colorectal cancer. Cancer Epidemiology, Biomarkers & Prevention, 10(8), 913–914.
Godley, P. A., Campbell, M. K., Gallagher, P., Martinson, F. E., Mohler, J. L., & Sandler, R. S. (1996). Biomarkers of essential fatty acid consumption and risk of prostatic carcinoma. Cancer Epidemiology, Biomarkers & Prevention, 5(11), 889–895.
Ramon, J. M., Bou, R., Romea, S., Alkiza, M. E., Jacas, M., Ribes, J., et al. (2000). Dietary fat intake and prostate cancer risk: a case–control study in Spain. Cancer Causes & Control, 11(8), 679–685.
Leitzmann, M. F., Stampfer, M. J., Michaud, D. S., Augustsson, K., Colditz, G. C., Willett, W. C., et al. (2004). Dietary intake of n−3 and n−6 fatty acids and the risk of prostate cancer. American Journal of Clinical Nutrition, 80(1), 204–216.
Simonsen, N., van’t Veer, P., Strain, J. J., Martin-Moreno, J. M., Huttunen, J. K., & Navajas, J. F. (1998). Adipose tissue omega-3 and omega-6 fatty acid content and breast cancer in the EURAMIC study. European Community Multicenter Study on Antioxidants, Myocardial Infarction, and Breast Cancer. American Journal of Epidemiology, 147(4), 342–352.
Maillard, V., Bougnoux, P., Ferrari, P., Jourdan, M. L., Pinault, M., Lavillonniere, F., et al. (2002). N−3 and N−6 fatty acids in breast adipose tissue and relative risk of breast cancer in a case–control study in Tours, France. International Journal of Cancer, 98(1), 78–83.
Williams, C. D., Whitley, B. M., Hoyo, C., Grant, D. J., Iraggi, J. D., Newman, K. A., et al. (2011). A high ratio of dietary n−6/n−3 polyunsaturated fatty acids is associated with increased risk of prostate cancer. Nutrition Research, 31(1), 1–8.
Kang, J. X. (2007). Fat-1 transgenic mice: a new model for omega-3 research. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 77(5–6), 263–267.
Hudert, C. A., Weylandt, K. H., Lu, Y., Wang, J., Hong, S., Dignass, A., et al. (2006). Transgenic mice rich in endogenous omega-3 fatty acids are protected from colitis. Proceedings of the National Academy of Sciences of the United States of America, 103(30), 11276–11281.
Xia, S., Lu, Y., Wang, J., He, C., Hong, S., Serhan, C. N., et al. (2006). Melanoma growth is reduced in fat-1 transgenic mice: impact of omega-6/omega-3 essential fatty acids. Proceedings of the National Academy of Sciences of the United States of America, 103(33), 12499–12504.
Berquin, I. M., Min, Y., Wu, R., Wu, J., Perry, D., Cline, J. M., et al. (2007). Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. Journal of Clinical Investigation, 117(7), 1866–1875.
Griffitts, J., Saunders, D., Tesiram, Y. A., Reid, G. E., Salih, A., Liu, S., et al. (2010). Non-mammalian fat-1 gene prevents neoplasia when introduced to a mouse hepatocarcinogenesis model: omega-3 fatty acids prevent liver neoplasia. Biochimica et Biophysica Acta, 1801(10), 1133–1144.
Jia, Q., Lupton, J. R., Smith, R., Weeks, B. R., Callaway, E., Davidson, L. A., et al. (2008). Reduced colitis-associated colon cancer in fat-1 (n−3 fatty acid desaturase) transgenic mice. Cancer Research, 68(10), 3985–3991.
Kang, J. X., Wang, J., Wu, L., & Kang, Z. B. (2004). Transgenic mice: fat-1 mice convert n−6 to n−3 fatty acids. Nature, 427(6974), 504.
Lim, K., Han, C., Dai, Y., Shen, M., & Wu, T. (2009). Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Molecular Cancer Therapeutics, 8(11), 3046–3055.
Lu, Y., Nie, D., Witt, W. T., Chen, Q., Shen, M., Xie, H., et al. (2008). Expression of the fat-1 gene diminishes prostate cancer growth in vivo through enhancing apoptosis and inhibiting GSK-3 beta phosphorylation. Molecular Cancer Therapeutics, 7(10), 3203–3211.
Nowak, J., Weylandt, K. H., Habbel, P., Wang, J., Dignass, A., Glickman, J. N., et al. (2007). Colitis-associated colon tumorigenesis is suppressed in transgenic mice rich in endogenous n−3 fatty acids. Carcinogenesis, 28(9), 1991–1995.
Weylandt, K. H., Krause, L. F., Gomolka, B., Chiu, C. Y., Bilal, S., Nadolny, A., et al. (2011). Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-alpha. Carcinogenesis, 32(6), 897–903.
Giovannucci, E., Rimm, E. B., Stampfer, M. J., Colditz, G. A., Ascherio, A., & Willett, W. C. (1994). Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Annals of Internal Medicine, 121(4), 241–246.
Wallace, J. M. (2002). Nutritional and botanical modulation of the inflammatory cascade—eicosanoids, cyclooxygenases, and lipoxygenases—as an adjunct in cancer therapy. Integrative Cancer Therapies, 1(1), 7–37.
Gogos, C. A., Ginopoulos, P., Salsa, B., Apostolidou, E., Zoumbos, N. C., & Kalfarentzos, F. (1998). Dietary omega-3 polyunsaturated fatty acids plus vitamin E restore immunodeficiency and prolong survival for severely ill patients with generalized malignancy: a randomized control trial. Cancer, 82(2), 395–402.
Okamoto, Y., Okano, K., Izuishi, K., Usuki, H., Wakabayashi, H., & Suzuki, Y. (2009). Attenuation of the systemic inflammatory response and infectious complications after gastrectomy with preoperative oral arginine and omega-3 fatty acids supplemented immunonutrition. World Journal of Surgery, 33(9), 1815–1821.
Wigmore, S. J., Barber, M. D., Ross, J. A., Tisdale, M. J., & Fearon, K. C. (2000). Effect of oral eicosapentaenoic acid on weight loss in patients with pancreatic cancer. Nutrition and Cancer, 36(2), 177–184.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kang, J.X., Liu, A. The role of the tissue omega-6/omega-3 fatty acid ratio in regulating tumor angiogenesis. Cancer Metastasis Rev 32, 201–210 (2013). https://doi.org/10.1007/s10555-012-9401-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-012-9401-9