
Abstract
Cancer metastasis is a complex, dynamic process that begins with dissemination of cells from the primary tumor and culminates in the formation of clinically detectable, overt metastases at one or more discontinuous secondary sites. Evidence from in vivo video microscopy as well as PCR and immunohistochemical studies suggest that cancer cell dissemination is an early event in tumor progression and that cells may persist in a potentially dormant state for a prolonged period. Similarly, the mechanisms by which these disseminated cells initiate growth and complete the process of metastatic colonization remain largely unknown. Understanding signal transduction pathways regulating this final step of metastasis is therefore critical for successful clinical management. While genetic mutations or epigenetic changes may be required for a cell or group of cells to separate and survive distant from the primary tumor, the microenvironment within secondary tissues plays a substantial role in influencing whether disseminated cells survive and proliferate. Our work is focused on using metastasis suppressor proteins to gain insight into why the majority of disseminated cells, which should be fully malignant, do not proliferate immediately at secondary sites. The translational goal of this work is to identify targets for inhibiting metastatic growth and prolonging disease-free survival.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Metastatic disease—a critical clinical problem
This year 560,000 Americans will die from cancer. The majority of these patients will die from the consequences of metastatic disease [1]. Surgery and adjuvant therapies effectively control many localized cancers yet options for treating metastatic disease are limited and, for certain cancers such as hormone refractory prostate, only marginal improvements in patient survival can be achieved with conventional chemotherapy. Although many prostate cancers are believed to be organ-confined at the time of definitive local therapy, the majority of patients likely harbor viable tumor cells that have escaped from the primary tumor and reside at secondary sites. The prostate cancer recurrence rate after radical prostatectomy or radiotherapy is approximately 20–40% [2]. Similarly, ovarian cancer may appear to be confined to the ovaries, but once properly staged with removal of the omentum and regional lymph nodes, 30% of ovarian cancer patients will have microscopic metastatic disease. Unfortunately, the vast majority of ovarian cancers will be diagnosed when there is already grossly metastatic disease present and despite aggressive adjuvant chemotherapy, the majority of these patients will eventually succumb to their disease. Patients with ovarian cancer who are left with minimal residual tumor implants following surgery respond better to chemotherapy and live longer, indicating that microscopic/minimal residual implants may have a different biology and exhibit unique molecular targets for therapeutic intervention. Prostate and ovarian cancers are just two examples of the wider need for rational biological and molecular targets that effectively identify patients at risk for relapse, and for discovering avenues for therapeutic intervention.
2 Metastatic colonization—an important clinical target
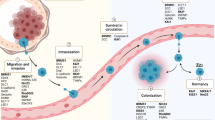
Cancer metastasis is a complex, dynamic process that begins with dissemination of cells from the primary tumor and culminates in the formation of clinically detectable, overt metastases at one or more discontinuous secondary sites. The impairment of any individual step in this cascade of in vivo events could interrupt the overall process. Metastatic colonization is the final step in this process and is defined as the lodging and subsequent growth of disseminated cancer cells into detectable metastases (Fig. 1) [3–5]. In addition to its well-characterized role in hematogenous and lymphatic routes of dissemination, metastatic colonization is also required when disseminated cells seed target sites within a body cavity, as in ovarian cancer, in which the majority of cancer cells disperse throughout the peritoneum to colonize surfaces of organs lined with mesothelium (Fig. 1). Multiple lines of evidence have shown that cancer cells can be detected at secondary sites even when the primary tumor was localized. Additionally experimental models have shown that the lodging and survival of single cells at secondary sites is a more efficient process than once believed [4]. Using in vivo video microscopy, Luzzi et al. observed that while 83% of B16F1 melanoma cells intravascularly injected into mice extravasated into the liver, the metastases that eventually formed originated from only 0.02% of cells originally injected [6]. Collectively, these data show that cancer cell dissemination is an early event and that cells may persist at secondary sites for extended periods of time before clinical presentation.

Metastatic colonization is the final step in the metastatic cascade. In order to become metastases, cells must proceed through an ordered set of changes beginning at the primary tumor. For hematogenous and lymphatic spread (bottom), epithelial tumor cells must invade through basement membrane, intravasate into the vasculature, and lodge at a secondary site such as the capillary beds of the lung (bottom right). To disseminate into a body cavity (top), epithelial tumor cells must be shed from the primary tumor, survive as disseminated cells, and adhere to and lodge in a secondary site such as on the mesothelium covering the surface of the omentum (top right)
Clinically and experimentally it is recognized that tumor formation and metastasis formation are distinct processes. This is illustrated by inherent differences in the biologies of specific cancers. For example, malignancies such as pancreatic cancer disseminate early in their natural history and give rise to relatively early metastases as demonstrated by the high incidence of occult metastases detected by immunohistochemistry and PCR in the lymph nodes, bone marrow, liver and peritoneal fluid [7]. In contrast, other cancers such as basal cell carcinomas in the skin are locally confined, invading normal structures of the epidermis, but rarely giving rise to distant metastases. Such differences raise challenging questions. What molecular and biological properties endow a cell with the capacity to leave a primary tumor and lodge at a distant site? What additional attributes are required for disseminated cells to survive, initiate growth, and form clinically significant metastases? Are additional mutational and epigenetic events required for disseminated cancer cells to survive and proliferate at secondary sites? What role do stochastic and societal interactions play in determining the response of cells to physical and environmental stresses encountered during the process of metastasis? This review will focus on the ways that a group of genes and proteins known as metastasis suppressors can be used to address these questions.
3 Metastasis suppressors—tools to modulate metastatic colonization
Proteins that regulate the ability of cancer cells to form metastases are playing a powerful role in identifying signaling pathways which can control metastatic growth and have the potential to uncover unique targets for therapeutic intervention and disease management. Since the first metastasis suppressor gene, NM23, was identified in 1988, 20 gene products with bona fide metastasis suppressor activity have been identified [8]. Historically, gene products that suppress metastasis have been defined on the basis of the ability of the ectopically expressed protein to impair the formation of macroscopic metastases without significantly impacting primary tumor growth in animal models [8]. Recently this definition has also been broadened to include gene products that suppress metastatic colonization in experimental metastasis assays. In these assays, no primary tumor forms, but instead cells are injected directly into the circulation or body cavity to colonize target organs, e.g. lung metastasis can be investigated by intravascular injection, and ovarian cancer metastasis is mimicked by intraperitoneal injection. Analogous to tumor suppressors, the class of gene products that impair the development of primary tumors, putative metastasis suppressors are identified by their reduced expression in metastatic tumor cell lines, compared with their expression in tumorigenic cell lines that are not capable of metastasizing in spontaneous metastasis animal models [3]. While loss of protein expression has been correlated with advancing tumor progression in clinical studies, the decreased activity of a protein, and not merely its presence or absence, is also an indicator of potential metastasis suppressors.
The metastasis suppressor proteins identified thus far represent a wide range of biological functions including stress-activated kinases, proteins that regulate gap junction formation and histidine kinases [8]. Metastasis suppressor proteins show both general and cell-type specific functions. While many of these proteins have been validated in multiple cell types, they also do not suppress metastasis in all contexts. For example, JNKK1/MKK4, hereafter referred to as JNKK1, has been identified as a suppressor of prostate and ovarian cancer metastasis, however JNKK1 is also required for development. Homozygous deletion of JNKK1 is lethal by embryonic day 14 [9] and accompanied by severe anemia, suggesting that JNKK1 is required for normal heptocyte proliferation and differentiation [10]. Data from in vivo studies in our own lab suggest that the biological context in which cancer cells reside plays an important role in the activation and function of these proteins [11, 12]. But what specific microenvironmental and or cellular factors determine the activation and biological outcome of a particular metastasis suppressor protein? Perhaps it is this aspect of metastasis suppressor proteins which is their greatest experimental utility. One can begin to dissect the complex cancer cell-host tissue interactions by injecting the cancer cells expressing the metastasis suppressor protein into different permissive host organs (i.e. organs in which the naïve cancer cells will grow). Outcomes using this approach will provide information regarding the tissue specificity of metastasis suppressor activation and function. Comparison of results from such studies can lead to a clearer understanding of the types of biological differences that have a real, measurable impact on metastatic ability. In the following sections we will discuss two examples that illustrate the importance of understanding both the molecular events downstream of a metastasis suppressor as well as the external and intracellular environments that dictate the function of a protein.
4 JNKK1/MKK4—all roads lead to suppression
The stress signaling kinase JNKK1 serves as an example of a metastasis suppressor protein that is able to regulate two signaling pathways to achieve the same in vivo outcome. JNKK1 is a mitogen activated protein (MAP) kinase that functions in the stress-activated protein kinase (SAPK) cascade. MAP kinases occupy a central position in cell growth, differentiation, and transformation (Fig. 2). To date, four MAP kinase modules have been well-characterized: extracellular signal-regulated protein kinase (ERK), ERK-5, c-Jun NH2-terminal protein kinase (JNK), and p38 [13]. Each cascade consists of a MAP3K, a MAP2K, and a MAPK. The ERK pathway is activated predominantly by mitogenic stimuli via Raf and MEK1, the MAP3K and MAP2K, respectively. The substrates of activated ERK include transcription factors such as c-Fos and Elk-1. In contrast, the JNK and p38 pathways are generally activated in response to stress stimuli.

MAP kinases are activated in signaling modules. MAP kinases activate their targets through a series of sequential phosphorylation events. In response to a stressor, a MAP3K such as MEKK1 phosphorylates a MAP2K such as JNKK1. The MAP2K then phosphorylates and activates a MAPK. JNKK1 can activate both of the stress-activated MAPKs, JNK and p38. The outcome of stress-activated protein kinase signaling depends on the stimulus and the cell-type, variously resulting in proliferation, apoptosis, or cell cycle arrest
JNKK1 is a dual-specificity serine/threonine kinase within the SAPK cascade, meaning that it is able to phosphorylate downstream MAPKs on both Tyrosine as well as Serine and Threonine residues. The SAPK pathway consists of the c-Jun NH2-terminal protein kinase (JNK) and p38 MAPK signaling modules [13]. The JNK signaling cascade consists of two MAP2Ks, JNKK1 and MKK7, while the p38 signaling cascade MAP2Ks include JNKK1, MKK3, and MKK6. The substrates of activated JNK and p38 include components of the AP-1 transcription factor, such as ATF-2 and c-Jun [13–15]. Therefore, activation of either of these pathways can initiate dramatic changes in gene expression. The biological outcome of MAPK activation depends on a variety of factors including cell type, cell environment, signal strength and duration, and subcellular localization of signaling proteins [16].
Using the AT6.1 rat prostatic cancer model in combination with positional cloning strategies we identified JNKK1 as a metastasis suppressor [17]. Ectopic expression of JNKK1 significantly reduced the number of overt surface lung metastases in a spontaneous metastasis assay [11]. Studies using the kinase inactive JNKK1 mutant (JNKK1-KR) further demonstrated that the kinase activity of JNKK1 is required for suppression of overt metastases and is sufficient to prolong animal survival. JNKK1 kinase activity was detected in prostate cells disseminated in the lung but not in cells of the primary tumor. While ectopic expression of MKK7, a JNK-specific kinase, suppressed the formation of metastases, MKK6, a p38-specific kinase, had no effect, suggesting that JNKK1 signals through JNK to suppress prostate cancer metastasis.
We also identified JNKK1 as a metastasis suppressor in a mouse model of ovarian cancer [12, 18]. When injected into the peritoneal cavity of mice, the human ovarian cancer cell line SKOv3ip.1 parallels many of characteristics of clinical ovarian cancer. Lesions have a papillary serous histology and a pattern metastatic of spread characteristic of clinical disease in which cells disseminate to peritoneal surfaces including the omentum and surfaces of the liver and mesentery of the bowel. Ectopically expressed JNKK1 in these cells significantly decreased the number of discrete metastatic lesions and increased the life span of the animals. In stark contrast to the prostate cancer model, however, ectopic expression of the p38-specific kinase MKK6 suppressed the formation of overt metastases while ectopic expression of the JNK-specific kinase MKK7 had no effect [12]. These data showed that JNKK1 suppresses metastatic colonization by signaling through p38 in SKOV3ip.1 cells.
While these data initially seem contradictory, understanding the molecular events downstream of the kinase points to a likely resolution of the conflict. At the same time, this interesting finding underscores the importance of epigenetic factors—both the differences that determine cell type specificity as well as features of the target microenviroment that activate stress kinase signaling—in determining the outcome of a cell that has left the primary tumor.
5 Downstream biochemistry—convergence on a common mechanism
One explanation for the similar biological outcome of JNKK1 activation in both of these model systems is the potential convergence of the downstream MAPKs on transcription of a common set of genes. A central consequence of MAPK activation is alterations in gene transcription [19]. Both JNK and p38 activate components of the AP-1 transcription factor, JNK by phosphorylating the AP-1 component c-Jun at Serines 63 and 73, and p38 by phosphorylating ATF-2 at Threonines 69 and 71, respectively [19, 20]. Because both proteins comprise the same transcription factor family, activating either one could have overlapping consequences on gene expression and a common cellular result—growth arrest of JNKK1 expressing cells.
AP-1 is not a single protein but rather a family of homodimeric or heterodimeric protein complexes formed by basic region-leucine zipper (bZIP) proteins of the Jun, Fos, Maf, and ATF families. The distinct composition of the AP-1 transcription factor complex influences the target genes. The originally identified dimers composing AP-1 were Jun:Jun, Jun:Fos, and Jun:Fra complexes that bind a seven base pair DNA consensus sequence known as a TRE [phorbol 12-O-tetradecanoate-13-acetate (TPA) response element] [21, 22]. Subsequently, ATF homodimers and ATF:Jun heterodimers were described. These preferentially bind cAMP responsive elements (CREs), an eight base pair DNA sequence that differs from TRE by one base. Therefore, the composition of AP-1 modulates the set of promoter elements that are preferentially bound when AP-1 is activated, influencing the set of target genes expressed in response to a given stimulus [21].
One recent study used DNA microarray technology and a high-throughput chromatin immunoprecipitation assay to validate promoters bound by c-Jun or ATF-2, in particular, in response to the DNA-damaging agent cisplatin in a human breast carcinoma cell line [23]. This study found 269 genes with promoters that were bound upon phosphorylation of ATF-2 and c-Jun following genotoxic stress. A subset of 121 genes was bound by both ATF-2 and c-Jun. This gene set includes candidates with established roles in cell survival and proliferation including DNA repair genes (such as RAD50, GADD45G, MSH2, MSH6, and ATM), apoptosis-associated genes (BCL2 and TRAF3), and regulators of cell cycle progression (CDK4, MYC, CDKN1B, CCND2, and CCNB1). Although this study did not evaluate protein expression for all genes identified by the screen, substantial evidence in the literature supports the finding that cell cycle regulation is an important mechanism by which JNK and p38 influence cell fate. Cyclin D1 [24], p53, p21Cip1, p16INK4A, p19ARF have all been identified as genes with protein products that are regulated by AP-1 [25], as well as genes encoding proteins involved in regulating the cytoskeleton or extracellular matrix [26, 27].
A rich and growing literature support a role for both p38 and JNK in cell survival, cell cycle regulation, and proliferation. While some reports have found that JNK activation is involved in Ras-induced oncogenic transformation and tumor development, other studies have found that JNK acts as a suppressor of transformation under these circumstances by eliminating transformed cells [28]. In addition to its known role in promoting apoptosis, JNK also has an established function in the G1/S transition as well as an emerging role in G2/M progression and cytokinesis [29]. The involvement of p38 in cell cycle progression is equally complex, activating cell cycle checkpoints in some systems and promoting progression through the cell cycle in others [30]. For example, p38 has been implicated in inducing the G2/M checkpoint, delaying entry into mitosis via Cdc25b inhibition. Furthermore, p38 activation can prevent anaphase entry by activating the spindle assembly checkpoint [31, 32]. Of particular interest is data demonstrating a role for p38 in arresting cells at cell cycle checkpoints following treatment with DNA damaging agents in the absence of p53 [33]. These reports in conjunction with our observations suggest that a single metastasis suppressor, JNKK1, can exert its effect on colonization through two different routes with the common mechanism of inhibiting proliferation of disseminated cells.
6 Metastasis and epigenetics—JNKK1 and cell type/microenvironment
The diversity of potential cellular responses due to JNKK1 signaling highlight a critical need to understand the role that cell type and extracellular environment have on a cancer cell. Many cancers exhibit a preference for metastasis formation within certain organs, for example breast and prostate cancers show a predilection for metastatic growth in the bone. Since the late 1800s biologists have been debating the mechanism of tumor cell trophism for secondary sites. In 1889 physician Stephen Paget proposed that a cancer cell, like a seed, requires a favorable “soil” provided by certain target organs in which to grow [34, 35]. This idea was promptly challenged in the 1920s by James Ewing, who argued that circulatory patterns accounted for the predilection of certain cancers to metastasize to specific organs. A considerable amount of evidence indicates that molecular factors present in specific organs can influence whether or not a particular tumor cell type will grow there. Although cancer cells are therefore said to “home” to specific organs, it is more likely, that this trophism is due to favorable growth conditions existing in the organ for the cell type [4]. This hypothesis could be interpreted as a combination of Ewing and Paget’s models for the susceptibility of secondary tissues to be colonized by metastatic cells. While mechanical factors and size restriction cause tumor cells or clusters to arrest in capillary beds adjacent to the primary tumor [4], the local environment including nutrient supply, chemokines and organ-specific molecular interactions may have a strong influence on the ability of a cell to survive and thrive in a particular location. It would follow, then, that the tissue where a disseminated cell arrests has a great deal to do with whether the cell is able to become a clinically detectable metastasis or remain indolent.
The extracellular signals that regulate the ability of disseminated cancer cells to avoid death and complete steps of metastatic colonization remain largely unknown. One group recently took a systems biology approach to test two different models for the mechanism of cell type specific sensitivities to apoptosis inducing TNFalpha in combination with IFN [36]. They hypothesized that the reason why some cells are more sensitive to apoptosis than others after the same treatment is either because different epithelial cell types have unique and separate transducers and effectors, or that, more simplistically, different cell types share a common effector network, termed “common processing,” but have different upstream mechanisms of transducing the signal. An algorithm derived from the kinase activities of apoptosis-insensitive HT-29 cells accurately predicted the apoptotic response of the sensitive HeLa cells within 92%, suggesting that different epithelial cells are likely to share common processing mechanisms. Therefore, cell type differences may be at the level of receiving input, rather than the downstream signaling components themselves. This could be interpreted to mean that cell extrinsic features, such as the extracellular environment and the mechanisms of cell–cell and cell–matrix interactions, may be responsible for the response of a given cell to a particular stimulus.
It remains unclear how disseminated cancer cells, which can persist at secondary sites, ultimately complete metastatic colonization. Our laboratory was the first to demonstrate biochemically the specific activation of a metastasis suppressor at a secondary site [11]. This observation raised the important question of how JNKK1 is activated in disseminated cancer cells. Once again, much can be learned from the incongruities between the prostate and ovarian cancer animal model systems. JNK and p38 can be activated by many external stimuli including DNA-alkylating chemotherapeutic agents, ultraviolet and ionizing radiation, and inflammatory cytokines. In addition, mechanical and chemical stresses such as morphological changes in the extracellular space and osmotic stress induce signaling through JNK and p38 pathways. After leaving the primary tumor, cancer cells must adapt to changing environments in order to survive and proliferate in secondary organ sites. The route of tumor spread, depending on tumor type, may be intravascular, lymphatic or by seeding of a body cavity, such as peritoneal spread in ovarian cancer. Cells that travel intravascularly may adhere to platelets or leukocytes in the bloodstream and form emboli that lodge in secondary organs. Disseminated cancer cells may also adhere to vascular endothelial cells and extravasate into the organ tissue and proliferate. All of these steps may act as potential stressors differentially activating MAP kinase family members.
Cell–cell or cell–matrix interactions by cancer cells with host tissue cells at target sites are likely to play and important role in cell survival and subsequent growth. Cell surface proteins, such as integrins and selectins, have been implicated in the metastatic process and may signal through the JNK and p38 pathways [37]. For example, evidence suggests that cell-extracellular interactions can be transduced to p38 by a mechanism that involves integrins and fibronectin [38]. Further evidence that JNK and p38 pathways play a role in cell–environmental interactions includes the finding that p38 may be required for TGF-beta-induced epithelial–mesenchymal transition [39]. The outcome of adhesion-mediated JNK and p38 activation may be increased transcription of target genes that regulate cell cycle and cell survival.
7 Concluding remarks
Even though research over the past decade has resulted in the improved treatment of primary tumors, there are great strides to be made in the treatment of metastatic disease [40]. Uncovering the reasons disseminated cells, which should be fully malignant, do not grow immediately at secondary sites may provide targets for inhibiting metastatic growth. Indolence of these cells can be temporary—what leads quiescent or non-proliferating cells to eventually grow into clinically relevant disease is still unknown. Metastasis suppressor proteins, such as JNKK1, are providing biochemical and molecular tools to dissect key steps in metastatic colonization. This clinically important last step of cancer progression is no longer a black box. Fundamental information on the mechanisms regulating metastatic colonization is required to make an impact in the clinic. If we understand the processes involved in quiescence perhaps we can explore therapeutic options that favor stimulating pathways that keep cancer cells dormant, or, conversely, inhibit pathways that lead to their eventual outgrowth.
References
Steeg, P. S. (2006). Tumor metastasis: Mechanistic insights and clinical challenges. Nature Medicine, 12, 895–904.
Ward, J. F., & Moul, J. W. (2005). Rising prostate-specific antigen after primary prostate cancer therapy. Nature Clinical Practice. Urology, 2, 174–182.
Steeg, P. (2003). Metastasis suppressors alter the signal transduction of cancer cells. Nature Reviews. Cancer, 3, 55–63.
Chambers, A. F., Groom, A. C., & MacDonald, I. C. (2002). Dissemination and growth of cancer cells in metastatic sites. Nature Reviews. Cancer, 2, 563–572.
Kauffman, E. C., Robinson, V. L., Stadler, W. M., Sokoloff, M. H., & Rinker-Schaeffer, C. W. (2003). Metastasis suppression: The evolving role of metastasis suppressor genes for regulating cancer cell growth at the secondary site. Journal of Urology, 169, 1122–1133.
Luzzi, K. J., MacDonald, I. C., Schmidt, E. E., Kerkvliet, N., Morris, V. L., Chambers, A. F., et al. (1998). Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. American Journal of Pathology, 153, 865–873.
Nakao, A., Fujii, T., Sugimoto, H., Kanazumi, N., Nomoto, S., Kodera, Y., et al. (2006). Oncological problems in pancreatic cancer surgery. World Journal of Gastroenterology, 12, 4466–4472.
Rinker-Schaeffer, C. W., O’Keefe, J. P., Welch, D. R., & Theodorescu, D. (2006). Metastasis suppressor proteins: Discovery, molecular mechanisms, and clinical application. Clinical Cancer Research, 12, 3882–3889.
Yang, D., Tournier, C., Wysk, M., Lu, H. T., Xu, J., Davis, R. J., et al. (1997). Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional activity. Proceedings of the National Academy of Sciences of the United States of America, 94, 3004–3009.
Ganiatsas, S., Kwee, L., Fujiwara, Y., Perkins, A., Ikeda, T., & Labow, M. A. (1998). SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proceedings of the National Academy of Sciences of the United States of America, 95, 6881–6886.
Vander Griend, D. J., Kocherginsky, M., Hickson, J. A., Stadler, W. M., Lin, A., & Rinker-Schaeffer, C. W. (2005). Suppression of metastatic colonization by the context-dependent activation of the c-Jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Research, 65, 10984–10991.
Hickson, J. A., Huo, D., Vander Griend, D. J., Lin, A., Rinker-Schaeffer, C. W., & Yamada, S. D. (2006). The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Research, 66, 2264–2270.
Kyriakis, J. M., & Avruch, J. (2001). Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiological Reviews, 81, 807–869.
Pearson, G., Robinson, F., Beers Gibson, T., Xu, B. E., Karandikar, M., Berman, K., et al. (2001). Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocrine Reviews, 22, 153–183.
Ip, Y. T., & Davis, R. J. (1998). Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Current Opinion in Cell Biology, 10, 205–219.
Lin, A. (2003). Activation of the JNK signaling pathway: Breaking the brake on apoptosis. Bioessays, 25, 17–24.
Yoshida, B. A., Dubauskas, Z., Chekmareva, M. A., Christiano, T. R., Stadler, W. M., & Rinker-Schaeffer, C. W. (1999). Mitogen-activated protein kinase kinase 4/stress-activated protein/Erk kinase 1 (MKK4/SEK1), a prostate cancer metastasis suppressor gene encoded by human chromosome 17. Cancer Research, 59, 5483–5487.
Yamada, S. D., Hickson, J. A., Hrobowski, Y., Vander Griend, D. J., Benson, D., Montag, A., et al. (2002). Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Research, 62, 6717–6723.
Davis, R. J. (1999). Signal transduction by the c-Jun N-terminal kinase. Biochemical Society Symposium, 64, 1–12.
Karin, M., Liu, Z., & Zandi, E. (1997). AP-1 function and regulation. Current Opinion in Cell Biology, 9, 240–246.
van Dam, H., & Castellazzi, M. (2001). Distinct roles of Jun:Fos and Jun:ATF dimers in oncogenesis. Oncogene, 20, 2453–2464.
Shaulian, E., & Karin, M. (2001). AP-1 in cell proliferation and survival. Oncogene, 20, 2390–2400.
Hayakawa, J., Mittal, S., Wang, Y., Korkmaz, K. S., Adamson, E., English, C., et al. (2004). Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Molecular Cell, 16, 521–535.
Lavoie, J. N., L’Allemain, G., Brunet, A., Muller, R., & Pouyssegur, J. (1996). Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. Journal of Biological Chemistry, 271, 20608–20616.
Shaulian, E., & Karin, M. (2002). AP-1 as a regulator of cell life and death. Natural Cell Biology, 4, E131–136.
Vogt, P. K. (2001). Jun, the oncoprotein. Oncogene, 20, 2365–2377.
Rinehart-Kim, J., Johnston, M., Birrer, M., & Bos, T. (2000). Alterations in the gene expression profile of MCF-7 breast tumor cells in response to c-Jun. International Journal of Cancer, 88, 180–190.
Kennedy, N. J., & Davis, R. J. (2003). Role of JNK in tumor development. Cell Cycle, 2, 199–201.
Shim, J., Lee, H., Park, J., Kim, H., & Choi, E. J. (1996). A non-enzymatic p21 protein inhibitor of stress-activated protein kinases. Nature, 381, 804–806.
MacCorkle, R. A., & Tan, T. H. (2005). Mitogen-activated protein kinases in cell-cycle control. Cell Biochemistry and Biophysics, 43, 451–461.
Mikhailov, A., Shinohara, M., & Rieder, C. L. (2005). The p38-mediated stress-activated checkpoint. A rapid response system for delaying progression through antephase and entry into mitosis. Cell Cycle, 4, 57–62.
Takenaka, K., Moriguchi, T., & Nishida, E. (1998). Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science, 280, 599–602.
Reinhardt, H. C., Aslanian, A. S., Lees, J. A., & Yaffe, M. B. (2007). p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell, 11, 175–189.
Paget, S. (1889). The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Reviews, 8, 98–101.
Paget, S. (1989). The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Reviews, 8, 98–101.
Miller-Jensen, K., Janes, K. A., Brugge, J. S., & Lauffenburger, D. A. (2007). Common effector processing mediates cell-specific responses to stimuli. Nature, 448, 604–608.
Guo, W., & Giancotti, F. G. (2004). Integrin signalling during tumour progression. Nature Reviews. Molecular Cell Biology, 5, 816–826.
Ossowski, L., & Aguirre-Ghiso, J. A. (2000). Urokinase receptor and integrin partnership: Coordination of signaling for cell adhesion, migration and growth. Current Opinion in Cell Biology, 12, 613–620.
Zavadil, J., & Bottinger, E. P. (2005). TGF-beta and epithelial-to-mesenchymal transitions. Oncogene, 24, 5764–5774.
Jemal, A., Murray, T., Ward, E., Samuels, A., Tiwari, R. C., Ghafoor, A., et al. (2005). Cancer statistics, 2005. CA Cancer Journal for Clinician, 55, 10–30.
Acknowledgements
We express our appreciation to Dr. Russell Szmulewitz for his critical and thoughtful reading of our manuscript. We thank Dr. Arieh Shalhav, Dr. Charles Brendler and the University of Chicago Section of Urology for their strong and unwavering support our metastasis suppressor protein studies. This work supported by The University of Chicago RESCUE Fund (CWR-S); DOD Ovarian Cancer Research Grant DAMD17-03-1-0169 (JH, DY), Grant RO1 CA 89569 (CWR-S, JH), DOD Ovarian Cancer Research Grant W81XWH-06-1-0041 (CWR-S), Gynecologic Cancer Foundation/Ann Schreiber Ovarian Cancer Research Grant (JH, CWR-S), support from the Department of Pathology (TL) and Graduate Training in Growth and Development T32 HD07009 (JLT).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Taylor, J., Hickson, J., Lotan, T. et al. Using metastasis suppressor proteins to dissect interactions among cancer cells and their microenvironment. Cancer Metastasis Rev 27, 67–73 (2008). https://doi.org/10.1007/s10555-007-9106-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-007-9106-7